The E. coli Global Regulator DksA Reduces Transcription during T4 Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phage and Strains
2.2. Quantification of Plaque Size
2.3. Burst Size Analysis
2.4. Purification of RNA
2.5. RNA-seq
2.6. Primer Extension Analyses
2.7. RT-qPCR
2.8. Proteins
2.9. In Vitro Transcription
3. Results
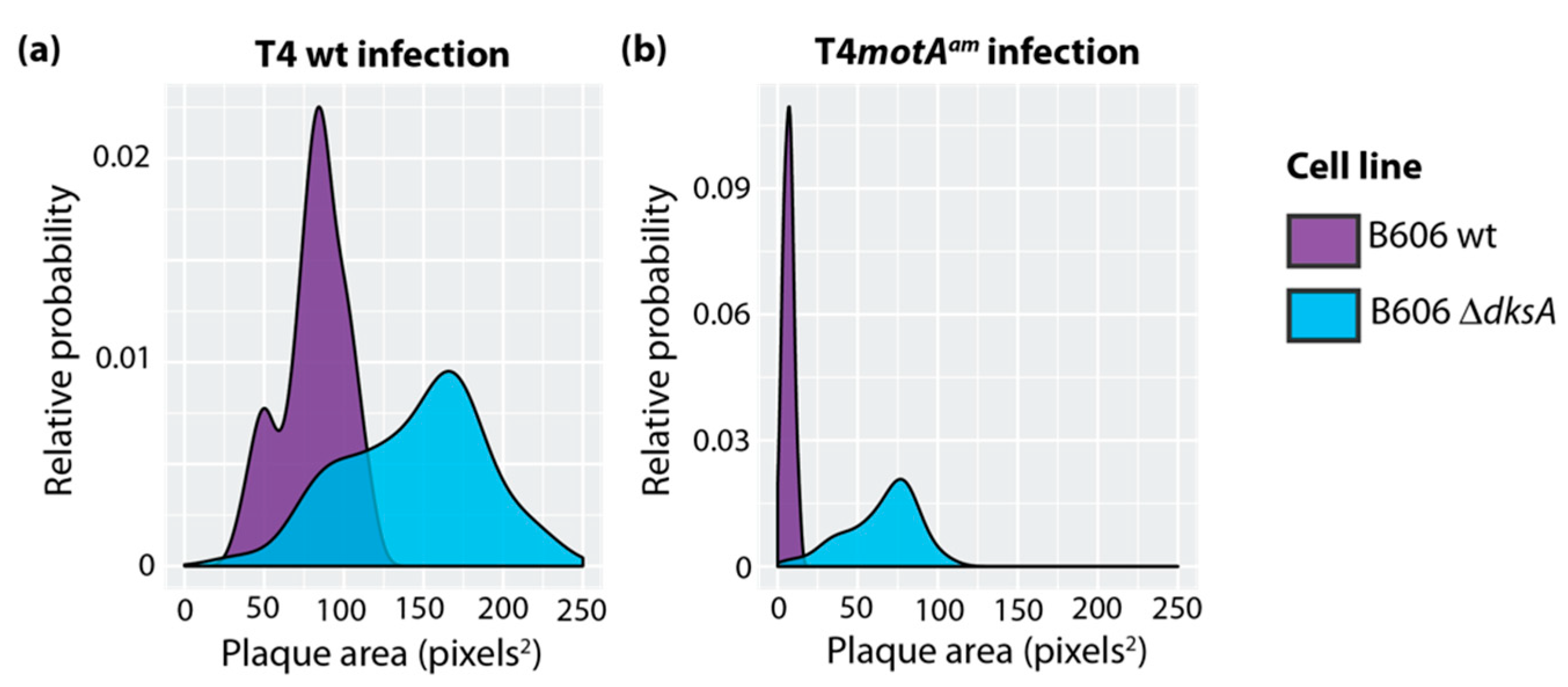
3.1. T4 Infections in the Absence of ppGpp or DksA Produce Larger Plaques
3.2. Deletion of dksA Results in an Increase in Phage Progeny per Infection
3.3. Deletion of dksA Results in a Shorter Latent Period in a T4motAam Infection
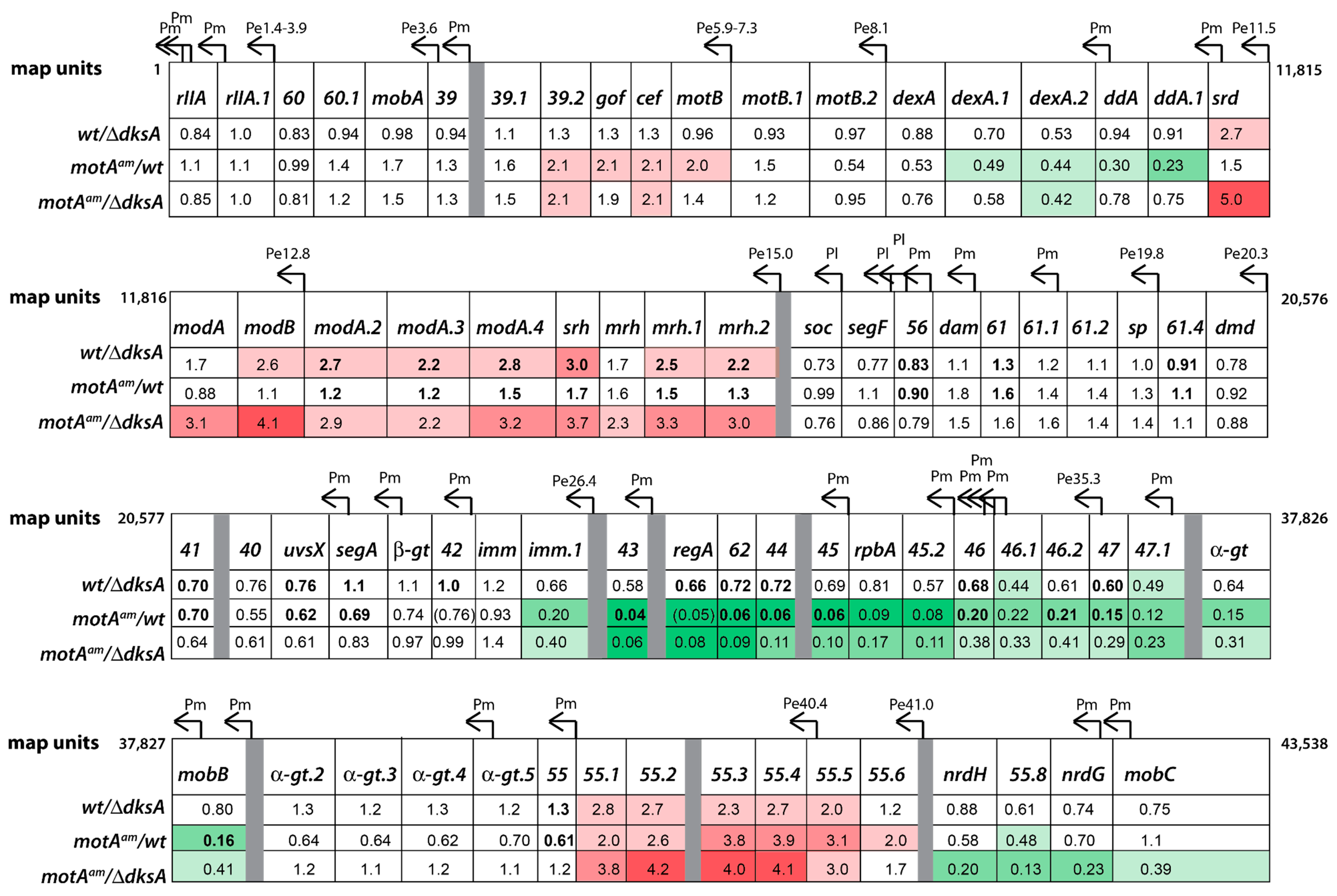
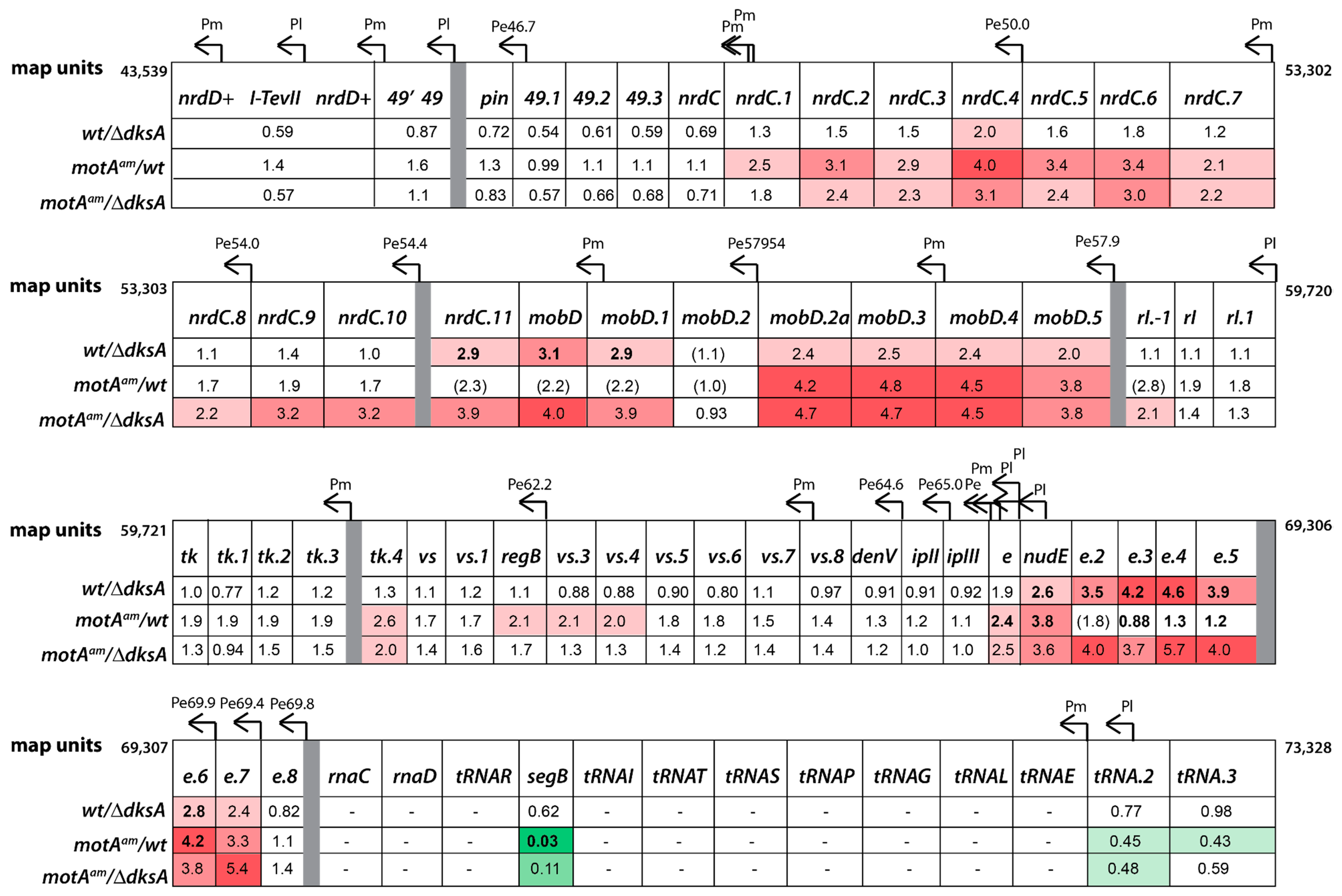
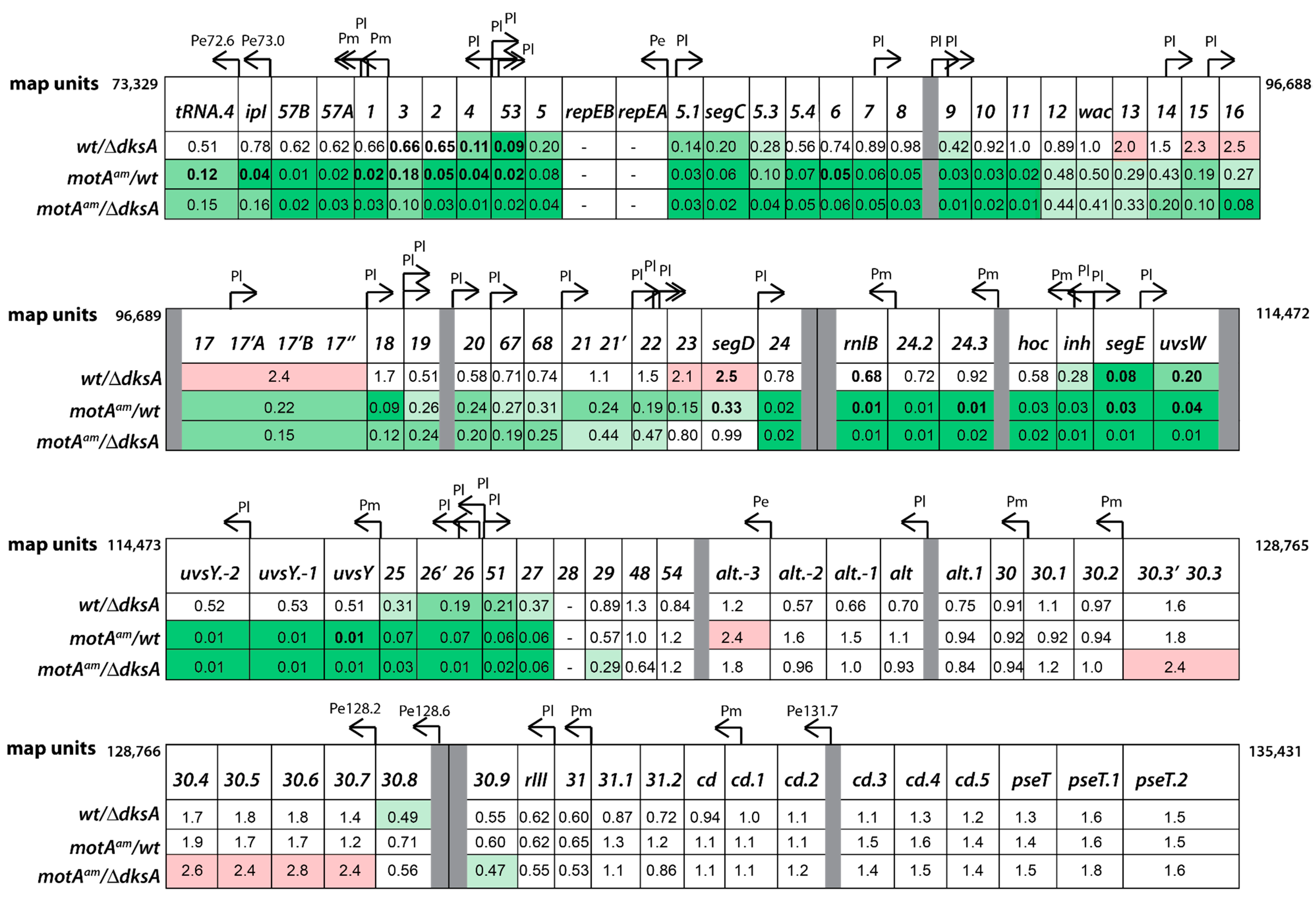
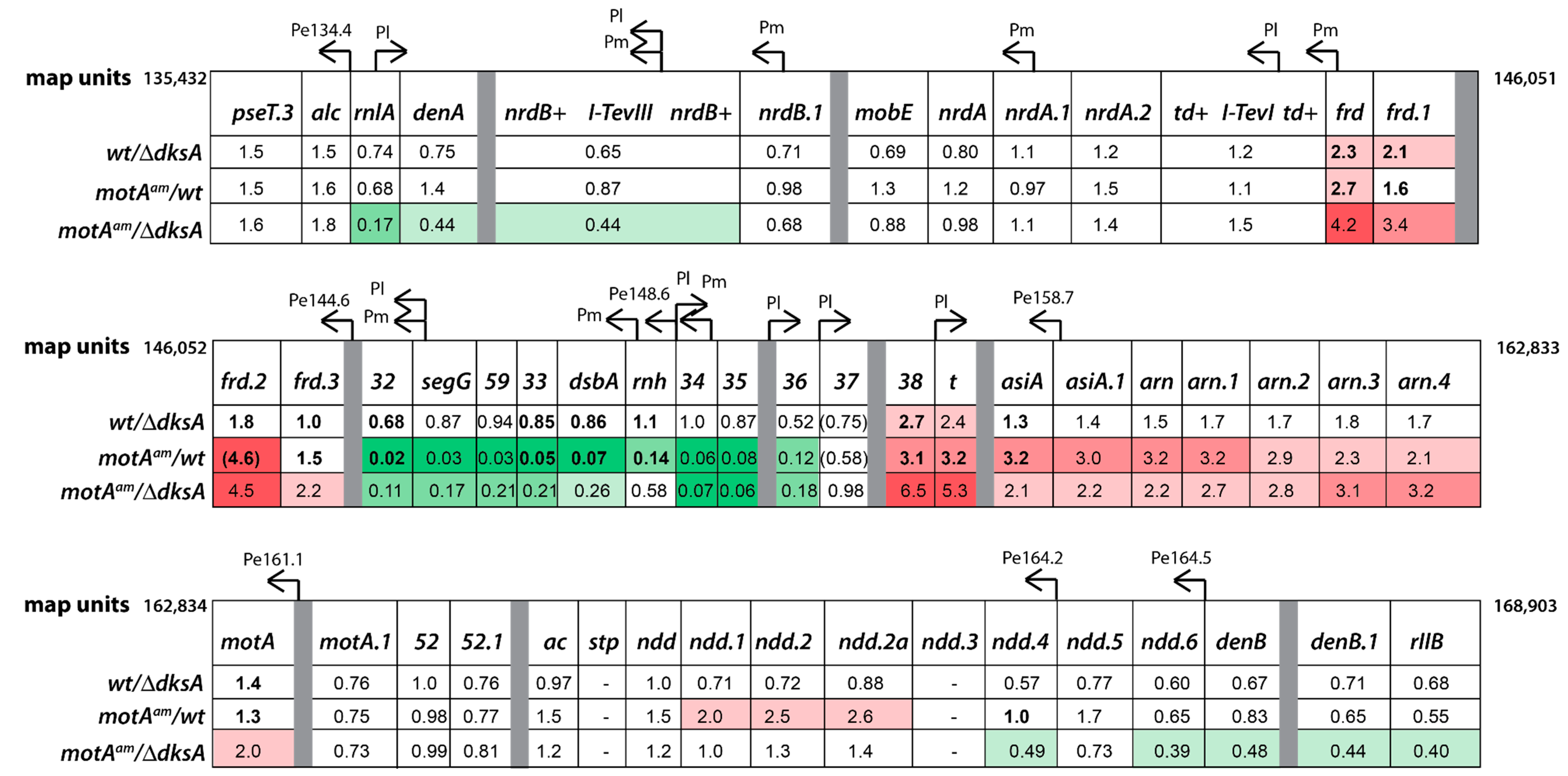
3.4. Global Transcriptome of the T4motAam Infection Reveals that Pm’s Are Differentially Affected
3.5. Reduction of Early Promoter Activity during T4 wt Infection in the Presence of DksA
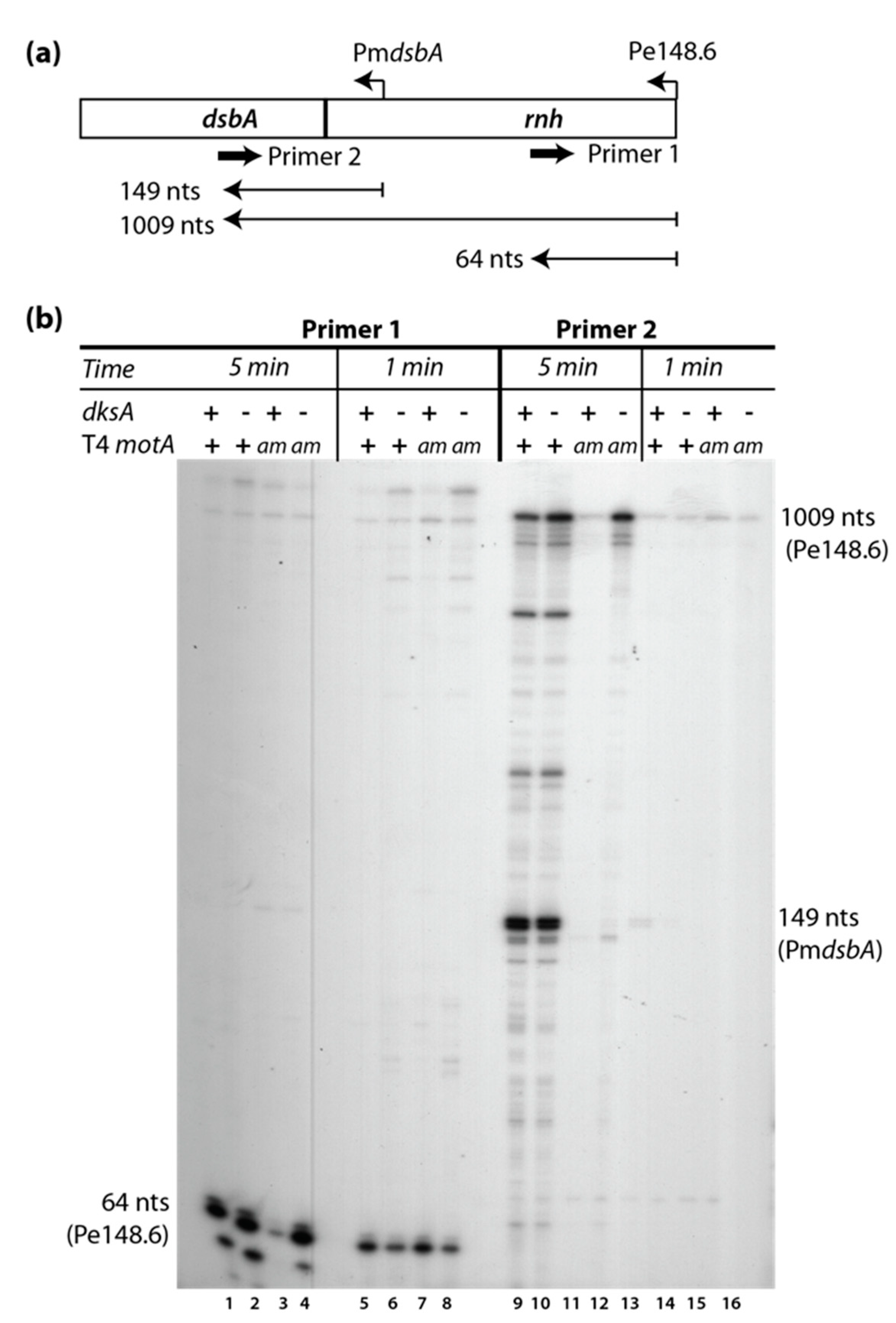
3.6. Middle Gene Expression is Partially Restored in a T4motAam Infection of B606 ∆dksA
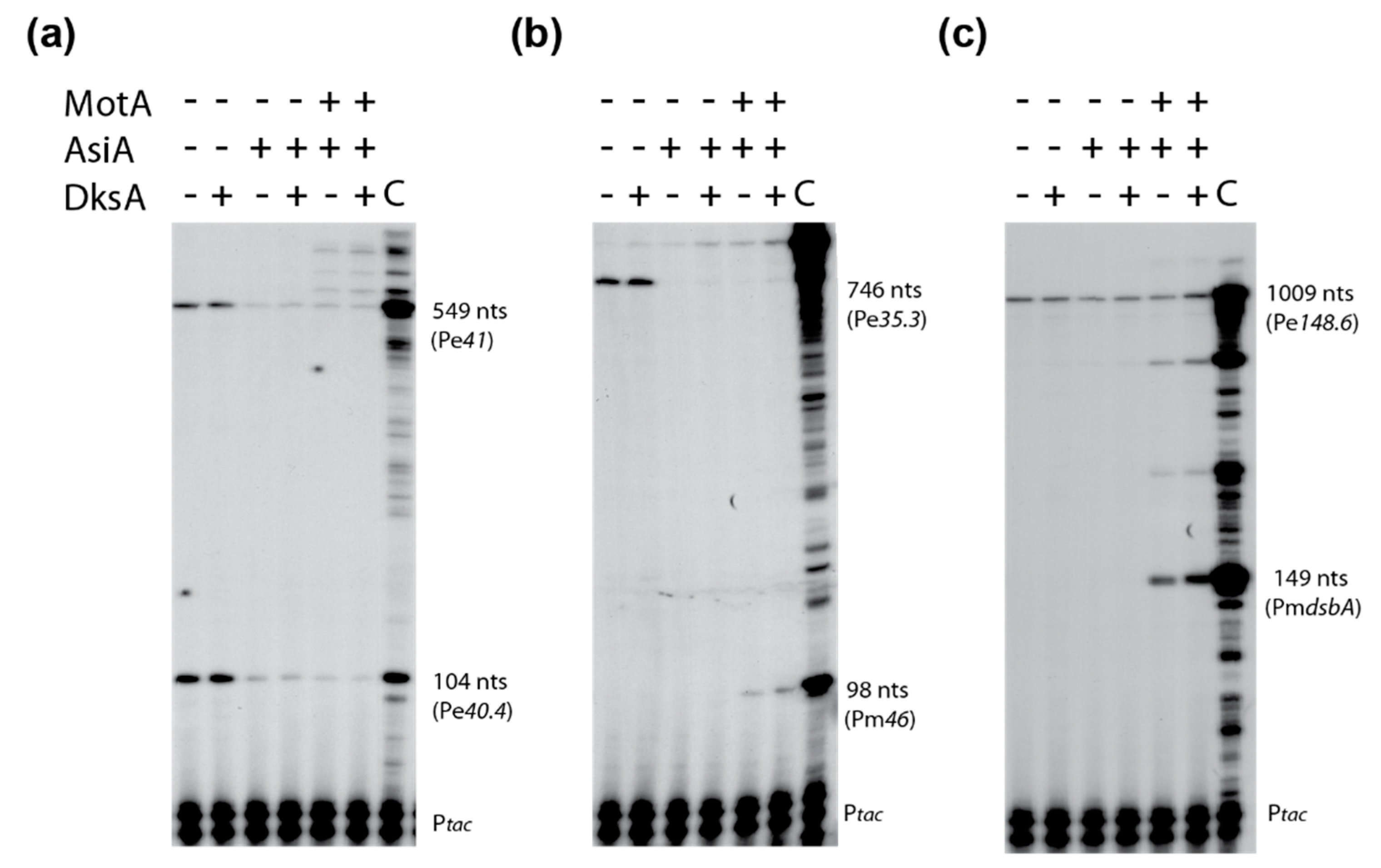
3.7. DksA Does Not Inhibit T4 Pe Transcription In Vitro
3.8. Deletion of ppGpp Has Only Modest Effects on T4 Transcription
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Lopez-Maury, L.; Marguerat, S.; Bahler, J. Tuning gene expression to changing environments: From rapid responses to evolutionary adaptation. Nat. Rev. Genet. 2009, 10, 68. [Google Scholar] [CrossRef]
- Clokie, M.R.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keen, E.C. A century of phage research: Bacteriophages and the shaping of modern biology. Bioessays 2015, 37, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Brussow, H.; Canchaya, C.; Hardt, W.D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed]
- Decker, K.B.; Hinton, D.M. Transcription regulation at the core: Similarities among bacterial, archaeal, and eukaryotic RNA polymerases. Annu. Rev. Microbiol. 2013, 67, 113–139. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.S. X-ray crystal structure of Escherichia coli RNA polymerase sigma70 holoenzyme. J. Biol. Chem. 2013, 288, 9126–9134. [Google Scholar] [CrossRef] [PubMed]
