1. Introduction
The World Health Organization stated in February 2016 that Zika infection was considered as a public health emergency of international concern [
1] opening a new chapter in the history of vector-borne diseases. Arboviruses are viruses transmitted among vertebrate hosts by arthropod vectors. Successful transmission of an arbovirus relies on a complex life cycle in the vector, which after midgut infection and dissemination, is released in saliva for active transmission to the vertebrate host [
2]. Arboviruses belong to nine families: Asfarviridae, Flaviviridae, Orthomyxoviridae, Reoviridae, Rhabdoviridae, the newly recognized Nyamiviridae (order Mononegavirales), and the families Nairoviridae, Phenuiviridae, and Peribunyaviridae in the new order, Bunyavirales. Most arboviruses possess an RNA genome and are mainly transmitted by mosquitoes [
3]. While acute infections in vertebrate hosts are typically self-limiting, arboviruses establish persistent infections in arthropods granting a central role as a viral reservoir to the vector [
4].
Arboviruses circulate primarily within an enzootic cycle involving zoophilic vector species and non-human hosts. Recurrent spillovers cause occasional infections of humans initiating an epidemic cycle. Arboviruses such as dengue (DENV;
Flavivirus, Flaviviridae), chikungunya (CHIKV;
Alphavirus, Togaviridae), Zika (ZIKV;
Flavivirus, Flaviviridae), and Yellow fever virus (YFV;
Flavivirus, Flaviviridae) do not need to amplify in wild animals to cause outbreaks in humans, which act simultaneously as amplifier, disseminator, and source of infection for the major vectors, the anthropophilic mosquitoes
Aedes aegypti and
Aedes albopictus [
5]. Thus, the success of these viruses comes from their feature to be mainly transmitted by human-biting mosquitoes strongly adapted to urban environments. The establishment of a new epidemic cycle is undoubtedly related to the introduction of a viremic vertebrate host (humans, animals) acting as a vehicle for importation of the virus into environments receptive to viral amplification. Other arboviruses such as West Nile virus (WNV;
Flavivirus, Flaviviridae) remain circulating within an enzootic cycle with sporadic spillovers causing human cases.
Many regions experience simultaneous circulation of different arboviruses [
6,
7], and co-infections in vectors were reported [
8]. These coinfections can present an opportunity for viruses to exchange genetic material. Impacts of such genetic events on virulence for vertebrate hosts are still unknown [
9]. Thus, being able to detect a wide range of arboviruses in thousands of field-collected mosquitoes in a single experiment can be a valuable tool to predict arboviral emergences in human populations. Indeed, similar methods were developed with success to screen tick-borne pathogens (bacteria, parasites, and viruses) and allowed the detection of expected and unexpected pathogens in large-scale epidemiological studies [
10,
11]. Therefore, we developed a high-throughput system based on real-time microfluidic PCR, which is able to detect 96 mosquito-borne viruses in 96 samples within one single run. With this method, we have screened: (1) Mosquitoes infected artificially using a feeding system to validate our tool, (2) mosquitoes collected in countries endemic for the major human arboviruses (e.g., Senegal, Cambodia, Brazil), and (3) mosquitoes collected during the Zika and Yellow fever outbreaks in the Americas (French Guiana, Guadeloupe, Brazil, Suriname). This method allowed for the detection of epidemic viruses (ZIKV, CHIKV, YFV) but also unexpected viruses (e.g., Trivittatus virus, TVTV,
Orthobunyavirus, Bunyaviridae) underlining the need of such a tool for early detection of emerging mosquito-borne viruses.
2. Materials and Methods
2.1. Mosquitoes
To test the ability of our assays to detect viruses present in pools of mosquitoes, 47 batches of three infected mosquitoes of the species,
Ae. aegypti,
Ae. albopictus, and
Cx. pipiens (infection performed by artificial feeding system), were provided by the Institut Pasteur (Paris). Six different viruses, single or double infections, were tested in a pilot study. Briefly, batches of 60 7–10-day-old females were challenged with an infectious blood meal containing 1.4 mL of washed rabbit erythrocytes, 700 μL of viral suspension, and 1 mM of adenosine 5’-triphosphate (ATP) as a phagostimulant [
12]. The blood meal was provided to mosquitoes at a titer of 10
7 focus-forming unit (FFU)/mL using a Hemotek membrane feeding system (Hemotek Ltd., Blackburn, UK). After 20 min, fully engorged females were transferred in cardboard containers and maintained with 10% sucrose until examination.
