Post-Exposure Protection in Mice against Sudan Virus by a Two Antibody Cocktail

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Macaque Immunization
2.2. Construction and Screening of the Anti-SUDV Antibody Gene Library
2.3. Library Packaging
2.4. Affinity and Cell Based Neutralization
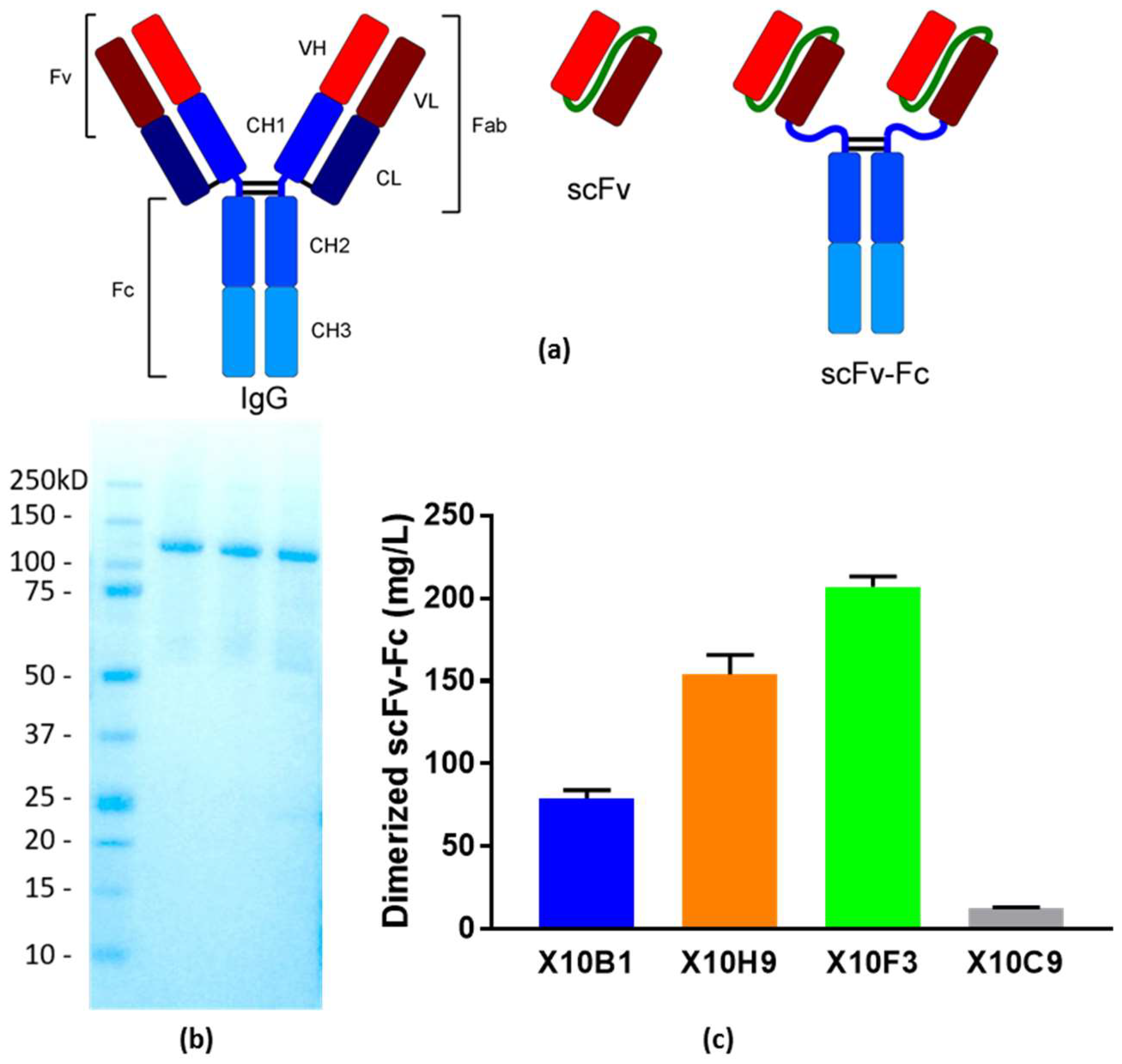
2.5. Cell-Free scFv-Fc Production and Purification
2.6. Murine Protection Studies
3. Results
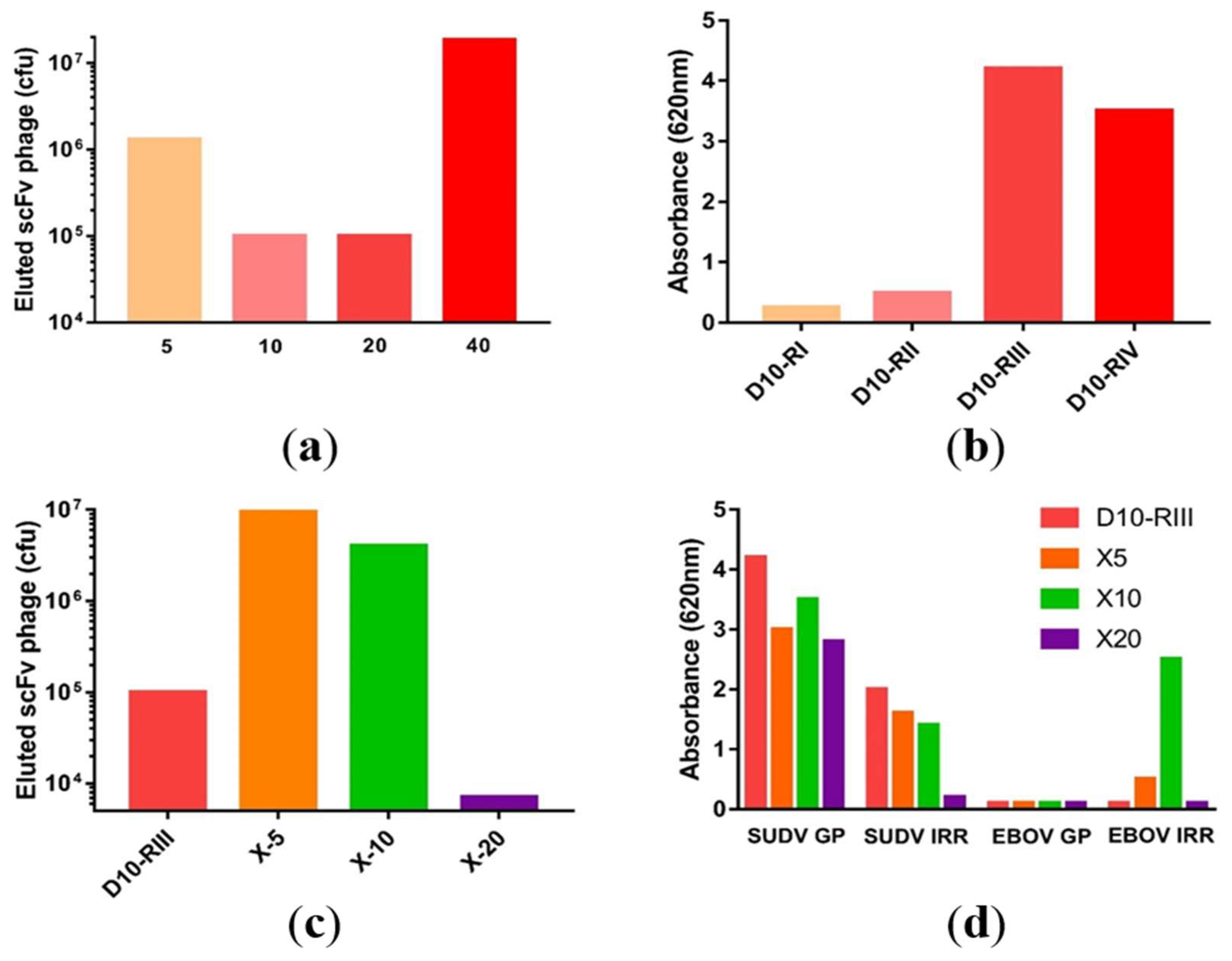
3.1. Macaque Immunization and Antibody Generation
3.2. Library Construction and Isolation of scFvs Specific to SUDV-GP
3.3. Antibody Recovery and Characterization
3.4. Cell-Free Production Platform for Rapid scFv-Fc Antibody Production
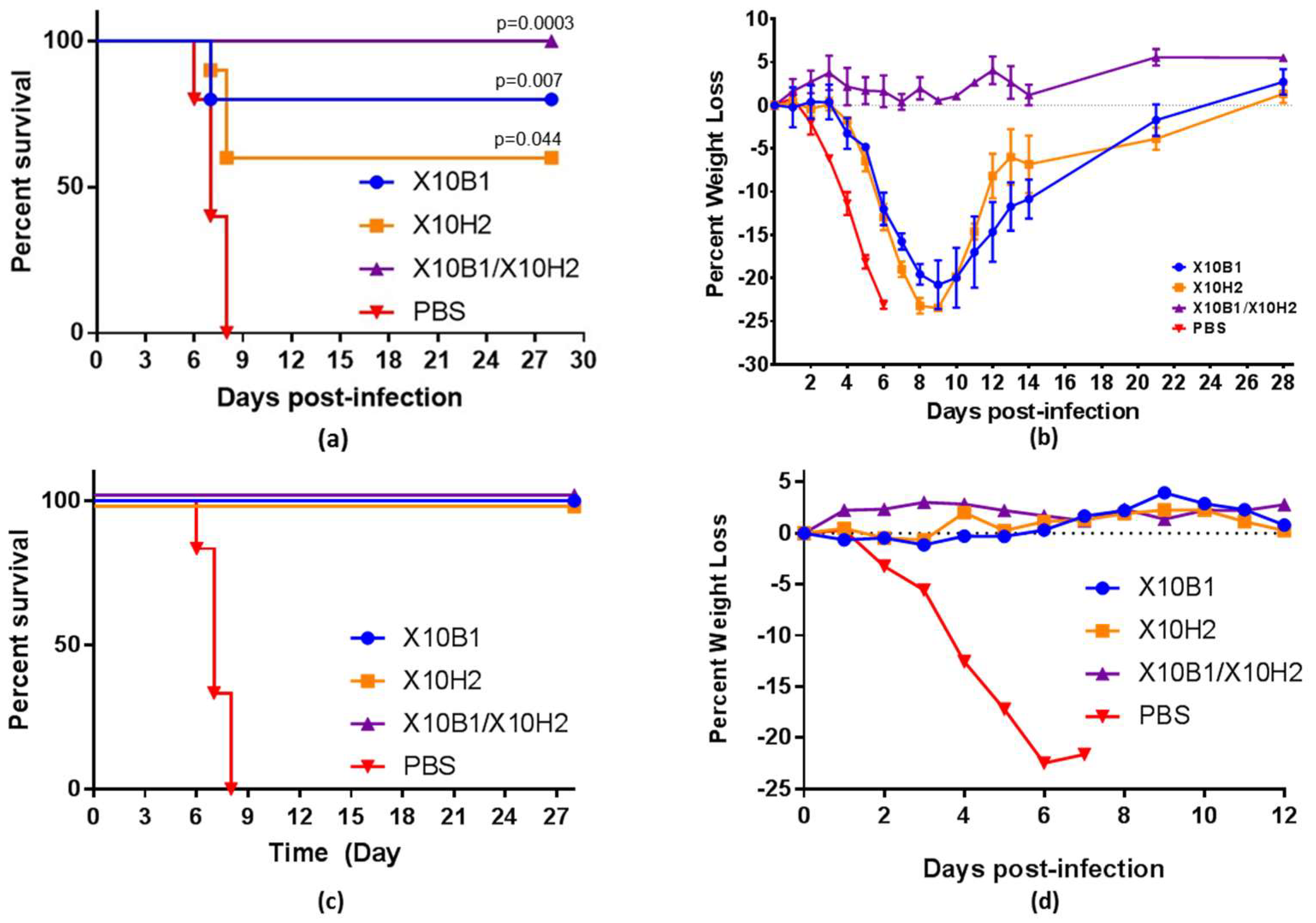
3.5. In Vivo Murine Protection
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kuhn, R. Togaviridae: The Viruses and Their Replication. In Fields Virology, 5th ed.; Knipe, D.M., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Feldmann, H.; Geisbert, T.W. Ebola haemorrhagic fever. Lancet 2011, 377, 849–862. [Google Scholar] [CrossRef]
- Feldmann, H.; Bugany, H.; Mahner, F.; Klenk, H.D.; Drenckhahn, D.; Schnittler, H.J. Filovirus-induced endothelial leakage triggered by infected monocytes/macrophages. J. Virol. 1996, 70, 2208–2214. [Google Scholar] [PubMed]
- Lee, J.E.; Saphire, E.O. Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 2009, 4, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Wong, G.; Audet, J.; Bello, A.; Fernando, L.; Alimonti, J.B.; Fausther-Bovendo, H.; Wei, H.; Aviles, J.; Hiatt, E.; et al. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature 2014, 514, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Froude, J.W.; Stiles, B.; Pelat, T.; Thullier, P. Antibodies for biodefense. MAbs 2011, 3, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Howell, K.A.; Qiu, X.; Brannan, J.M.; Bryan, C.; Davidson, E.; Holtsberg, F.W.; Wec, A.Z.; Shulenin, S.; Biggins, J.E.; Douglas, R.; et al. Antibody Treatment of Ebola and Sudan Virus Infection via a Uniquely Exposed Epitope within the Glycoprotein Receptor-Binding Site. Cell Rep. 2016, 15, 1514–1526. