Genetic Analysis of Avian Coronavirus Infectious Bronchitis Virus in Yellow Chickens in Southern China over the Past Decade: Revealing the Changes of Genetic Diversity, Dominant Genotypes, and Selection Pressure

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Isolation and Propagation
2.2. Primers for S1, M, N, and E Genes Amplification
2.3. RNA Extraction and Amplification of S1, E, M, and N Genes
2.4. Gene Sequencing, Alignments, and Phylogenetic Analysis
2.5. Recombination Detection
2.6. Analysis of Entropy of Amino Acid Sequences
2.7. Analysis of Positive Selection
2.8. Prediction of N-Glycosylation Sites
2.9. Analysis of Molecular Evolutionary Rate, the Most Recent Common Ancestor, and Population Size
3. Results
3.1. Alignment Analysis of S1, E, M, and N Gene Sequences
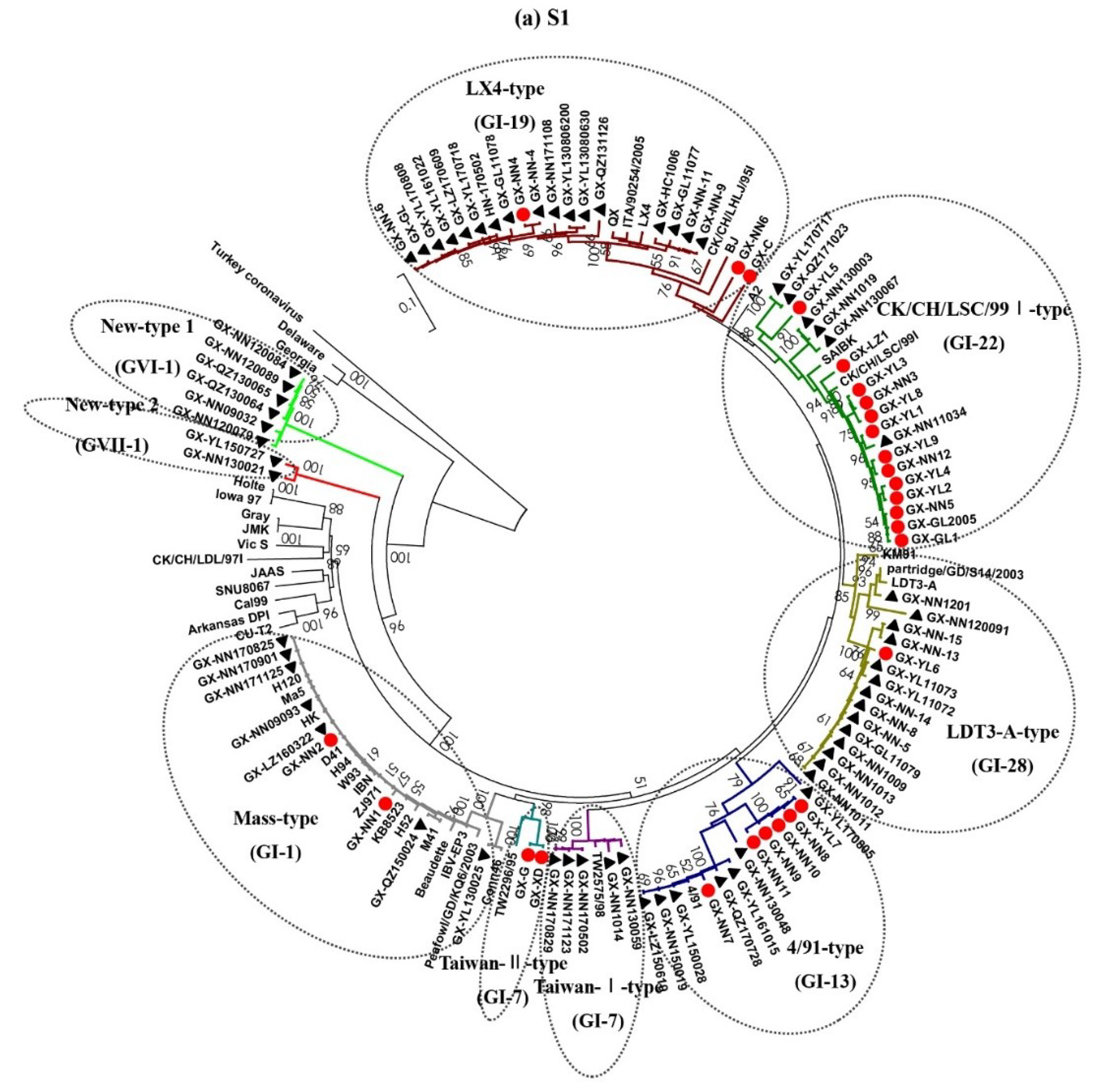
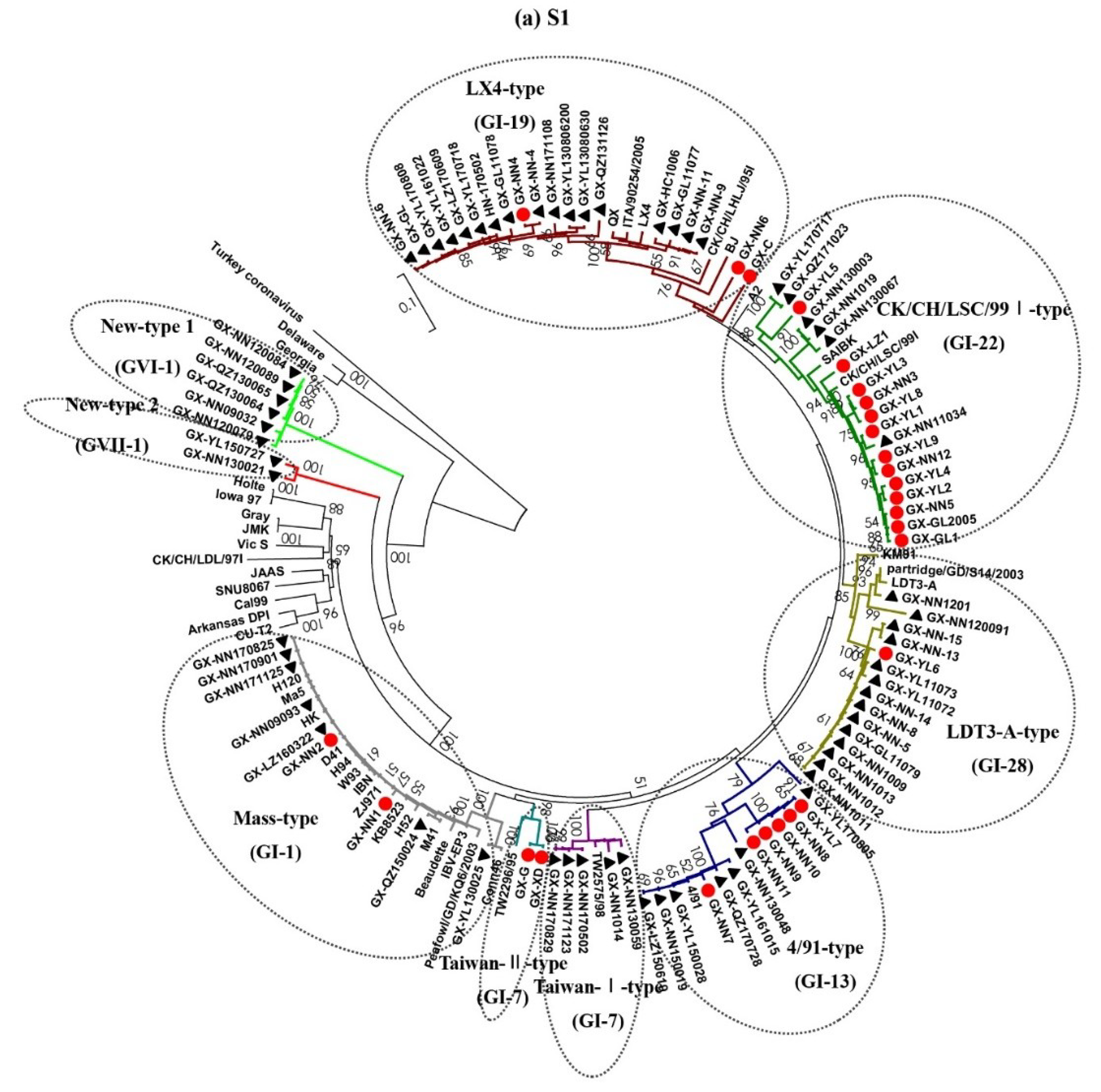
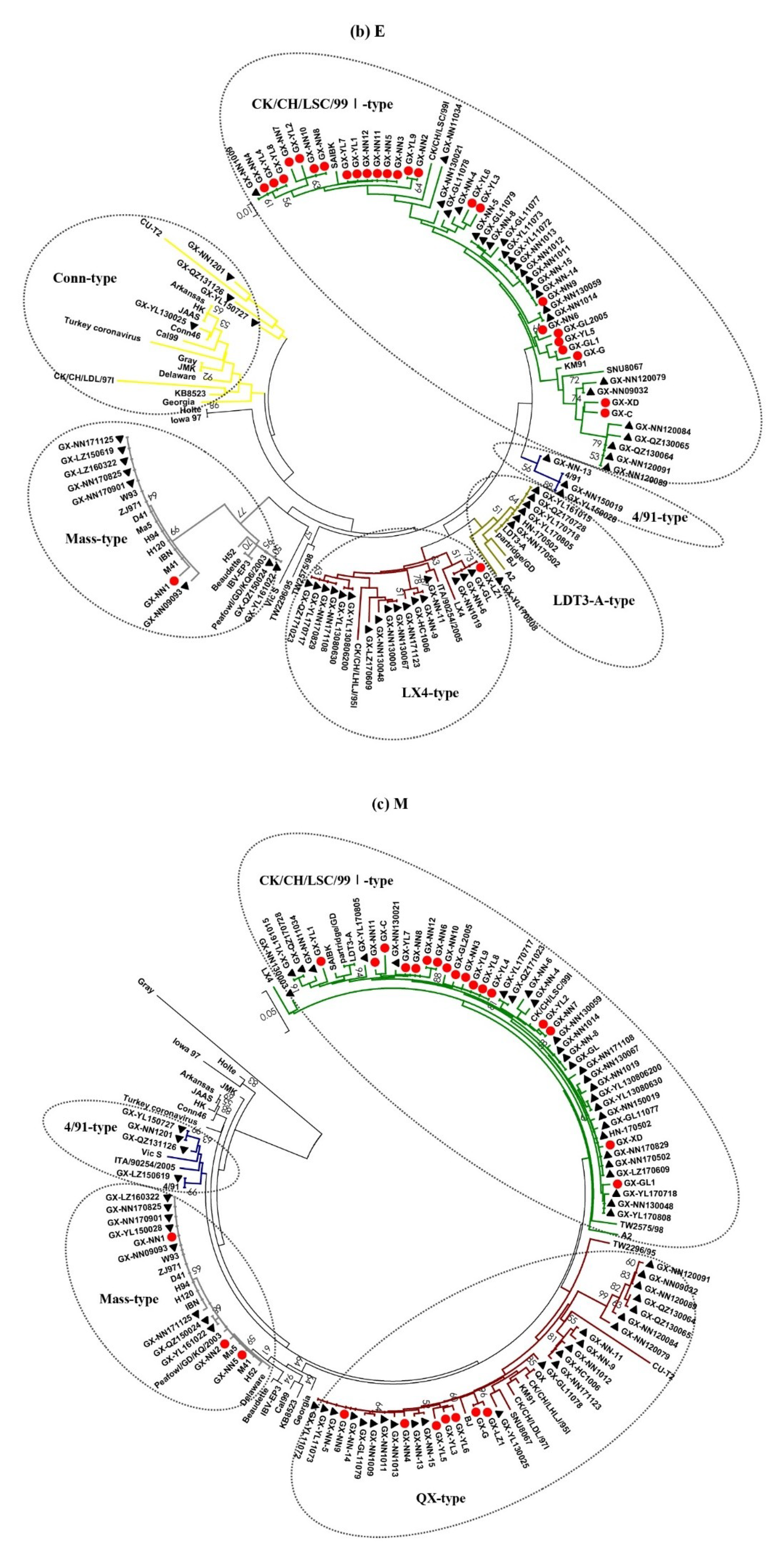
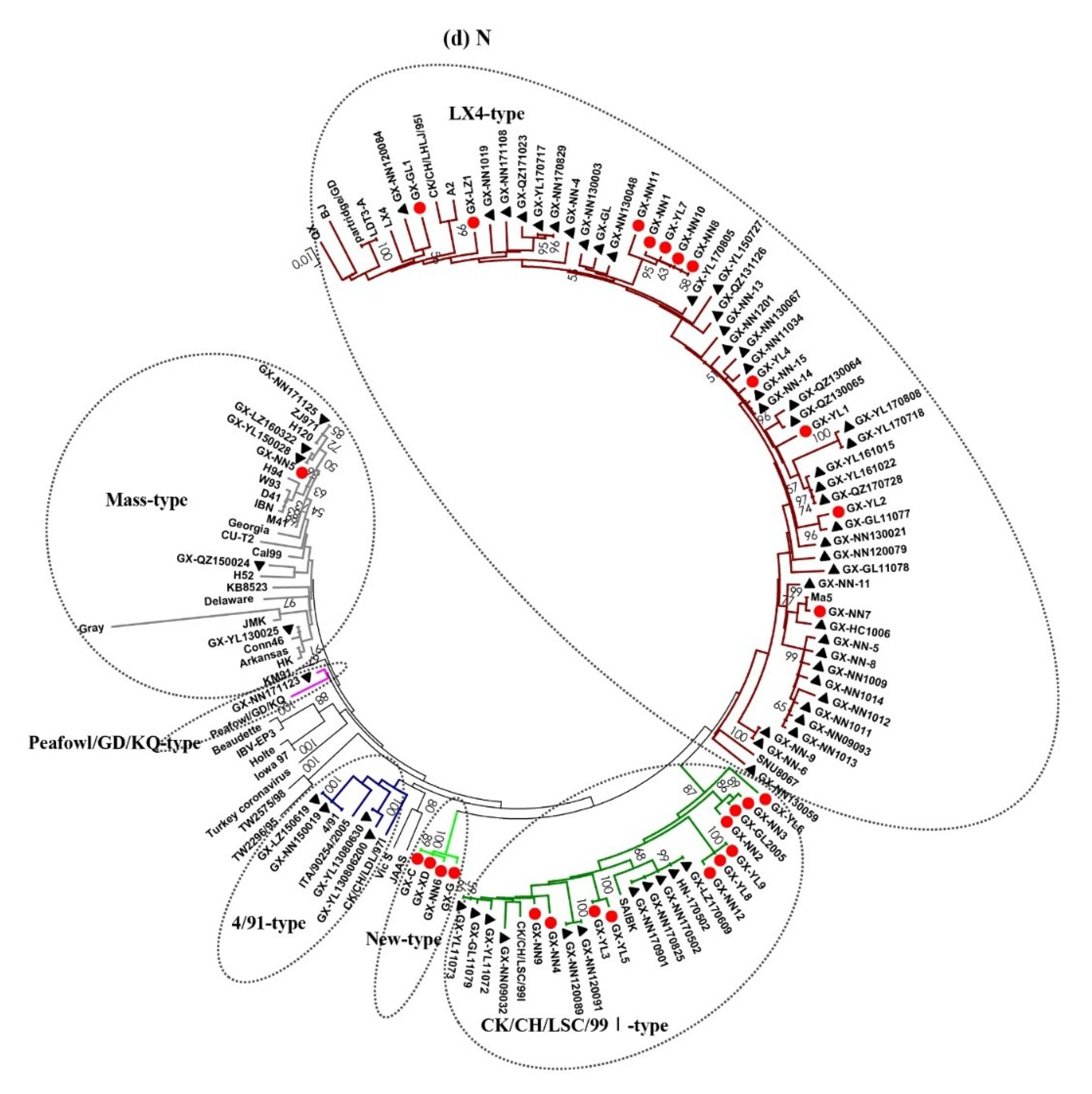
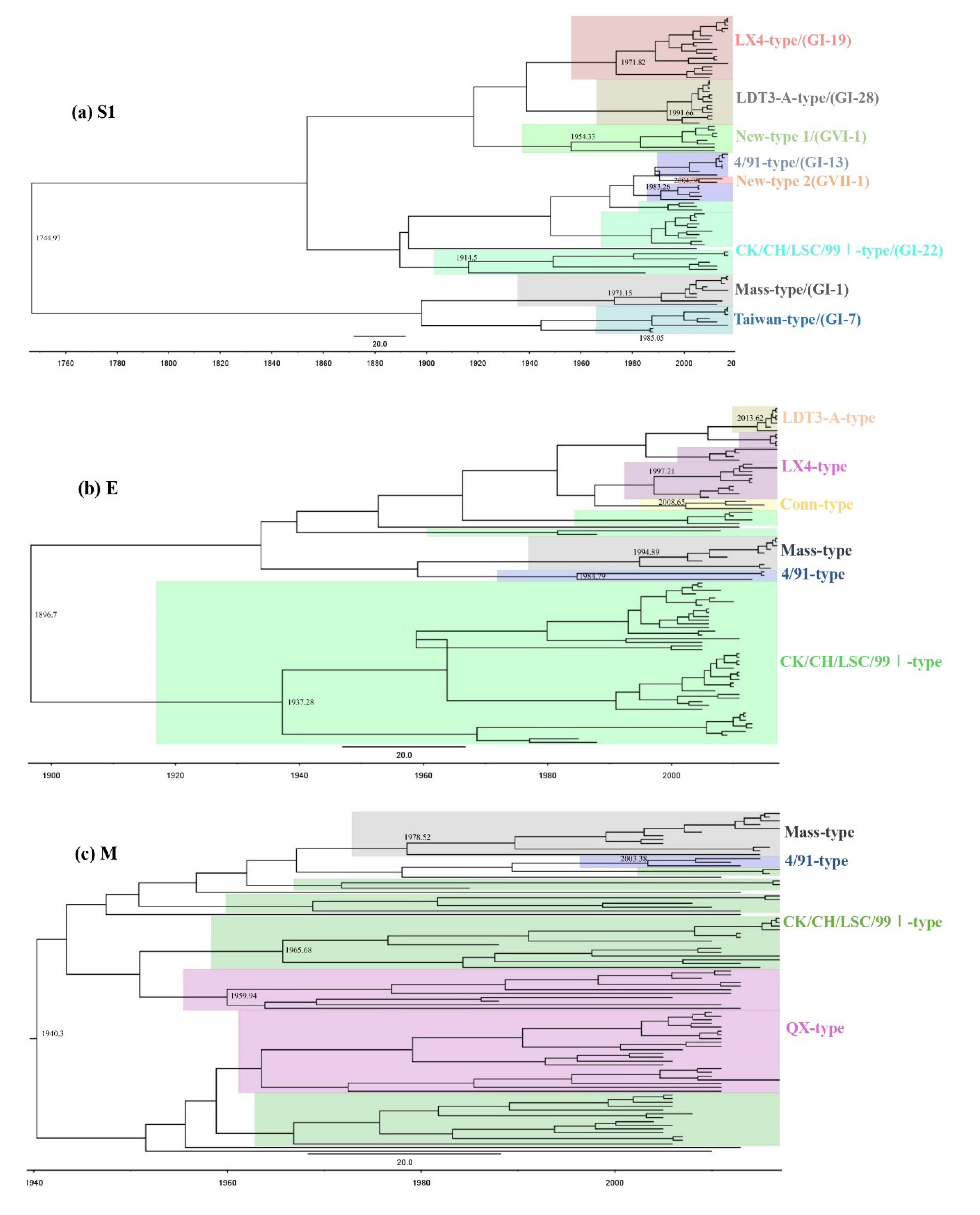
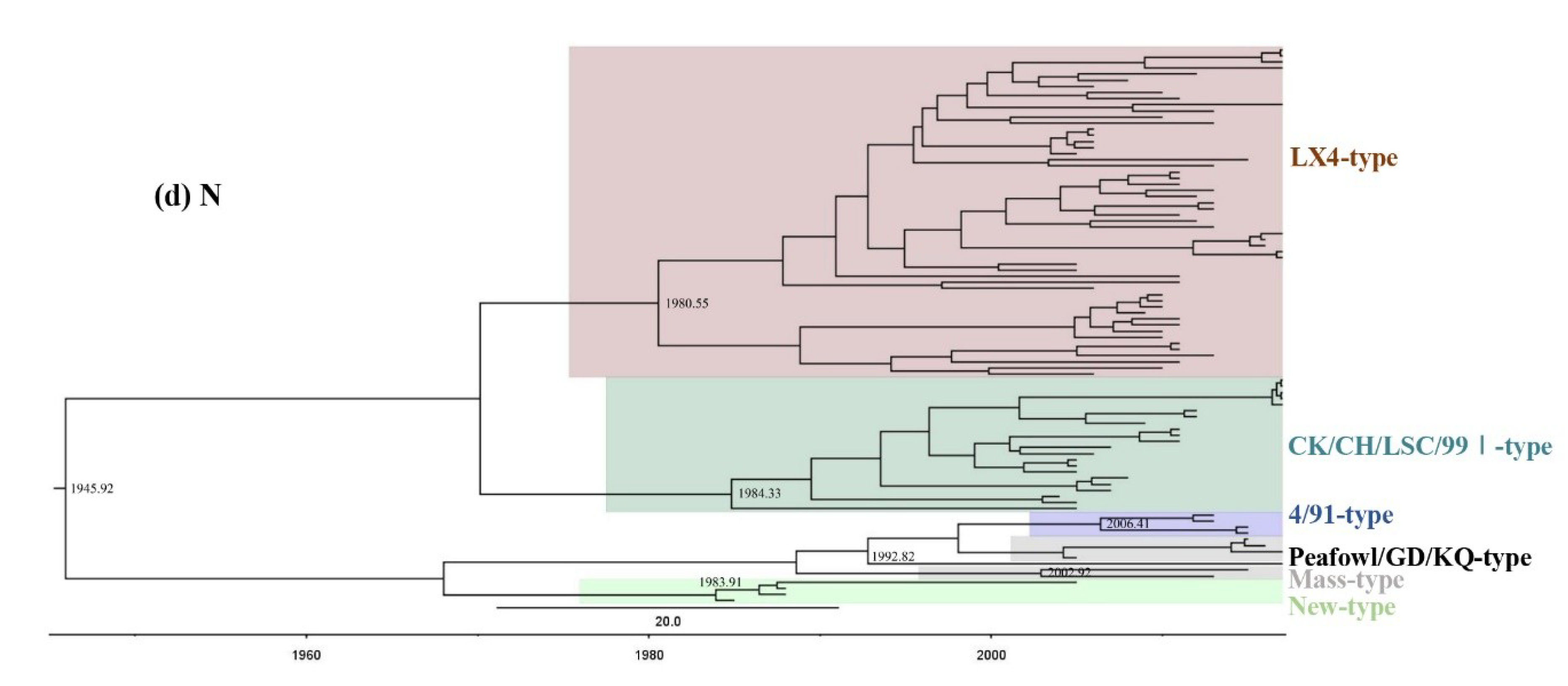
3.2. Phylogenetic Analysis
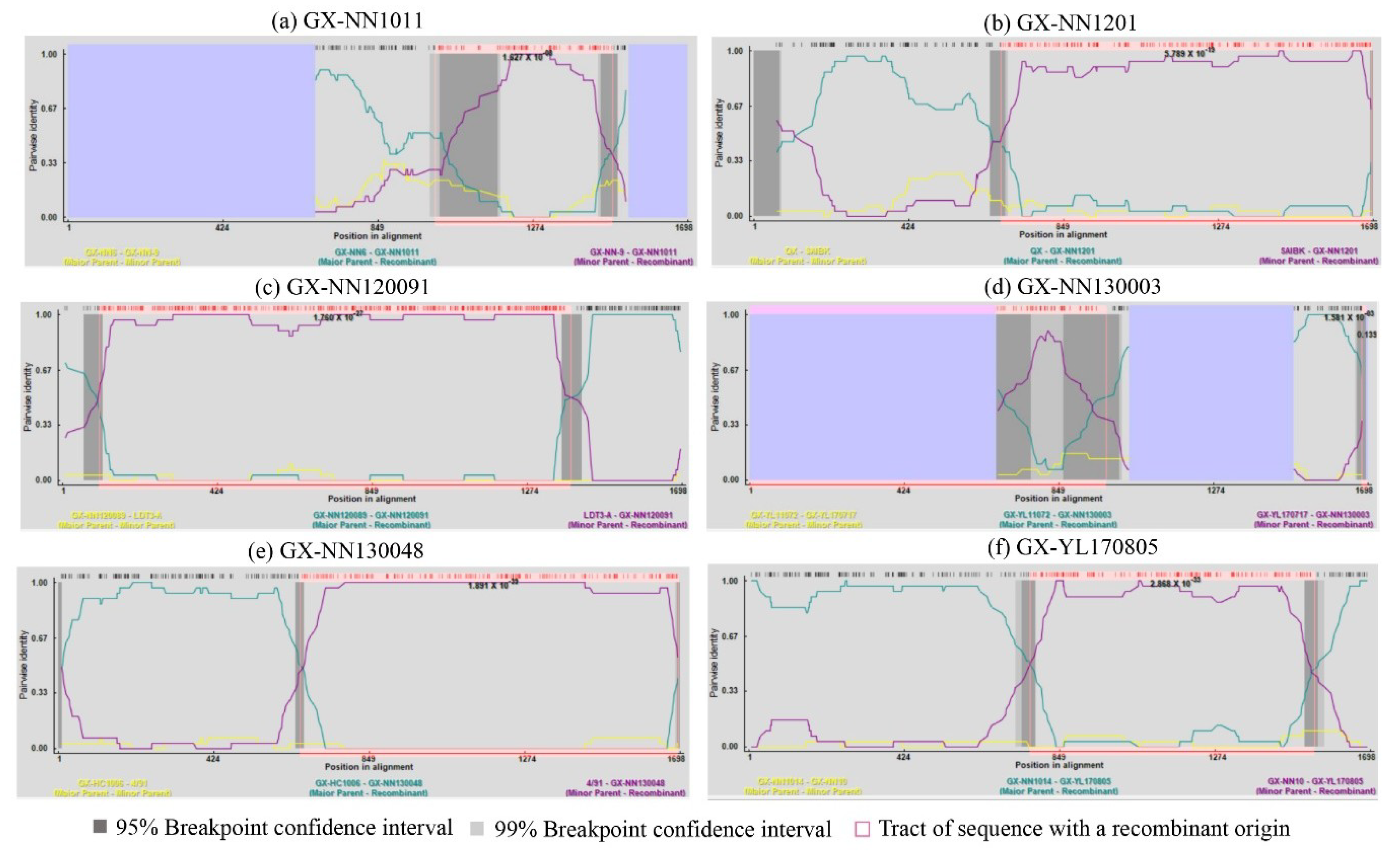
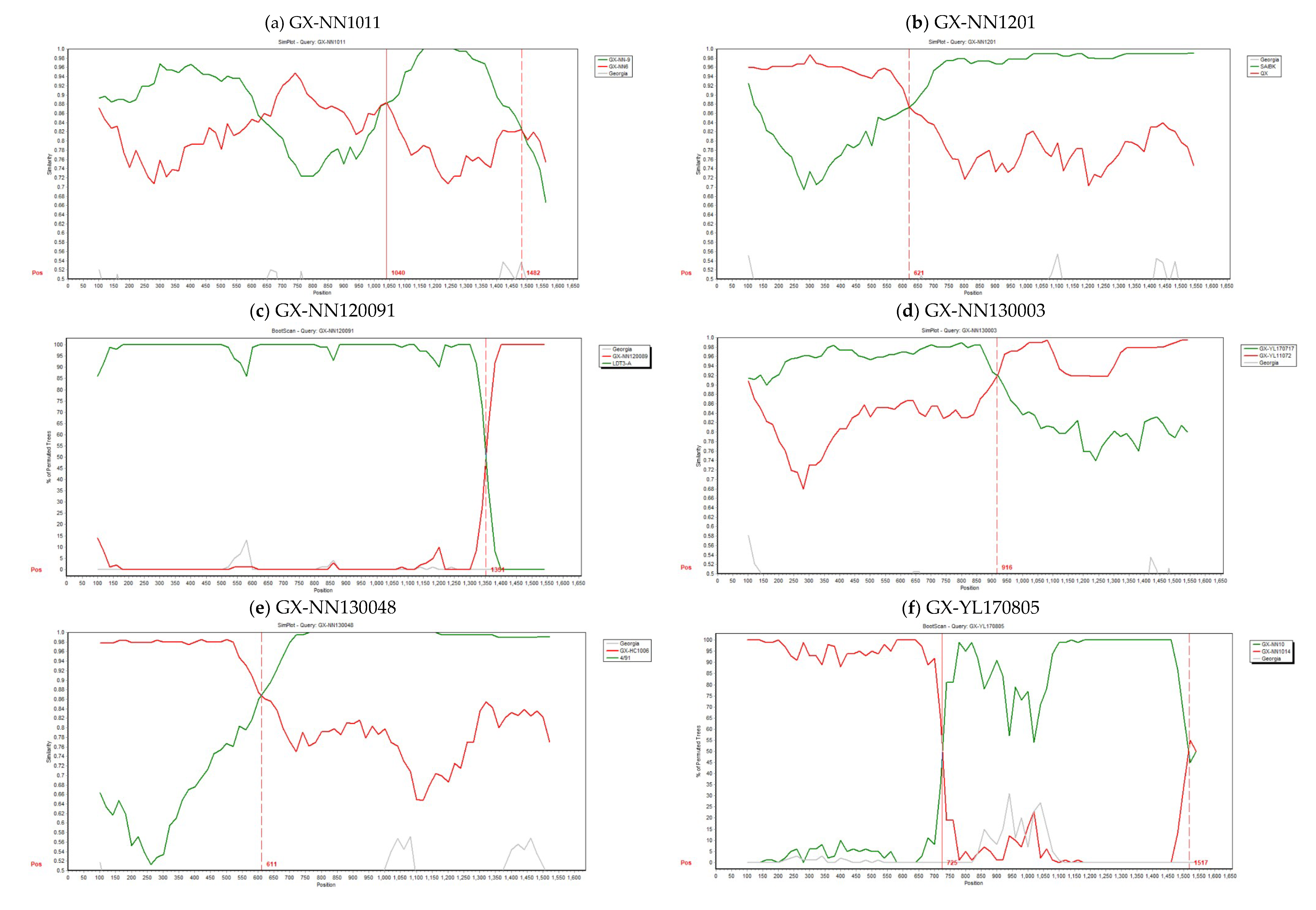
3.3. Analysis of Recombinants
3.4. Analysis of Entropy of Amino Acid Sequences
3.5. Positive Selection of the S1, E, M, and N Proteins
3.6. Prediction of N-Linked Glycosylation Sites in S1, E, M, and N Proteins of IBVs by NetNGlyc Server
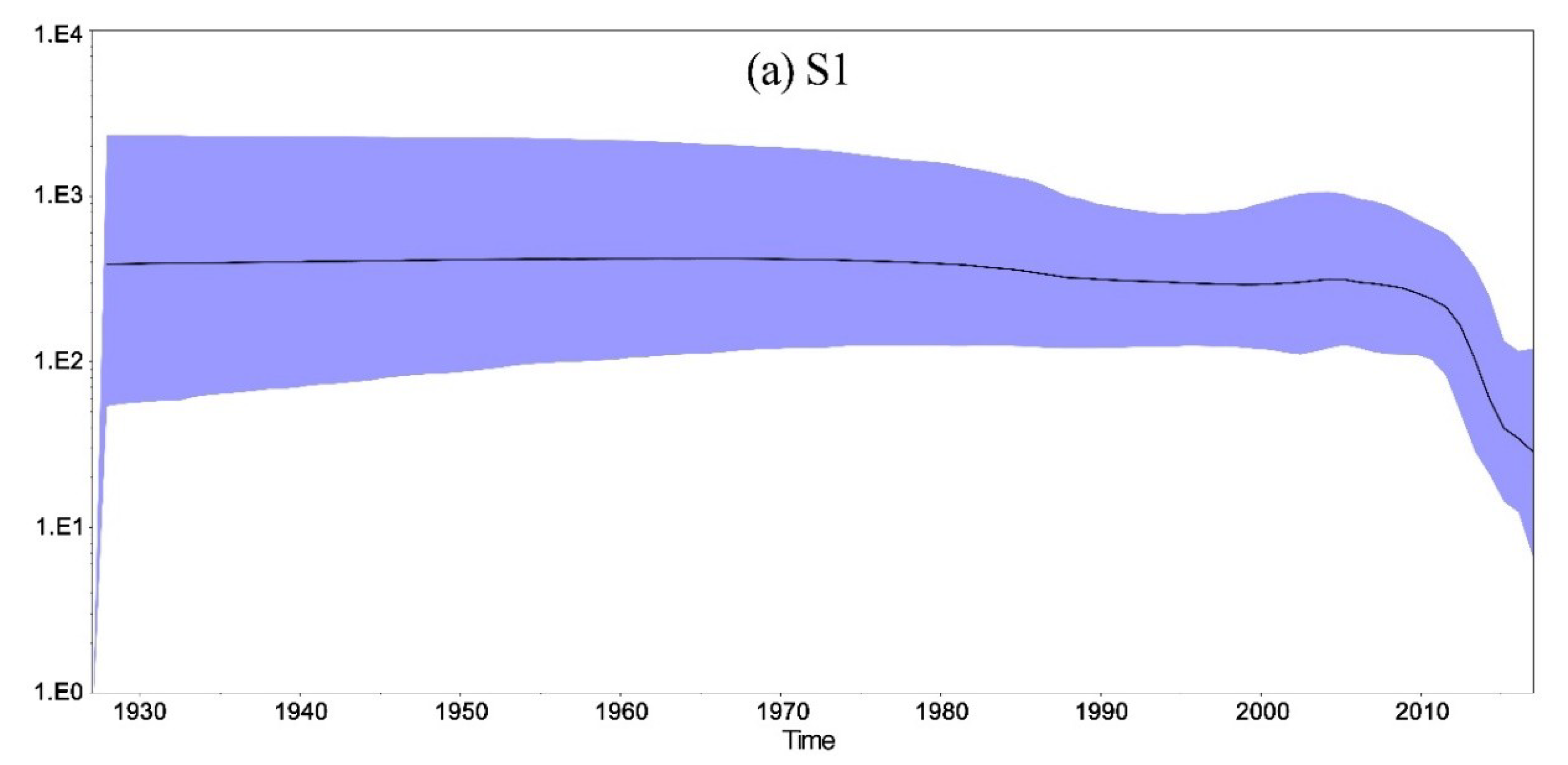
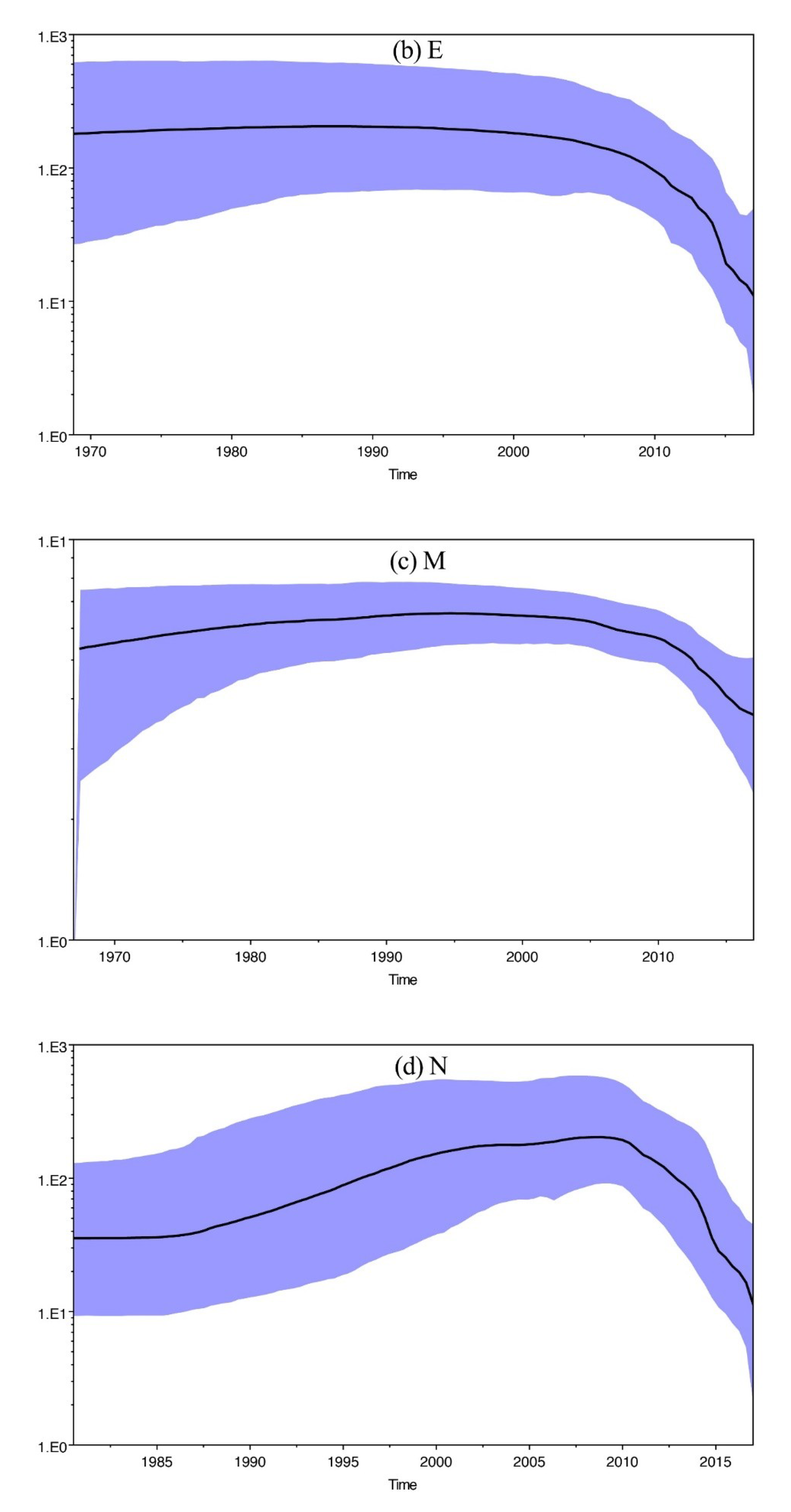
3.7. Molecular Evolutionary Rate, the Most Recent Common Ancestor, and Population Size Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, M.; Wang, X.Y.; Wei, P.; Chen, Q.Y.; Wei, Z.J.; Mo, M.L. Serotype and genotype diversity of infectious bronchitis viruses isolated during 1985–2008 in Guangxi, China. Arch. Virol. 2012, 157, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Mo, M.L.; Huang, B.C.; Fan, W.S.; Wei, Z.J.; Wei, T.C.; Li, K.R.; Wei, P. Continuous evolution of avian infectious bronchitis virus resulting in different variants co-circulating in southern China. Arch. Virol. 2013, 158, 1783–1786. [Google Scholar] [CrossRef] [PubMed]
- Mo, M.L.; Huang, B.C.; Wei, P.; Wei, T.C.; Chen, Q.Y.; Wang, X.Y.; Li, M.; Fan, W.S. Complete Genome Sequences of Two Chinese Virulent Avian Coronavirus Infectious Bronchitis Virus Variants. J. Virol. 2012, 86, 10903–10904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, M.L.; Li, M.; Huang, B.C.; Fan, W.S.; Wei, P.; Wei, T.C.; Cheng, Q.Y.; Wei, Z.J.; Lang, Y.H. Molecular characterization of major structural protein genes of avian coronavirus infectious bronchitis virus isolates in southern China. Viruses 2013, 5, 3007–3020. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.Y.; Xue, Y.; Wang, F.; Chen, F.; Shu, D.M.; Xie, Q.M. Analysis of S1 gene of avian infectious bronchitis virus isolated in southern China during 2011–2012. Virus Genes. 2014, 49, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.S.; Li, H.M.; He, Y.N.; Tang, N.; Zhang, L.H.; Wang, H.Y.; Zhong, L.; Chen, J.C.; Wei, T.C.; Huang, T.; et al. Immune protection conferred by three commonly used commercial live attenuated vaccines against the prevalent local strains of avian infectious bronchitis virus in southern China. J. Vet. Med. Sci. 2018, 80, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Han, Z.X.; Chen, Y.Q.; Zhao, W.J.; Sun, J.F.; Zhao, Y.; Liu, S.W. Characterization of the complete genome, antigenicity, pathogenicity, tissue tropism, and shedding of a recombinant avian infectious bronchitis virus with a ck/CH/LJL/140901-like backbone and an S2 fragment from a 4/91-like virus. Virus Res. 2018, 244, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, D. Coronavirus avian infectious bronchitis virus. Vet. Res. 2007, 38, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.M.; Kwon, H.J.; Choi, K.S.; Kim, J.H. Comparative genomics of QX-like infectious bronchitis viruses in Korea. Arch. Virol. 2017, 162, 1237–1250. [Google Scholar] [CrossRef]