- Saecker, R.M.; Record, M.T., Jr.; Dehaseth, P.L. Mechanism of bacterial transcription initiation: RNA polymerase—Promoter binding, isomerization to initiation-competent open complexes, and initiation of RNA synthesis. J. Mol. Biol. 2011, 412, 754–771. [Google Scholar] [CrossRef] [PubMed]
- Parshin, A.; Shiver, A.L.; Lee, J.; Ozerova, M.; Schneidman-Duhovny, D.; Gross, C.A.; Borukhov, S. DksA regulates RNA polymerase in Escherichia coli through a network of interactions in the secondary channel that includes Sequence Insertion 1. Proc. Natl. Acad. Sci. USA 2015, 112, E6862–E6871. [Google Scholar] [CrossRef] [PubMed]
- Zenkin, N.; Yuzenkova, Y. New Insights into the Functions of Transcription Factors that Bind the RNA Polymerase Secondary Channel. Biomolecules 2015, 5, 1195–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perederina, A.; Svetlov, V.; Vassylyeva, M.N.; Tahirov, T.H.; Yokoyama, S.; Artsimovitch, I.; Vassylyev, D.G. Regulation through the secondary channel—Structural framework for ppGpp-DksA synergism during transcription. Cell 2004, 118, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Mechold, U.; Potrykus, K.; Murphy, H.; Murakami, K.S.; Cashel, M. Differential regulation by ppGpp versus pppGpp in Escherichia coli. Nucleic Acids Res. 2013, 41, 6175–6189. [Google Scholar] [CrossRef] [PubMed]
- Ross, W.; Sanchez-Vazquez, P.; Chen, A.Y.; Lee, J.H.; Burgos, H.L.; Gourse, R.L. ppGpp Binding to a Site at the RNAP-DksA Interface Accounts for Its Dramatic Effects on Transcription Initiation during the Stringent Response. Mol. Cell 2016, 62, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.J.; Barker, M.M.; Ross, W.; Schneider, D.A.; Webb, C.; Foster, J.W.; Gourse, R.L. DksA: A critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell 2004, 118, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, S.T.; Lemke, J.J.; Vrentas, C.E.; Gaal, T.; Ross, W.; Gourse, R.L. Effects of DksA, GreA, and GreB on transcription initiation: Insights into the mechanisms of factors that bind in the secondary channel of RNA polymerase. J. Mol. Biol. 2007, 366, 1243–1257. [Google Scholar] [CrossRef] [PubMed]
- Roghanian, M.; Zenkin, N.; Yuzenkova, Y. Bacterial global regulators DksA/ppGpp increase fidelity of transcription. Nucleic Acids Res. 2015, 43, 1529–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molodtsov, V.; Sineva, E.; Zhang, L.; Huang, X.; Cashel, M.; Ades, S.E.; Murakami, K. Allosteric effector ppGpp potentiates the inhibition of transcript initiation by DksA. Mol. Cell 2018, 69, 828–839.e5. [Google Scholar] [CrossRef] [PubMed]