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Parren, P.W.; Sanchez, A.; Rensink, I.; Rodriguez, L.L.; Khan, A.S.; Peters, C.J.; Burton, D.R. Recombinant human monoclonal antibodies to Ebola virus. J. Infect. Dis. 1999, 179 (Suppl. 1), S235–S239. [Google Scholar] [CrossRef] [PubMed]
- Cross, R.W.; Mire, C.E.; Feldmann, H.; Geisbert, T.W. Post-exposure treatments for Ebola and Marburg virus infections. Nat. Rev. Drug Discov. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Koellhoffer, J.F.; Zak, S.E.; Frei, J.C.; Liu, N.; Long, H.; Ye, W.; Nagar, K.; Pan, G.; Chandran, K.; et al. Synthetic antibodies with a human framework that protect mice from lethal Sudan ebolavirus challenge. ACS Chem. Biol. 2014, 9, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Wec, A.Z.; Herbert, A.S.; Murin, C.D.; Nyakatura, E.K.; Abelson, D.M.; Fels, J.M.; He, S.; James, R.M.; de La Vega, M.A.; Zhu, W.; et al. Antibodies from a Human Survivor Define Sites of Vulnerability for Broad Protection against Ebolaviruses. Cell 2017, 169, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Ohimain, E.I. Recent advances in the development of vaccines for Ebola virus disease. Virus Res. 2016, 211, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Marzi, A.; Feldmann, H. Ebola virus vaccines: An overview of current approaches. Expert Rev. Vaccines 2014, 13, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Bradfute, S.B.; Dye, J.M., Jr.; Bavari, S. Filovirus vaccines. Hum. Vaccines 2011, 7, 701–711. [Google Scholar] [CrossRef]