- Kusters, J.G.; Jager, E.J.; Lenstra, J.A.; Koch, G.; Posthumus, W.P.; Meloen, R.H.; van der Zeijst, B.A. Analysis of an immunodominant region of infectious bronchitis virus. J. Immunol. 1989, 143, 2692–2698. [Google Scholar]
- Cavanagh, D.; Davis, P.J.; Cook, J.A.; Li, D.; Kant, A.; Koch, G. Location of the amino acid differences in the S1 spike glycoprotein subunit of closely related serotypes of infectious bronchitis virus. Avian Pathol. 1992, 21, 33–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, J.O.; Shubin, R.A.; Sussman, M.A.; Casteel, N.; Stohlman, S.A. Monoclonal antibodies to the matrix (E1) glycoprotein of mouse hepatitis virus protect mice from encephalitis. Virology 1989, 168, 162–167. [Google Scholar] [CrossRef]
- Ambepitiya Wickramasinghe, I.N.; van Beurden, S.J.; Weerts, E.A.; Verheije, M.H. The avian coronavirus spike protein. Virus Res. 2014, 194, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.Y.; Chen, T.; Zhang, X.; Shao, G.M.; Cao, Y.; Chen, D.K.; Lin, W.C.; Chen, F.; Xie, Q.M. Molecular characteristic and pathogenicity analysis of a virulent recombinant avian infectious bronchitis virus isolated in China. Poult. Sci. 2018, 97, 3519–3531. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.Y.; Wang, F.; Xue, Y.; Zhou, Q.F.; Chen, F.; Bi, Y.Z.; Xie, Q.M. Epidemiology and characterization of avian infectious bronchitis virus strains circulating in southern China during the period from 2013–2015. Sci. Rep. 2017, 7, 6576. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Chen, H.W. Infectious bronchitis virus variants: Molecular analysis and pathogenicity investigation. Int. J. Mol. Sci. 2017, 18, 2030. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.W.; Ren, M.T.; Sheng, J.; Ma, T.X.; Han, Z.X.; Zhao, Y.; Sun, J.F.; Liu, S.W. Genetic and biological characteristics of four novel recombinant avian infectious bronchitis viruses isolated in China. Virus Res. 2019, 263, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Valastro, V.; Holmes, E.C.; Britton, P.; Fusaro, A.; Jackwood, M.W.; Cattoli, G.; Monne, I. S1 gene-based phylogeny of infectious bronchitis virus: An attempt to harmonize virus classification. Infect. Genet. Evol. 2016, 39, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Bande, F.; Arshad, S.S.; Omar, A.R.; Hair-Bejo, M.; Mahmuda, A.; Nair, V. Global distributions and strain diversity of avian infectious bronchitis virus: A review. Anim. Health Res. Rev. 2017, 18, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Marandino, A.; Vagnozzi, A.; Craig, M.I.; Tomas, G.; Techera, C.; Panzera, Y.; Vera, F.; Perez, R. Genetic and antigenic heterogeneity of infectious bronchitis virus in South America: Implications for control programmes. Avian Pathol. 2019, 48, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Irvine, R.M.; Cox, W.J.; Ceeraz, V.; Reid, S.M.; Ellis, R.J.; Jones, R.M.; Errington, J.; Wood, A.M.; McVicar, C.; Clark, M.I. Detection of IBV QX in commercial broiler flocks in the UK. Vet. Rec. 2010, 167, 877–879. [Google Scholar] [CrossRef] [PubMed]