- Potrykus, K.; Wegrzyn, G.; Hernandez, V.J. Multiple mechanisms of transcription inhibition by ppGpp at the lambdap(R) promoter. J. Biol. Chem. 2002, 277, 43785–43791. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, L.U.; Gummesson, B.; Joksimovic, P.; Farewell, A.; Nystrom, T. Identical, independent, and opposing roles of ppGpp and DksA in Escherichia coli. J. Bacteriol. 2007, 189, 5193–5202. [Google Scholar] [CrossRef] [PubMed]
- Kanjee, U.; Ogata, K.; Houry, W.A. Direct binding targets of the stringent response alarmone (p)ppGpp. Mol. Microbiol. 2012, 85, 1029–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henard, C.A.; Tapscott, T.; Crawford, M.A.; Husain, M.; Doulias, P.T.; Porwollik, S.; Liu, L.; McClelland, M.; Ischiropoulos, H.; Vazquez-Torres, A. The 4-cysteine zinc-finger motif of the RNA polymerase regulator DksA serves as a thiol switch for sensing oxidative and nitrosative stress. Mol. Microbiol. 2014, 91, 790–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinella, D.; Potrykus, K.; Murphy, H.; Cashel, M. Effects on Growth by Changes of the Balance between GreA, GreB, and DksA Suggest Mutual Competition and Functional Redundancy in Escherichia coli. J. Bacteriol. 2012, 194, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Laptenko, O.; Lee, J.; Lomakin, I.; Borukhov, S. Transcript cleavage factors GreA and GreB act as transient catalytic components of RNA polymerase. EMBO J. 2003, 22, 6322–6334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinton, D.M. Transcriptional control in the prereplicative phase of T4 development. Virol. J. 2010, 7, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiduschek, E.P.; Kassavetis, G.A. Transcription of the T4 late genes. Virol. J. 2010, 7, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, T.; Vanhouwe, G.; Epstein, R.H. Isolation and Characterization of Conditional Lethal Mutations in the Mot Gene of Bacteriophage-T4. J. Mol. Biol. 1978, 126, 551–570. [Google Scholar] [CrossRef]
- Studier, F.W.; Rosenberg, A.H.; Dunn, J.J.; Dubendorff, J.W. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990, 185, 60–89. [Google Scholar] [PubMed]