In ZIKV-endemic and -epidemic regions from South America, Africa, and Asia (Brazil, French Guiana, Guadeloupe, Suriname, Senegal, Cambodia), adult mosquitoes were collected, identified using morphological characters, and dissected to separate abdomen from the remaining body parts (RBP) (See Tables 1–6 for details). Abdomens of the same species were grouped by pools of 20–30 individuals in cryovials, and RBP were stored individually at −80 °C until further analysis.
2.2. RNA Extraction
Total RNAs were extracted from each pool using the Nucleospin RNA II extraction kit (Macherey-Nagel, Hoerdt, France). Pools were ground in 350 µL Lysis Buffer and 3.5 µL β-mercaptoethanol using the homogenizer Precellys®24 Dual (Bertin, France) at 5500 rpm for 20 s. Total RNA per pool was eluted in 50 µL of RNase free water and stored at −80 °C until use.
When pools of abdomens were positive for virus, the RBP (head/thorax) of individual mosquitoes composing each pool were homogenized in 300 µL of DMEM with 10% fetal calf serum using the homogenizer Precellys®24 Dual (Bertin, France) at 5500 rpm for 20 s. Then, total RNAs were extracted from 100 µL of homogenates using the Nucleospin RNA II extract kit (Macherey-Nagel, Germany) and 200 µL were conserved at −80 °C for attempts to isolate the virus. Total RNA per sample was eluted in 50 µL of RNase free water and stored at −80 °C until use.
2.3. Reverse Transcription and cDNA Pre-Amplification
RNAs were transcribed to cDNA by reverse transcription using the qScript cDNA Supermix kit according to the manufacturer’s instructions (Quanta Biosciences, Beverly, USA). Briefly, the reaction was performed in a final volume of 5 µL containing 1 µL of qScript cDNA supermix 5X, 1 µL of RNA, and 3 µL of RNase free water; with one cycle at 25 °C for 5 min, one cycle at 42 °C for 30 min, and one final cycle at 85 °C for 5 min.
For cDNA pre-amplification, the Perfecta Preamp Supermix (Quanta Biosciences, Beverly, USA) was used according to the manufacturer’s instructions. All primers were pooled to a final concentration of 200 nM each. The reaction was performed in a final volume of 5 μL containing 1 μL Perfecta Preamp 5×, 1.25 μL pooled primers, 1.5 µL distilled water, and 1.25 μL cDNA, with one cycle at 95 °C for 2 min, 14 cycles at 95 °C for 10 s, and 3 min at 60 °C. At the end of the cycling program, the reactions were 1:5 diluted. Pre-amplified cDNAs were stored at −20 °C until use.
2.4. Assay Design
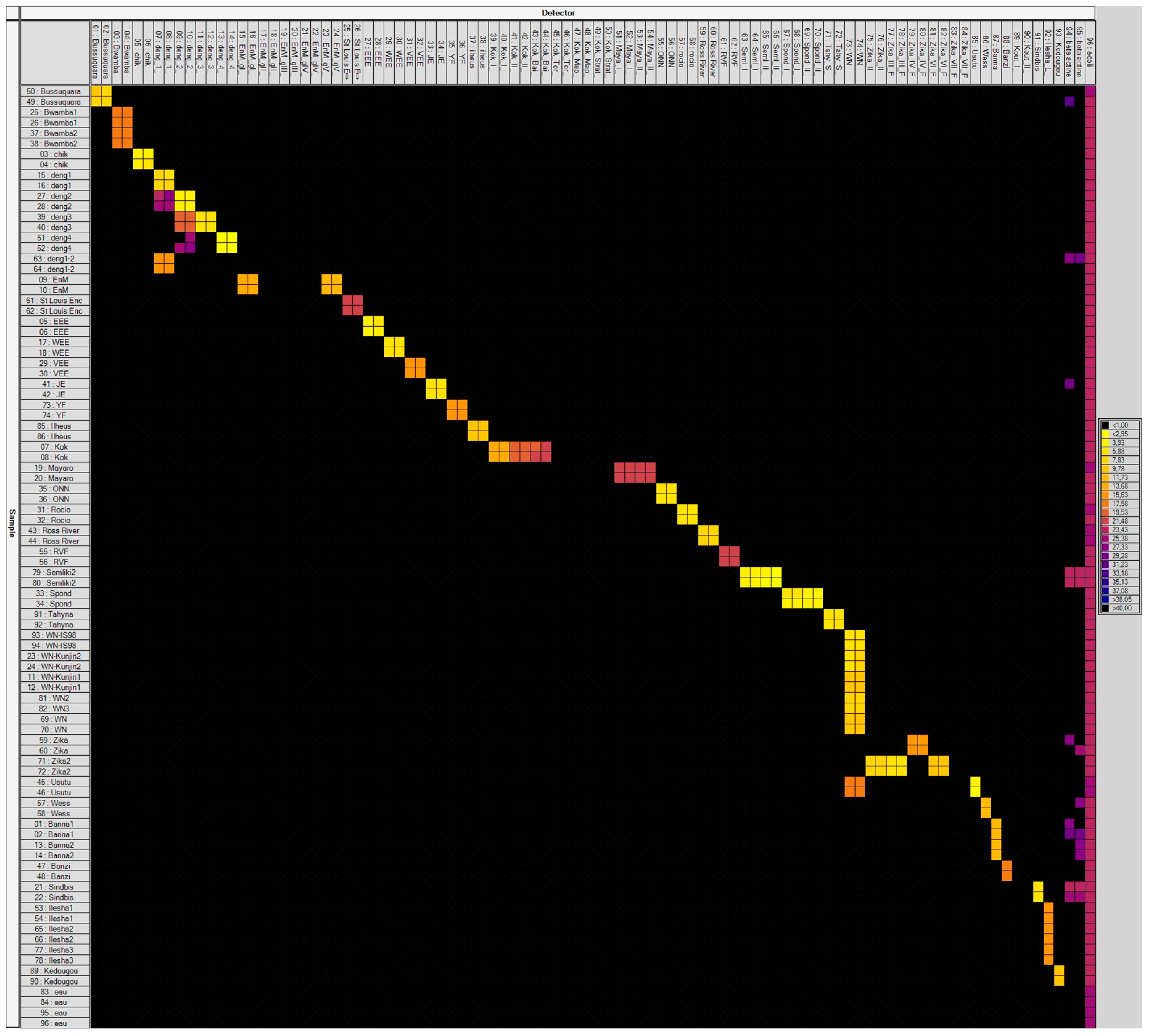