- Naguib, M.M.; Hoeper, D.; Arafa, A.; Setta, A.M.; Abed, M.; Monne, I.; Beer, M.; Harder, T.C. Full genome sequence analysis of a newly emerged QX-like infectious bronchitis virus from Sudan reveals distinct spots of recombination. Infect. Genet. Evol. 2016, 46, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Massi, P.; Tucciarone, C.M.; Barbieri, I.; Tosi, G.; Fiorentini, L.; Ciccozzi, M.; Lavazza, A.; Cecchinato, M.; Moreno, A. Think globally, act locally: Phylodynamic reconstruction of infectious bronchitis virus (IBV) QX genotype (GI-19 lineage) reveals different population dynamics and spreading patterns when evaluated on different epidemiological scales. PLoS ONE 2017, 12, e0184401. [Google Scholar] [CrossRef] [PubMed]
- Hamadan, A.M.; Ghalyanchilangeroudi, A.; Hashemzadeh, M.; Hosseini, H.; Karimi, V.; Yahyaraeyat, R.; Najafi, H. Genotyping of avian infectious bronchitis viruses in Iran (2015–2017) reveals domination of IS-1494 like virus. Virus Res. 2017, 240, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Khanh, N.P.; Tan, S.W.; Yeap, S.K.; Satharasinghe, D.A.; Hair-Bejo, M.; Bich, T.N.; Omar, A.R. Molecular Characterization of QX-Like and Variant Infectious Bronchitis Virus Strains in Malaysia Based on Partial Genomic Sequences Comprising the S-3a/3b-E-M-Intergenic Region-5a/5b-N Gene Order. Avian Dis. 2017, 61, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, M.W.; Lee, D.H. Different evolutionary trajectories of vaccine-controlled and non-controlled avian infectious bronchitis viruses in commercial poultry. PLoS ONE 2017, 12, e0176709. [Google Scholar] [CrossRef] [PubMed]
- Toro, H.; van Santen, V.L.; Jackwood, M.W. Genetic diversity and selection regulates evolution of infectious bronchitis virus. Avian Dis. 2012, 56, 449–455. [Google Scholar] [CrossRef] [PubMed]
- McKinley, E.T.; Hilt, D.A.; Jackwood, M.W. Avian coronavirus infectious bronchitis attenuated live vaccines undergo selection of subpopulations and mutations following vaccination. Vaccine 2008, 26, 1274–1284. [Google Scholar] [CrossRef]
- Van Santen, V.L.; Toro, H. Rapid selection in chickens of subpopulations within ArkDPI-derived infectious bronchitis virus vaccines. Avian Pathol. 2008, 37, 293–306. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhang, H.; Zhao, J.; Zhong, Q.; Jin, J.H.; Zhang, G.Z. Evolution of infectious bronchitis virus in China over the past two decades. J. Gen. Virol. 2016, 97, 1566–1574. [Google Scholar] [CrossRef]
- Li, H.J.; Wang, P.K.; Lin, L.L.; Shi, M.Y.; Gu, Z.M.; Huang, T.; Mo, M.L.; Wei, T.C.; Zhang, H.M.; Wei, P. The emergence of the infection of subgroup J avian leucosis virus escalated the tumour incidence in commercial Yellow chickens in Southern China in recent years. Transbound Emerg. Dis. 2019, 66, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Liang, Y.D.; Wei, F.Y. Current status of poultry production in Guangxi and prospect of poultry industry in China. China Poult. 2013, 35, 2–6. (In Chinese) [Google Scholar]
- Wei, P. Challenge and development opportunity of quality chicken industry in China: A review about 9 issues on the topic. China Poult. 2019, 41, 1–6. (In Chinese) [Google Scholar]
- Wu, X.; Yang, X.; Xu, P.W.; Zhou, L.; Zhang, Z.K.; Wang, H.N. Genome sequence and origin analyses of the recombinant novel IBV virulent isolate SAIBK2. Virus Genes 2016, 52, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; He, X.; Yao, K.C.; Du, L.J.; Liu, P.; Yan, Q.G.; Wen, Y.P.; Cao, S.J.; Han, X.F.; Huang, Y. Phylogenetic and antigenic analysis of avian infectious bronchitis virus in southwestern China, 2012–2016. Infect. Genet. Evol. 2016, 45, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Mo, M.L.; Hong, S.M.; Kwon, H.J.; Kim, I.H.; Song, C.S.; Kim, J.H. Genetic Diversity of Spike, 3a, 3b and E Genes of Infectious Bronchitis Viruses and Emergence of New Recombinants in Korea. Viruses 2013, 5, 550–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Carranza, C.; Astolfi-Ferreira, C.S.; Santander Parra, S.H.; Nunez, L.F.N.; Penzes, Z.; Chacon, J.L.; Sesti, L.; Chacon, R.D.; Piantino Ferreira, A.J. Genetic characterisation and analysis of infectious bronchitis virus isolated from Brazilian flocks between 2010 and 2015. Brit. Poult. Sci. 2017, 58, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods. 