- Jeong, H.; Barbe, V.; Lee, C.H.; Vallenet, D.; Yu, D.S.; Choi, S.H.; Couloux, A.; Lee, S.W.; Yoon, S.H.; Cattolico, L.; et al. Genome sequences of Escherichia coli B strains REL606 and BL21(DE3). J. Mol. Biol. 2009, 394, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Kang, P.J.; Craig, E.A. Identification and characterization of a new Escherichia coli gene that is a dosage-dependent suppressor of a dnaK deletion mutation. J. Bacteriol. 1990, 172, 2055–2064. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Kalman, M.; Ikehara, K.; Zemel, S.; Glaser, G.; Cashel, M. Residual guanosine 3′,5′-bispyrophosphate synthetic activity of relA null mutants can be eliminated by spoT null mutations. J. Biol. Chem. 1991, 266, 5980–5990. [Google Scholar] [PubMed]
- Dressman, H.K.; Drake, J.W. Lysis and lysis inhibition in bacteriophage T4: RV mutations reside in the holin t gene. J. Bacteriol. 1999, 181, 4391–4396. [Google Scholar] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Yin, J. Imaging the propagation of viruses. Biotechnol. Bioeng. 1996, 52, 438–442. [Google Scholar] [CrossRef]
- Racine, J.S. RStudio: A Platform-Independent IDE for R and Sweave. J. Appl. Econ. 2012, 27, 167–172. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis. In Use R; Springer: Berlin, Germany, 2009. [Google Scholar]
- Carson, K. Single-Step Growth. In Molecular Biology of Bacteriophage T4; Karam, J.D., Kreuzer, K.N., Mosig, G., Hall, D.H., Eiserling, F.A., Black, L.W., Spicer, E.K., Kutter, E., Carlson, K., Miller, E.S., Eds.; American Society of Microbiology: Washington, DC, USA, 1994; pp. 434–437. [Google Scholar]
- Hinton, D.M. Transcript analyses of the uvsX-40-41 region of bacteriophage T4. Changes in the RNA as infection proceeds. J. Biol. Chem. 1989, 264, 14432–14439. [Google Scholar] [PubMed]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- James, T.D.; Cashel, M.; Hinton, D.M. A Mutation within the beta Subunit of Escherichia coli RNA Polymerase Impairs Transcription from Bacteriophage T4 Middle Promoters. J. Bacteriol. 2010, 192, 5580–5587. [Google Scholar] [CrossRef] [PubMed]
- Hinton, D.M. Transcription from a bacteriophage T4 middle promoter using T4 motA protein and phage-modified RNA polymerase. J. Biol. Chem. 1991, 266, 18034–18044. [Google Scholar] [PubMed]