Mosquito-borne viruses (MBV), their targeted genes, and the corresponding primers/probe sets are listed in
Table S1. For a total of 64 viruses including 149 genotypes/serotypes, primers and probes were specifically designed. Indeed, selection was based on specific constraints of temperature of annealing (60 °C for primers and 70 °C for probes); primers/probe sets published in the literature were included if they fit into these criteria. Each primer/probe set was validated using a dilution range of several cDNA positive controls (when available) (
Table S1), by real-time PCR on a LightCycler
® 480 (LC480) (Roche Applied Science, Penzberg, Germany). Real-time PCR assays were performed in a final volume of 12 µL using the LightCycler
® 480 Probe Master Mix 1X (Roche Applied Science, Germany) with primers and probes at 200 nM and 2 µL of control cDNA (virus reference material) or DNA (Plasmid). Thermal cycling conditions were as follows: 95 °C for 5 min, 45 cycles at 95 °C for 10 s, and 60 °C for 15 s, and one final cooling cycle at 40 °C for 10 s.
2.5. High-Throughput Real-Time PCR
The BioMark™ real-time PCR system (Fluidigm, South San Francisco, CA, USA) was used for high-throughput microfluidic real-time PCR amplification using the 96.96 dynamic arrays (Fluidigm, South San Francisco, CA, USA). These chips dispensed 96 PCR mixes and 96 samples into individual wells, after which on-chip microfluidics assembled PCR reactions in individual chambers prior to thermal cycling resulting in 9216 individual reactions. Real-time PCRs were performed using FAM- and black hole quencher (BHQ1)-labeled TaqMan probes with TaqMan Gene Expression Master Mix in accordance with manufacturer’s instructions (Applied Biosystems, Foster City, CA, USA). Thermal cycling conditions were as follows: 2 min at 50 °C, 10 min at 95 °C, followed by 40 cycles of 2-step amplification of 15 sec at 95 °C, and 1 min at 60 °C. Data were acquired on the BioMark™ real-time PCR system and analyzed using the Fluidigm real-time PCR Analysis software to obtain C
t values (see Michelet et al. 2014 for more details [
10]). Primers and probes were evaluated for their specificity against cDNA reference samples. One negative water control was included per chip. To determine if factors present in the sample could inhibit the PCR,
Escherichia coli strain EDL933 DNA was added to each sample as an internal inhibition control, using primers and probes specific for the
E. coli eae gene [
13].