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.C.; Deng, Q.M.; Zhai, G.S.; He, C.W.; Li, H.Q.; Zhang, Y.Q.; Zeng, R.L.; Mo, M.L.; Huang, T.; Wei, P. Re-emergence of a genotype VIII virulent Newcastle disease virus isolated from Chinese game fowl after 13years. Transbound Emerg. Dis. 2019, 66, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.J.; Gao, M.Y.; Xu, Q.Q.; Xu, Y.; Zhao, Y.; Chen, Y.Q.; Zhang, T.T.; Wang, Q.L.; Han, Z.X.; Li, H.X.; et al. Origin and evolution of LX4 genotype infectious bronchitis coronavirus in China. Vet. Microbiol. 2017, 198, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Q.; Jiang, L.; Zhao, W.J.; Liu, L.L.; Zhao, Y.; Shao, Y.H.; Li, H.X.; Han, Z.X.; Liu, S.W. Identification and molecular characterization of a novel serotype infectious bronchitis virus (GI-28) in China. Vet. Microbiol. 2017, 198, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.W.; Han, Z.X.; Jiang, L.; Sun, J.F.; Zhao, Y.; Liu, S.W. Genetic diversity of avian infectious bronchitis virus in China in recent years. Infect. Genet. Evol. 2018, 66, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Tsai, C.T. Genetic grouping for the isolates of avian infectious bronchitis virus in Taiwan. Arch. Virol. 1996, 141, 1677–1688. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.P.; Wang, C.H. Development of attenuated vaccines from Taiwanese infectious bronchitis virus strains. Vaccine 2006, 24, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.Q.; Han, Z.X.; Wang, Q.L.; Zhang, T.T.; Gao, M.Y.; Zhao, Y.; Shao, Y.H.; Li, H.X.; Kong, X.G.; Liu, S.W. Emergence of novel nephropathogenic infectious bronchitis viruses currently circulating in Chinese chicken flocks. Avian Pathol. 2016, 45, 54–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.K.; Zhou, Y.S.; Wang, H.N.; Zeng, F.Y.; Yang, X.; Zhang, Y.; Zhang, A.Y. Molecular detection and smoothing spline clustering of the IBV strains detected in China during 2011–2012. Virus Res. 2016, 211, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Zou, N.L.; Zhao, F.F.; Wang, Y.P.; Liu, P.; Cao, S.J.; Wen, X.T.; Huang, Y. Genetic analysis revealed LX4 genotype strains of avian infectious bronchitis virus became predominant in recent years in Sichuan area, China. Virus Genes 2010, 41, 202–209. [Google Scholar] [CrossRef]
- Luo, H.B.; Qin, J.P.; Chen, F.; Xie, Q.M.; Bi, Y.Z.; Cao, Y.C.; Xue, C.Y. Phylogenetic analysis of the S1 glycoprotein gene of infectious bronchitis viruses isolated in China during 2009–2010. Virus Genes 2012, 44, 19–23. [Google Scholar] [CrossRef]
- Xu, G.; Liu, X.Y.; Zhao, Y.; Chen, Y.; Zhao, J.; Zhang, G.Z. Characterization and analysis of an infectious bronchitis virus strain isolated from southern China in 2013. Virol. J. 2016, 13, 40. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.C.; Chu, P.Y.; Chang, M.Y.; Hsiao, K.L.; Lin, J.H.; Liu, H.F. Spatial temporal dynamics and molecular evolution of re-emerging rabies virus in Taiwan. Int. J. Mol. Sci. 2016, 17, 392. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.W.; Chen, J.F.; Chen, J.D.; Kong, X.G.; Shao, Y.H.; Han, Z.X.; Feng, L.; Cai, X.H.; Gu, S.L.; Liu, M. Isolation of avian infectious bronchitis coronavirus from domestic peafowl (Pavo cristatus) and teal (Anas). J. Gen. Virol. 2005, 86, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.K.; Leung, C.Y.; Gilbert, M.; Joyner, P.H.; Ng, E.M.; Tse, T.M.; Guan, Y.; Peiris, J.S.; Poon, L.L. Avian coronavirus in wild aquatic birds. J. Virol. 2011, 85, 12815–12820. [Google Scholar] [CrossRef] [PubMed]