- Guild, N.; Gayle, M.; Sweeney, R.; Hollingsworth, T.; Modeer, T.; Gold, L. Transcriptional activation of bacteriophage T4 middle promoters by the motA protein. J. Mol. Biol. 1988, 199, 241–258. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C-T method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, J.S.; Hinton, D.M. An N-terminal mutation in the bacteriophage T4 motA gene yields a protein that binds DNA but is defective for activation of transcription. J. Bacteriol. 1996, 178, 6133–6139. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.L.; James, T.D.; Knipling, L.; Waddell, M.B.; White, S.; Hinton, D.M. Architecture of the Bacteriophage T4 Activator MotA/Promoter DNA Interaction during Sigma Appropriation. J. Biol. Chem. 2013, 288, 27607–27618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinton, D.M.; March-Amegadzie, R.; Gerber, J.S.; Sharma, M. Bacteriophage T4 middle transcription system: T4-modified RNA polymerase; AsiA, a sigma 70 binding protein; and transcriptional activator MotA. Methods Enzymol. 1996, 274, 43–57. [Google Scholar] [PubMed]
- Potrykus, K.; Vinella, D.; Murphy, H.; Szalewska-Palasz, A.; D’Ari, R.; Cashel, M. Antagonistic regulation of Escherichia coli ribosomal RNA rrnB P1 promoter activity by GreA and DksA. J. Biol. Chem. 2006, 281, 15238–15248. [Google Scholar] [CrossRef] [PubMed]
- Gallet, R.; Kannoly, S.; Wang, I.N. Effects of bacteriophage traits on plaque formation. BMC Microbiol. 2011, 11, 181. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T.; Culler, R.R. Optimizing bacteriophage plaque fecundity. J. Theor. Biol. 2007, 249, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Benson, K.H.; Kreuzer, K.N. Role of MotA transcription factor in bacteriophage T4 DNA replication. J. Mol. Biol. 1992, 228, 88–100. [Google Scholar] [CrossRef]
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Ruger, W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef] [PubMed]
- Stitt, B.; Hinton, D.M. Regulation of Middle-Mode Transcription. In Molecular Biology of Bacteriophage T4; Karam, J.D., Ed.; American Society for Microbiology: Washington, DC, USA, 1994; pp. 142–160. [Google Scholar]
- Truncaite, L.; Piesiniene, L.; Kolesinskiene, G.; Zajanckauskaite, A.; Driukas, A.; Klausa, V.; Nivinskas, R. Twelve new MotA-dependent middle promoters of bacteriophage T4: Consensus sequence revised. J. Mol. Biol. 2003, 327, 335–346. [Google Scholar] [CrossRef]