2.6. Validation of the Results by Real-Time PCR, Virus Isolation, and Genome Sequencing
When cDNA of pools of abdomens were detected positive for viruses, the cDNAs of RBP (head/thorax) of individual mosquitoes composing each pool were screened by real-time PCRs on a LightCycler
® 480 (LC480) (Roche Applied Science, Penzberg, Germany). Real-time PCR assay targeting the virus of interest (see primers/probe sets in
Table S1) was performed in a final volume of 12 µL using the LightCycler
® 480 Probe Master Mix 1X (Roche Applied Science, Germany), with primers and probes at 200 nM and 2 µL of control DNA. Thermal cycling conditions were as follows: 95 °C for 5 min, 45 cycles at 95 °C for 10 s, and 60 °C for 15 s, and one final cooling cycle at 40 °C for 10 s.
When a positive sample was confirmed, virus isolation was attempted in Vero and C6/36 cells. Then, total RNA was extracted using the Nucleospin RNA II extract kit (Macherey-Nagel, Hoerdt, France) following the manufacturer instructions and full genome sequencing was attempted. For ZIKV, 12 overlapping amplicons were produced using the reverse transcriptase Platinum Taq High Fidelity polymerase enzyme (Thermo Fisher Scientific, Waltham, MA, USA) and specific primers (
Table S2). PCR products were pooled in equimolar proportions. After Qubit quantification using Qubit
® dsDNA HS Assay Kit and Qubit 2.0 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA), amplicons were fragmented (sonication) into fragments of 200 bp long. Libraries were built adding barcode for sample identification, and primers to fragmented DNA using AB Library Builder System (Thermo Fisher Scientific, Waltham, MA, USA). To pool the barcoded samples equimolarly, a quantification step by the 2100 Bioanalyzer instrument (Agilent Technologies, Santa Clara, California, USA) was performed. An emulsion PCR of the pools and loading on 520 chip was done using the automated Ion Chef instrument (Thermo Fisher Scientific, Waltham, MA, USA). Sequencing was performed using the S5 Ion torrent technology (Thermo Fisher Scientific, Waltham, MA, USA) following manufacturer’s instructions. Consensus sequence was obtained after removing the 30 first and last nucleotides of each read, trimming reads depending on quality (reads with quality over >99%) and length (reads over 100 pb were kept), and mapping them on a reference (KY415987, most similar sequence after Blastn) using CLC genomics workbench software 11.0.1 (Qiagen, Hilden, Germany). A de novo contig was also produced to ensure that the consensus sequence was not affected by the reference sequence.
4. Discussion
In this study, we developed a new high-throughput virus-detection assay based on microfluidic PCRs able to detect 64 MBVs in mosquitoes. Only four primer sets demonstrated cross-reactivity with viruses from the same genus or serotype. Moreover, specificity of 54 assays was not fully tested in the absence of their respective positive controls. Nevertheless, these designs did not show any cross-reaction with RNA positive controls from other viruses. Subsequently, we used this newly developed assay to perform a large epidemiological survey screening in six countries/territories during the last Zika pandemic. This new method has allowed the detection of (i) three human infecting arboviruses, ZIKV, YFV, and CHIKV, in mosquitoes and (ii) other unexpected viruses such as TVTV.
The efficiency of our tool was the first requirement; we used artificially infected mosquitoes to detect different viruses (DENV1-4, CHIKV, WNV, ZIKV) offered in single and dual infections. Our assays can target DENV with however cross-reactions between serotypes. Caused by one of the four serotypes (DENV1-4), dengue is the most important arboviral disease worldwide [
14]. It is widely accepted that a subsequent infection with a second serotype can produce more severe symptoms [
15]. This situation becomes challenging when multiple serotypes co-circulate [
16]. Mosquitoes co-infected with different DENV serotypes are occasionally detected [
17].
Aedes aegypti and
Ae. albopictus are urban vectors of DENV responsible for most epidemic outbreaks in Asia, Latin America, the Caribbean, and Pacific islands [
14]. Co-infected
Aedes mosquitoes are capable of transmitting multiple arboviruses during one bite [
9]. Dual DENV detections in mosquitoes may be a sign of co-circulation of DENV and then may help in predicting co-infections in humans. Diagnosis of dengue infections cannot be based on clinical symptoms as dengue disease shares common symptoms with other arboviral diseases [
18]. To discriminate dengue serotypes, viral isolation and viral RNA detection remain the gold standard methods but should be performed during patient viremia (within five days after the onset of fever). Less constraining and costly, mass viral screening of mosquitoes in surveillance and epidemic contexts can be an advantageous substitute.