- Li, L.L.; Xue, C.Y.; Chen, F.; Qin, J.P.; Xie, Q.M.; Bi, Y.Z.; Cao, Y.C. Isolation and genetic analysis revealed no predominant new strains of avian infectious bronchitis virus circulating in South China during 2004–2008. Vet. Microbiol. 2010, 143, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zhao, W.J.; Han, Z.X.; Chen, Y.Q.; Zhao, Y.; Sun, J.F.; Li, H.X.; Shao, Y.H.; Liu, L.L.; Liu, S.W. Genome characterization, antigenicity and pathogenicity of a novel infectious bronchitis virus type isolated from south China. Infect. Genet. Evol. 2017, 54, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; (Guangxi University, Nanning, China). Sequence analysis of structural genes and serotype identification of Guangxi infectious bronchitis virus isolate from 2015 to 2017. Personal communication, 2018. [Google Scholar]
- Abozeid, H.H.; Paldurai, A.; Khattar, S.K.; Afifi, M.A.; El-Kady, M.F.; El-Deeb, A.H.; Samal, S.K. Complete genome sequences of two avian infectious bronchitis viruses isolated in Egypt: Evidence for genetic drift and genetic recombination in the circulating viruses. Infect. Genet. Evol. 2017, 53, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Fraga, A.P.; Graf, T.; Pereira, C.S.; Ikuta, N.; Kazantzi Fonseca, A.S.; Lunge, V.R. Phylodynamic analysis and molecular diversity of the avian infectious bronchitis virus of chickens in Brazil. Infect. Genet. Evol. 2018, 61, 77–83. [Google Scholar] [CrossRef]
- Zhou, H.S.; Zhang, M.H.; Tian, X.; Shao, H.X.; Qian, K.; Ye, J.Q.; Qin, A.J. Identification of a novel recombinant virulent avian infectious bronchitis virus. Vet. Microbiol. 2017, 199, 120–127. [Google Scholar] [CrossRef]
- Han, Z.X.; Zhao, W.J.; Chen, Y.Q.; Xu, Q.Q.; Sun, J.F.; Zhang, T.T.; Zhao, Y.; Liang, S.L.; Gao, M.Y.; Wang, Q.L.; et al. Genetic, antigenic, and pathogenic characteristics of avian infectious bronchitis viruses genotypically related to 793/B in China. Vet. Microbiol. 2017, 203, 125–135. [Google Scholar] [CrossRef]
- Schikora, B.M.; Shih, L.M.; Hietala, S.K. Genetic diversity of avian infectious bronchitis virus California variants isolated between 1988 and 2001 based on the S1 subunit of the spike glycoprotein. Arch. Virol. 2003, 148, 115–136. [Google Scholar] [CrossRef]
- Tang, X.C.; Li, G.; Vasilakis, N.; Zhang, Y.; Shi, Z.L.; Zhong, Y.; Wang, L.F.; Zhang, S.Y. Differential stepwise evolution of SARS coronavirus functional proteins in different host species. BMC Evol. Biol. 2009, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- McKinley, E.T.; Jackwood, M.W.; Hilt, D.A.; Kissinger, J.C.; Robertson, J.S.; Lemke, C.; Paterson, A.H. Attenuated live vaccine usage affects accurate measures of virus diversity and mutation rates in avian coronavirus infectious bronchitis virus. Virus Res. 2011, 158, 225–234. [Google Scholar] [CrossRef] [PubMed]
- An, D.J.; Lim, S.I.; Choe, S.E.; Kim, K.S.; Cha, R.M.; Cho, I.S.; Song, J.Y.; Hyun, B.H.; Park, B.K. Evolutionary dynamics of classical swine fever virus in south Korea: 1987–2017. Vet. Microbiol. 2018, 225, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.M.; Li, H.P.; Wu, X.Z.; Zhong, Y.X.; Zhang, K.Q.; Zhang, Y.P.; Boerwinkle, E.; Fu, Y.X. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol. Biol. 2004, 4, 21. [Google Scholar] [CrossRef]