- Sozhamannan, S.; Stitt, B.L. Effects on mRNA degradation by Escherichia coli transcription termination factor Rho and pBR322 copy number control protein Rop. J. Mol. Biol. 1997, 268, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Sozhamannan, S.; Morris, J.G.; Stitt, B.L. Instability of pUC19 in Escherichia coli transcription termination factor mutant, rho026. Plasmid 1999, 41, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Luke, K.; Radek, A.; Liu, X.; Campbell, J.; Uzan, M.; Haselkorn, R.; Kogan, Y. Microarray analysis of gene expression during bacteriophage T4 infection. Virology 2002, 299, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Sanson, B.; Uzan, M. Post-transcriptional controls in bacteriophage T4: Roles of the sequence-specific endoribonuclease RegB. FEMS Microbiol. Rev. 1995, 17, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Uzan, M. Bacteriophage T4 RegB endoribonuclease. Methods Enzymol. 2001, 342, 467–480. [Google Scholar] [PubMed]
- Barker, M.M.; Gaal, T.; Josaitis, C.A.; Gourse, R.L. Mechanism of regulation of transcription initiation by ppGpp. I. Effects of ppGpp on transcription initiation in vivo and in vitro. J. Mol. Biol. 2001, 305, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Travers, A.A. Promoter sequence for stringent control of bacterial ribonucleic acid synthesis. J. Bacteriol. 1980, 141, 973–976. [Google Scholar] [PubMed]
- Haugen, S.P.; Ross, W.; Gourse, R.L. Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat. Rev. Microbiol. 2008, 6, 507–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Mooney, R.A.; Grass, J.A.; Sivaramakrishnan, P.; Herman, C.; Landick, R.; Wang, J.D. DksA guards elongating RNA polymerase against ribosome-stalling-induced arrest. Mol. Cell 2014, 53, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Kolmsee, T.; Delic, D.; Agyenim, T.; Calles, C.; Wagner, R. Differential stringent control of Escherichia coli rRNA promoters: Effects of ppGpp, DksA and the initiating nucleotides. Microbiology 2011, 157, 2871–2879. [Google Scholar] [CrossRef] [PubMed]
- Henard, C.A.; Bourret, T.J.; Song, M.; Vazquez-Torres, A. Control of redox balance by the stringent response regulatory protein promotes antioxidant defenses of Salmonella. J. Biol. Chem. 2010, 285, 36785–36793. [Google Scholar] [CrossRef] [PubMed]