- Froude, J.W.; Pelat, T.; Miethe, S.; Zak, S.E.; Wec, A.Z.; Chandran, K.; Brannan, J.M.; Bakken, R.R.; Hust, M.; Thullier, P.; et al. Generation and characterization of protective antibodies to Marburg virus. MAbs 2017, 9, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Pushko, P.; Bray, M.; Ludwig, G.V.; Parker, M.; Schmaljohn, A.; Sanchez, A.; Jahrling, P.B.; Smith, J.F. Recombinant RNA replicons derived from attenuated Venezuelan equine encephalitis virus protect guinea pigs and mice from Ebola hemorrhagic fever virus. Vaccine 2000, 19, 142–153. [Google Scholar] [CrossRef]
- Hevey, M.; Negley, D.; Pushko, P.; Smith, J.; Schmaljohn, A. Marburg virus vaccines based upon alphavirus replicons protect guinea pigs and nonhuman primates. Virology 1998, 251, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Pelat, T.; Hust, M.; Thullier, P. Obtention and engineering of non-human primate (NHP) antibodies for therapeutics. Mini Rev. Med. Chem. 2009, 9, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, A.; Kugler, J.; Wilke, S.; Schirrmann, T.; Hust, M. Construction of human antibody gene libraries and selection of antibodies by phage display. Methods Mol. Biol. 2014, 1060, 215–243. [Google Scholar] [PubMed]
- Rülker, T.; Voß, L.; Thullier, P.; O’Brien, L.M.; Pelat, T.; Perkins, S.D.; Langermann, C.; Schirrmann, T.; Dübel, S.; Marschall, H.J.; et al. Isolation and characterisation of a human-like antibody fragment (scFv) that inactivates VEEV in vitro and in vivo. PLoS ONE 2012, 7, e37242. [Google Scholar] [CrossRef] [PubMed]
- Schutte, M.; Thullier, P.; Pelat, T.; Wezler, X.; Rosenstock, P.; Hinz, D.; Kirsch, M.I.; Hasenberg, M.; Frank, R.; Schirrmann, T.; et al. Identification of a putative Crf splice variant and generation of recombinant antibodies for the specific detection of Aspergillus fumigatus. PLoS ONE 2009, 4, e6625. [Google Scholar] [CrossRef] [PubMed]