- Kuroda, M.; Niwa, S.; Sekizuka, T.; Tsukagoshi, H.; Yokoyama, M.; Ryo, A.; Sato, H.; Kiyota, N.; Noda, M.; Kozawa, K.; et al. Molecular evolution of the VP1, VP2, and VP3 genes in human rhinovirus species C. Sci. Rep. 2015, 5, 8185. [Google Scholar] [CrossRef]
- Shaha, M.; Chakraborty, S.; Hossain, M.S.; Hashem, A.; Salimullah, M. Molecular evolution and genomics of hepatitis B virus subgenotype C2 strain predominant in the chronic patients in Bangladesh. Virus Dis. 2018, 29, 486–490. [Google Scholar] [CrossRef]
- Franzo, G.; Naylor, C.J.; Lupini, C.; Drigo, M.; Catelli, E.; Listorti, V.; Pesente, P.; Giovanardi, D.; Morandini, E.; Cecchinato, M. Continued use of IBV 793B vaccine needs reassessment after its withdrawal led to the genotype’s disappearance. Vaccine 2014, 32, 6765–6767. [Google Scholar] [CrossRef]
- Franzo, G.; Tucciarone, C.M.; Blanco, A.; Nofrarias, M.; Biarnes, M.; Cortey, M.; Majo, N.; Catelli, E.; Cecchinato, M. Effect of different vaccination strategies on IBV QX population dynamics and clinical outbreaks. Vaccine 2016, 34, 5670–5676. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IBV Isolates | Years of Isolation | Days of Age | Locations a | GenBank Accession Numbers | |||
|---|---|---|---|---|---|---|---|
| S1 | N | M | E | ||||
| GX-NN09032 | 2009 | N/A | Nanning | JX292013 | JX567013 | JX567012 | KJ872796 |
| GX-NN09093 | 2009 | 25 | Nanning | JX292011 | JX273225 | JX273198 | KJ940533 |
| GX-GL | 2010 | N/A | Guilin | JX292008 | JX273221 | JX014370 | KJ872786 |
| GX-HC1006 | 2010 | N/A | Hechi | JX292009 | JX273229 | JX273194 | KJ940510 |
| GX-NN1009 | 2010 | N/A | Nanning | JX292000 | JX273213 | JX014366 | KJ872793 |
| GX-NN1011 | 2010 | N/A | Nanning | JX292001 | JX273216 | JX014371 | KJ940528 |
| GX-NN1012 | 2010 | N/A | Nanning | JX292002 | JX273217 | JX014372 | KJ940529 |
| GX-NN1013 | 2010 | N/A | Nanning | JX292003 | JX273218 | JX014373 | KJ940530 |
| GX-NN1014 | 2010 | N/A | Nanning | JX292004 | JX273215 | JX014367 | KJ940531 |
| GX-NN1019 | 2010 | N/A | Nanning | JX292005 | JX273219 | JX014368 | KJ872794 |
| GX-GL11077 | 2011 | 17 | Guilin | JX291992 | JX273212 | JX273186 | KJ940507 |
| GX-GL11078 | 2011 | 11 | Guilin | JX291993 | JX273209 | JX273187 | KJ940508 |
| GX-GL11079 | 2011 | N/A | Guilin | JX291994 | JX273208 | JX273188 | KJ940509 |
| GX-NN-4 | 2011 | 32 | Nanning | JX291983 | JX273199 | JX273176 | KJ940515 |
| GX-NN-5 | 2011 | 77 | Nanning | JX291984 | JX273200 | JX273177 | KJ940517 |
| GX-NN-6 | 2011 | 18 | Nanning | JX291985 | JX273207 | JX273178 | KJ940519 |
| GX-NN-8 | 2011 | 27 | Nanning | JX291986 | JX273201 | JX273179 | KJ940520 |
| GX-NN-9 | 2011 | 38 | Nanning | JX291987 | JX273202 | JX273180 | KJ940521 |
| GX-NN-11 | 2011 | 45 | Nanning | JX291988 | JX273203 | JX273182 | KJ940523 |
| GX-NN-13 | 2011 | 39 | Nanning | JX291989 | JX273204 | JX273183 | KJ940525 |
| GX-NN-14 | 2011 | 34 | Nanning | JX291990 | JX273205 | JX273184 | KJ940526 |
| GX-NN-15 | 2011 | 15 | Nanning | JX291991 | JX273206 | JX273185 | KJ940527 |
| GX-NN11034 | 2011 | 12 | Nanning | JX291999 | JX273223 | JX273191 | KJ940535 |
| GX-YL11072 | 2011 | 15 | Yulin | JX291995 | JX273210 | JX273189 | KJ940544 |
| GX-YL11073 | 2011 | 14 | Yulin | JX291996 | JX273211 | JX273190 | KJ940545 |
| GX-NN1201 | 2012 | 14 | Nanning | JX436331 | JX567014 | JX567012 | KJ940532 |
| GX-NN120079 | 2012 | 35 | Nanning | KJ999803 | KF996273 | KF996277 | KM278995 |
| GX-NN120084 | 2012 | 103 | Nanning | KJ999804 | KF996274 | KF996278 | KM278996 |
| GX-NN120089 | 2012 | 43 | Nanning | KJ999805 | KF996275 | KF996279 | KM278997 |
| GX-NN120091 | 2012 | 46 | Nanning | KJ999806 | KF996276 | KF996280 | KM278998 |