- Henard, C.A.; Vazquez-Torres, A. DksA-dependent resistance of Salmonella enterica serovar Typhimurium against the antimicrobial activity of inducible nitric oxide synthase. Infect. Immun. 2012, 80, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Payne, S.M. Induction of expression of hfq by DksA is essential for Shigella flexneri virulence. Mol. Microbiol. 2006, 62, 469–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.I.; Goulian, M. A network of regulators promotes dehydration tolerance in Escherichia coli. Environ. Microbiol. 2018, 20, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Crawford, M.A.; Henard, C.A.; Tapscott, T.; Porwollik, S.; McClelland, M.; Vazquez-Torres, A. DksA-Dependent Transcriptional Regulation in Salmonella Experiencing Nitrosative Stress. Front. Microbiol. 2016, 7, 444. [Google Scholar] [CrossRef] [PubMed]
- Holley, C.L.; Zhang, X.J.; Fortney, K.R.; Ellinger, S.; Johnson, P.; Baker, B.; Liu, Y.L.; Janowicz, D.M.; Katz, B.P.; Munson, R.S.; et al. DksA and (p)ppGpp Have Unique and Overlapping Contributions to Haemophilus ducreyi Pathogenesis in Humans. Infect. Immun. 2015, 83, 3281–3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkens, K.R.W. Transcription from early promoters. In Molecular Biology of Bacteriophage T4; Karam, J.D., Kreuzer, K.N., Mosig, G., Hall, D.H., Eiserling, F.A., Black, L.W., Spicer, E.K., Kutter, E., Carlson, K., Miller, E.S., Eds.; American Society for Microbiology: Washington, DC, USA, 1994; pp. 132–141. [Google Scholar]
- Webb, C.; Moreno, M.; Wilmes-Riesenberg, M.; Curtiss, R., 3rd; Foster, J.W. Effects of DksA and ClpP protease on sigma S production and virulence in Salmonella typhimurium. Mol. Microbiol. 1999, 34, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potrykus, K.; Murphy, H.; Philippe, N.; Cashel, M. ppGpp is the major source of growth rate control in E. coli. Environ. Microbiol. 2011, 13, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Young, K.D. A cAMP-independent carbohydrate-driven mechanism inhibits tnaA expression and TnaA enzyme activity in Escherichia coli. Microbiology 2014, 160, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Traxler, M.F.; Summers, S.M.; Nguyen, H.T.; Zacharia, V.M.; Hightower, G.A.; Smith, J.T.; Conway, T. The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Mol. Microbiol. 2008, 68, 1128–1148. [Google Scholar] [CrossRef] [PubMed]