- Miethe, S.; Rasetti-Escargueil, C.; Avril, A.; Liu, Y.; Chahboun, S.; Korkeala, H.; Mazuet, C.; Popoff, M.R.; Pelat, T.; Thullier, P.; et al. Development of Human-Like scFv-Fc Neutralizing Botulinum Neurotoxin E. PLoS ONE 2015, 10, e0139905. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.D.R. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Springs Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Rondot, S.; Koch, J.; Breitling, F.; Dubel, S. A helper phage to improve single-chain antibody presentation in phage display. Nat. Biotechnol. 2001, 19, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Soltes, G.; Hust, M.; Ng, K.K.; Bansal, A.; Field, J.; Stewart, D.I.; Dubel, S.; Cha, S.; Wiersma, E.J. On the influence of vector design on antibody phage display. J. Biotechnol. 2007, 127, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Moe, J.B.; Lambert, R.D.; Lupton, H.W. Plaque assay for Ebola virus. J. Clin. Microbiol. 1981, 13, 791–793. [Google Scholar] [PubMed]
- Zawada, J.F.; Yin, G.; Steiner, A.R.; Yang, J.; Naresh, A.; Roy, S.M.; Gold, D.S.; Heinsohn, H.G.; Murray, C.J. Microscale to manufacturing scale-up of cell-free cytokine production—A new approach for shortening protein production development timelines. Biotechnol. Bioeng. 2011, 108, 1570–1578. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Garces, E.D.; Yang, J.; Zhang, J.; Tran, C.; Steiner, A.R.; Roos, C.; Bajad, S.; Hudak, S.; Penta, K.; et al. Aglycosylated antibodies and antibody fragments produced in a scalable in vitro transcription-translation system. mAbs 2012, 4, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Abrahams, C.; Embry, M.; Yu, A.; Kahana, J.; Brown, M.; Narla, R.K.; Barnes, L.; Schwartz, E.; Boylan, J.; et al. Targeting CD74 with Novel Antibody Drug Conjugates (ADCs) for the Treatment of B-Cell Non-Hodgkin’s Lymphoma (NHL). Blood 2016, 128, 464. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Affinity (nM) | Neutralization Screen 1 | |||