| GX-NN130003 | 2013 | 22 | Nanning | KJ999794 | KF975404 | KF996281 | KM278999 |
| GX-YL130025 | 2013 | 15 | Yulin | KJ999795 | KF975405 | KF996283 | KM279001 |
| GX-NN130048 | 2013 | 22 | Nanning | KJ999796 | KF975406 | KJ940503 | KM279002 |
| GX-YL13080630 | 2013 | 30 | Yulin | KJ999797 | KF975407 | KF996286 | KM279003 |
| GX-YL130806200 | 2013 | 200 | Yulin | KJ999798 | KJ940498 | KJ940501 | KM279004 |
| GX-NN130059 | 2013 | 15 | Nanning | KJ999799 | KF975408 | KF996285 | KM279005 |
| GX-QZ131126 | 2013 | 80 | Qinzhou | KJ999800 | KJ940499 | KJ940502 | KM279006 |
| GX-QZ130064 | 2013 | 35 | Qinzhou | KJ999801 | KJ940500 | KJ940504 | KM279007 |
| GX-QZ130065 | 2013 | 37 | Qinzhou | KT149876 | KT188789 | KT188788 | KM279008 |
| GX-NN130067 | 2013 | 52 | Nanning | KJ999802 | KJ940497 | KJ940505 | KM279009 |
| GX-NN130021 | 2013 | 21 | Nanning | KP085589 | KF834569 | KF996282 | KM279000 |
| GX-NN150019 | 2015 | 20 | Nanning | MK887049 | MK887118 | MK887095 | MK887072 |
| GX-QZ150024 | 2015 | 13 | Qinzhou | MK887057 | MK887126 | MK887103 | MK887080 |
| GX-YL150028 | 2015 | 20 | Yulin | MK887060 | MK887129 | MK887106 | MK887083 |
| GX-LZ150619 | 2015 | 19 | Liuzhou | MK887046 | MK887115 | MK887092 | MK887069 |
| GX-YL150727 | 2015 | 73 | Yulin | MK887061 | MK887130 | MK887107 | MK887084 |
| GX-LZ160322 | 2016 | 10 | Liuzhou | MK887047 | MK887116 | MK887093 | MK887070 |
| GX-YL161022 | 2016 | 15 | Yulin | MK887063 | MK887132 | MK887109 | MK887086 |
| GX-YL161015 | 2016 | 100 | Yulin | MK887062 | MK887131 | MK887108 | MK887085 |
| HN-170502 | 2017 | 90 | Hainan | MK887045 | MK887114 | MK887091 | MK887068 |
| GX-LZ170609 | 2017 | 43 | Liuzhou | MK887048 | MK887117 | MK887094 | MK887071 |
| GX-YL170717 | 2017 | 105 | Yulin | MK887064 | MK887133 | MK887110 | MK887087 |
| GX-YL170718 | 2017 | 24 | Yulin | MK887065 | MK887134 | MK887111 | MK887088 |
| GX-QZ170728 | 2017 | 194 | Qinzhou | MK887058 | MK887127 | MK887104 | MK887081 |
| GX-YL170805 | 2017 | 24 | Yulin | MK887066 | MK887135 | MK887112 | MK887089 |
| GX-YL170808 | 2017 | 140 | Yulin | MK887067 | MK887136 | MK887113 | MK887090 |
| GX-NN170502 | 2017 | 118 | Nanning | MK887050 | MK887119 | MK887096 | MK887073 |
| GX-NN170829 | 2017 | 27 | Nanning | MK887052 | MK887121 | MK887098 | MK887075 |
| GX-NN170825 | 2017 | 11 | Nanning | MK887051 | MK887120 | MK887097 | MK887074 |
| GX-QZ171023 | 2017 | 9 | Qinzhou | MK887059 | MK887128 | MK887105 | MK887082 |
| GX-NN170901 | 2017 | 20 | Nanning | MK887053 | MK887122 | MK887099 | MK887076 |
| GX-NN171108 | 2017 | 52 | Nanning | MK887054 | MK887123 | MK887100 | MK887077 |
| GX-NN171123 | 2017 | 60 | Nanning | MK887055 | MK887124 | MK887101 | MK887078 |
| GX-NN171125 | 2017 | 42 | Nanning | MK887056 | MK887125 | MK887102 | MK887079 |
| Potential Recombinant | Breakpoints | Major Parent (Similarity) | Minor Parent (Similarity) | Detection Method a | |
|---|---|---|---|---|---|
| Beginning | Ending | ||||
| GX-NN1011 | 1040 | 1482 | GX-NN6 (LX4-type) (91.1%) | GX-NN-9 (LX4-type) (95.1%) | RDP, GENECONV, MaxChi, Chimera, SiScan, 3Seq |
| GX-NN1201 | 621 | 1612 | QX (LX4-type) (96%) | SAIBK (CK/CH/LSC/99I-type) (98.5%) | RDP, GENECONV, Bootscan, MaxChi, Chimera, SiScan, 3Seq |
| GX-NN120091 | 100 | 1351 | GX-NN120089 (New-type 1) (99.2%) | LDT3-A (99.1%) | RDP, GENECONV, Bootscan, MaxChi, SiScan, 3Seq |
| GX-NN130003 | 1 | 916 | GX-YL11072 (LDT3-A-type) (96.9%) | GX-YL170717 (CK/CH/LSC/99I-type) (95.8%) | MaxChi, Chimera, SiScan, 3Seq |
| GX-NN130048 | 611 | 1616 | GX-HC1006 (LX4-type) (98.1%) | 4/91 (99.6%) | RDP, GENECONV, Bootscan, MaxChi, SiScan, 3Seq |