- Shokri, A.; Veide, A.; Larsson, G. RelA1 gene control of Escherichia coli lipid structure and cell performance during glucose limited fed-batch conditions. Appl. Microbiol. Biotechnol. 2006, 73, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Santangelo, T.J.; Artsimovitch, I. Termination and antitermination: RNA polymerase runs a stop sign. Nat. Rev. Microbiol. 2011, 9, 319–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grainger, D.C. Structure and function of bacterial H-NS protein. Biochem. Soc. Trans. 2016, 44, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.S.; Beckett, E.; Bae, S.J.; Navarre, W.W. The 5.5 Protein of Phage T7 Inhibits H-NS through Interactions with the Central Oligomerization Domain. J. Bacteriol. 2011, 193, 4881–4892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, C.H.; Wang, H.C.; Ko, T.P.; Chang, Y.C.; Wang, A.H.J. The T4 Phage DNA Mimic Protein Arn Inhibits the DNA Binding Activity of the Bacterial Histone-like Protein H-NS. J. Biol. Chem. 2014, 289, 27046–27054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagemans, J.; Delattre, A.S.; Uytterhoeven, B.; de Smet, J.; Cenens, W.; Aertsen, A.; Ceyssens, P.J.; Lavigne, R. Antibacterial phage ORFans of Pseudomonas aeruginosa phage LUZ24 reveal a novel MvaT inhibiting protein. Front. Microbiol. 2015, 6, 1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patterson-West, J.; James, T.D.; Fernández-Coll, L.; Iben, J.R.; Moon, K.; Knipling, L.; Cashel, M.; Hinton, D.M. The E. coli Global Regulator DksA Reduces Transcription during T4 Infection. Viruses 2018, 10, 308. https://doi.org/10.3390/v10060308
Patterson-West J, James TD, Fernández-Coll L, Iben JR, Moon K, Knipling L, Cashel M, Hinton DM. The E. coli Global Regulator DksA Reduces Transcription during T4 Infection. Viruses. 2018; 10(6):308. https://doi.org/10.3390/v10060308
Chicago/Turabian StylePatterson-West, Jennifer, Tamara D. James, Llorenç Fernández-Coll, James R. Iben, Kyung Moon, Leslie Knipling, Michael Cashel, and Deborah M. Hinton. 2018. "The E. coli Global Regulator DksA Reduces Transcription during T4 Infection" Viruses 10, no. 6: 308. https://doi.org/10.3390/v10060308
APA StylePatterson-West, J., James, T. D., Fernández-Coll, L., Iben, J. R., Moon, K., Knipling, L., Cashel, M., & Hinton, D. M. (2018). The E. coli Global Regulator DksA Reduces Transcription during T4 Infection. Viruses, 10(6), 308. https://doi.org/10.3390/v10060308