|---|---|---|---|---|
| Antibody | SUDV | EBOV | SUDV | EBOV |
| X10B1 | 17.3 | 13.3 | +++ | + |
| X10B6 | 8.3 | 5.9 | +++ | - |
| X10H2 | 7.0 | ND 2 | +++ | + |
| X10F3 | 61.0 | ND 2 | +++ | + |
| X10C9 | 12.0 | ND 2 | +++ | - |
| X10H4 | 8.5 | 6.3 | +++ | - |
| X10G8 | 42.0 | ND 2 | +++ | ND 2 |
| X10H11 | 20.0 | 26.1 | +++ | + |
| X10H12 | 8.9 | 13.1 | + | - |
| X20C3 | 14.0 | 8.5 | - | - |
| X20D6 | 18.7 | 32.0 | +++ | ND 2 |
| X20F12 | 14.0 | 12.4 | - | - |
| X20A4 | 9.0 | 5.9 | - | ND 2 |
| X20A9 | 29.0 | 9.0 | - | ND 2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Froude, J.W.; Herbert, A.S.; Pelat, T.; Miethe, S.; Zak, S.E.; Brannan, J.M.; Bakken, R.R.; Steiner, A.R.; Yin, G.; Hallam, T.J.; et al. Post-Exposure Protection in Mice against Sudan Virus by a Two Antibody Cocktail. Viruses 2018, 10, 286. https://doi.org/10.3390/v10060286
Froude JW, Herbert AS, Pelat T, Miethe S, Zak SE, Brannan JM, Bakken RR, Steiner AR, Yin G, Hallam TJ, et al. Post-Exposure Protection in Mice against Sudan Virus by a Two Antibody Cocktail. Viruses. 2018; 10(6):286. https://doi.org/10.3390/v10060286
Chicago/Turabian StyleFroude, Jeffrey W., Andrew S. Herbert, Thibaut Pelat, Sebastian Miethe, Samantha E. Zak, Jennifer M. Brannan, Russell R. Bakken, Alexander R. Steiner, Gang Yin, Trevor J. Hallam, and et al. 2018. "Post-Exposure Protection in Mice against Sudan Virus by a Two Antibody Cocktail" Viruses 10, no. 6: 286. https://doi.org/10.3390/v10060286
APA StyleFroude, J. W., Herbert, A. S., Pelat, T., Miethe, S., Zak, S. E., Brannan, J. M., Bakken, R. R., Steiner, A. R., Yin, G., Hallam, T. J., Sato, A. K., Hust, M., Thullier, P., & Dye, J. M. (2018). Post-Exposure Protection in Mice against Sudan Virus by a Two Antibody Cocktail. Viruses, 10(6), 286. https://doi.org/10.3390/v10060286