| GX-YL170805 | 725 | 1517 | GX-NN1014 (Taiwan-I-type) (97.8%) | GX-NN10 (4/91-type) (98.5%) | RDP, GENECONV, Bootscan, MaxChi, Chimera, SiScan, 3Seq |
| Model | S1 Protein | E Protein | M Protein | N Protein | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SLAC | FEL | IFEL | SLAC | FEL | IFEL | SLAC | FEL | IFEL | SLAC | FEL | IFEL | |
| Mean dN/dS | 0.283 | 0.266 | 0.146 | 0.147 | ||||||||
| Numbers of positive selection site | 13 | 13 | 15 | 1 | 8 | 7 | 5 | 5 | 6 | 6 | 11 | 9 |
| Numbers of neutral selection site | 366 | 277 | 287 | 85 | 65 | 69 | 136 | 119 | 127 | 246 | 196 | 209 |
| Numbers of purifying selection site | 178 | 267 | 255 | 33 | 46 | 43 | 85 | 102 | 93 | 157 | 202 | 191 |
| Positively selected sites (aa) | 19, 39, 106, 119, 155, 503 | 119 | 12, 16, 17, 47, 151 | 7, 10, 235, 342, 345 | ||||||||
| Protein | Position | Sequence | Potential | Agreement | N-Glyc Result | N-linked Gylcosylation Sites/Strains | Percentage/% |
|---|---|---|---|---|---|---|---|
| S1 | 239/240 a 427/428 b | NFSD NITL | 0.5840–0.5851 0.5598–0.6254 | (7/9) (7/9) | + + | 14 (3/91) | 3.30 |
| 15 (3/91) | 3.30 | ||||||
| 16 (1/91) | 1.10 | ||||||
| 17 (3/91) | 3.30 | ||||||
| 18 (18/91) | 19.78 | ||||||
| 19 (29/91) | 31.87 | ||||||
| 20 (15/91) | 16.48 | ||||||
| 21 (17/91) | 18.68 | ||||||
| 22 (2/91) | 2.20 | ||||||
| E | 11/12 5/6 c | NGSF NKSL | 0.5228–0.5336 0.6297–0.7593 | (6/9) (9/9) | + ++ | 1 (2/91) | 1.10 |
| 2 (49/91) | 53.85 | ||||||
| 3 (40/91) | 43.96 | ||||||
| M | 3/4/6 | NCTL | 0.6007–0.7913 | (7/9)/ (9/9) | + ++ | 1 (7/91) | 7.69 |
| 2 (84/91) | 92.31 | ||||||
| N | 32 d | NASW | 0.5564–0.7239 | (6/9)/ (9/9) | + ++ | 0 (1/91) | 1.10 |
| 1(38/91) | 41.96 | ||||||
| 2 (51/91) | 56.04 | ||||||
| 3 (1/91) | 1.10 |
| Structural Genes | Mutation/Evolutionary Rates (Substitutions/Site/Year) | |
|---|---|---|
| Mean | 95 % HPD | |
| S1 | 4.6 × 10−3 | 3.7 × 10−3–5.5 × 10−3 |
| E | 4.3 × 10−3 | 2.8 × 10−3–5.8 × 10−3 |
| M | 3.9 × 10−3 | 1.2 × 10−3–4.6 × 10−3 |
| N | 3.7 × 10−3 | 2.7 × 10−3–4.7 × 10−3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, W.; Tang, N.; Dong, Z.; Chen, J.; Zhang, W.; Zhao, C.; He, Y.; Li, M.; Wu, C.; Wei, T.; et al. Genetic Analysis of Avian Coronavirus Infectious Bronchitis Virus in Yellow Chickens in Southern China over the Past Decade: Revealing the Changes of Genetic Diversity, Dominant Genotypes, and Selection Pressure. Viruses 2019, 11, 898. https://doi.org/10.3390/v11100898
Fan W, Tang N, Dong Z, Chen J, Zhang W, Zhao C, He Y, Li M, Wu C, Wei T, et al. Genetic Analysis of Avian Coronavirus Infectious Bronchitis Virus in Yellow Chickens in Southern China over the Past Decade: Revealing the Changes of Genetic Diversity, Dominant Genotypes, and Selection Pressure. Viruses. 2019; 11(10):898. https://doi.org/10.3390/v11100898
Chicago/Turabian StyleFan, Wensheng, Ning Tang, Zhihua Dong, Jiming Chen, Wen Zhang, Changrun Zhao, Yining He, Meng Li, Cuilan Wu, Tianchao Wei, and et al. 2019. "Genetic Analysis of Avian Coronavirus Infectious Bronchitis Virus in Yellow Chickens in Southern China over the Past Decade: Revealing the Changes of Genetic Diversity, Dominant Genotypes, and Selection Pressure" Viruses 11, no. 10: 898. https://doi.org/10.3390/v11100898
APA StyleFan, W., Tang, N., Dong, Z., Chen, J., Zhang, W., Zhao, C., He, Y., Li, M., Wu, C., Wei, T., Huang, T., Mo, M., & Wei, P. (2019). Genetic Analysis of Avian Coronavirus Infectious Bronchitis Virus in Yellow Chickens in Southern China over the Past Decade: Revealing the Changes of Genetic Diversity, Dominant Genotypes, and Selection Pressure. Viruses, 11(10), 898. https://doi.org/10.3390/v11100898