In the same way, WNV assays cross-reacted with the phylogenetically-related USUV. WNV is a flavivirus responsible of neuro-invasive disease in Europe and North America [
19]. Diagnosis of WNV infection remains challenging and human cases are usually underestimated. On the other hand, USUV has spread over Europe during the last 20 years causing bird mortalities and some rare human cases [
20]. Human infections are rare and often asymptomatic, and neurological disorders can be described [
20]. While WNV has circulated in Europe since the 1960s, USUV shares the same geographical distribution and also the same vectors,
Culex pipiens. Our tool did not succeed in distinguishing the two viruses, and therefore it needs more improvements.
By screening 17,958 mosquitoes collected in six countries/territories for 64 different MBVs, we succeeded in detecting ZIKV, YFV, CHIKV, and TVTV in mosquitoes.
The Zika outbreak was unexpected; the first human cases outside endemic regions in Africa were reported in Yap island in 2007 where the outbreak was poorly publicized despite the two third of the population affected [
21]. Few years later, ZIKV hit French Polynesia [
22] where the first notification of severe symptoms associated to ZIKV infections were done, Guillain–Barre syndrome [
23] and microcephaly in new-born [
24]. After ZIKV reached the American continent in 2015 [
25], phylogenetic analysis indicated that the circulating ZIKV belonged to the Asian clade [
26,
27]. Our tool was able to detect ZIKV in pools of abdomen of
Ae. aegypti and
Cx. pipiens from Guadeloupe and French Guiana. However, when analyzing disseminated viral particles in head and thorax, only
Ae. aegypti was found infected, corroborating the main role of this species in ZIKV transmission and limiting
Cx. quinquefasciatus and
Cx. pipiens as a vector [
28]. It is widely admitted that viral dissemination beyond the midgut can be a clue attesting the mosquito susceptibility to a virus. However, viral dissemination in mosquitoes depends on mosquito collection date; it increases over time after the infectious blood meal [
3]. In our study, it was not possible to have information on the physiological age of mosquitoes and when they were infected.
Our mass screening tool has detected YFV in five mosquito species:
Aedes scapularis, Aedes taeniorhynchus, Haemagogus janthinomys, Haemagogus leucocelaenus, and
Sabethes chloropterus. The species
Hg. janthinomys and
Hg. leucocelaenus are considered as the main vectors of YFV in Brazil [
29,
30] while
Aedes scapularis, Aedes taeniorhynchus, and
Sabethes chloropterus only play a secondary role [
31]. Other viruses preliminarily detected in Brazilian mosquitoes were TVTV and CHIKV. While CHIKV continues to cause sporadic cases in Brazil after the massive outbreak in 2015, TVTV was first isolated from
Aedes trivittatus in USA in 1948, and has never been detected outside North America where it is mainly distributed [
32]. Consequences of TVTV infections on humans remain unknown [
33]. Nevertheless, the presence of CHIKV and TVTV in tested mosquitoes was not confirmed.
This study demonstrates the feasibility of high-throughput screening methods to detect diverse MBVs in field-collected mosquitoes. Performing 9216 real-time PCRs in one run took four hours, and the cost was around
$10 per reaction from sample homogenization to virus detection by real-time PCR [
10,
11]. Nevertheless, the instrument is costly and requires some specific conditions of use. It is therefore recommended to identify focal points where this technology could be developed and to improve the conditions for transporting biological samples from the field to allow an optimal viral screening. Another main advantage of our tool is the adaptability of the system by adding new sets of primers and probes targeting newly emergent viruses in contrast to arrays with fixed panels of probes. Indeed, because the number of YFV cases was unusually high since January 2016 [
34], we added specific detections of YFV strains circulating in South America to screen field-collected mosquitoes from Brazil, French Guiana, Suriname, and Guadeloupe. In conclusion, our method designed to specifically identify MBVs in mosquitoes can be used to screen other types of samples such as human and/or animal blood or organs [
35]. We demonstrated the usefulness of this new screening method, which represents a powerful, cost-effective, and rapid system to track MBVs all around the world and could be easily customized to any viral emergence.

 ,
, 




{kind=link}
{kind=link}
{kind=link}