Targeted Chromatinization and Repression of HIV-1 Provirus Transcription with Repurposed CRISPR/Cas9

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. dCas9 Constructs
2.3. Lentivirus Packaging
2.4. Transductions and Infections
2.5. HEK293T Transfections
2.6. JLat Reactivation
2.7. HIV-1 RNA and DNA Quantification
2.8. Chromatin Immunoprecipitation Assay
2.9. Statistical Analysis
3. Results
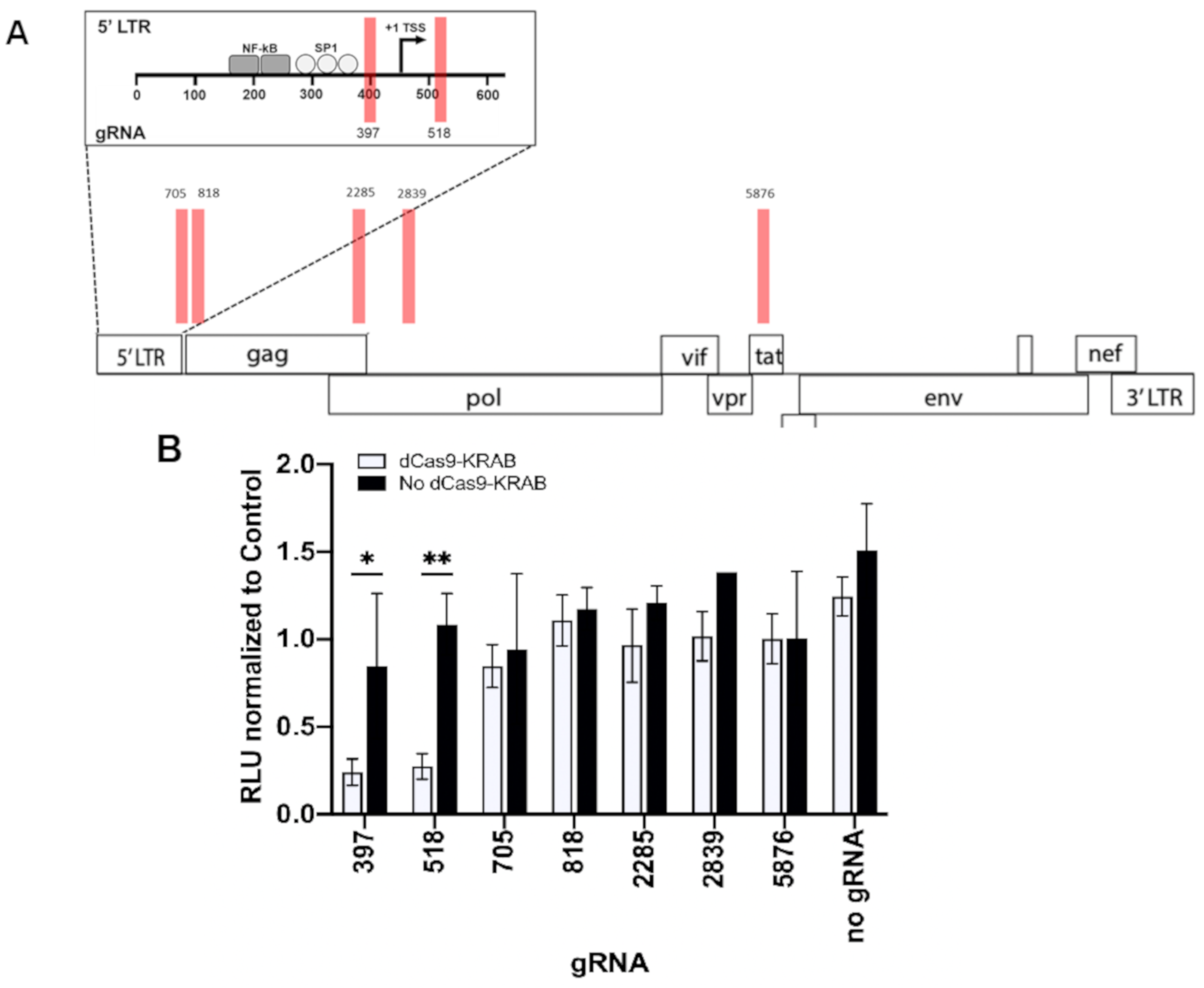
3.1. Repressing HIV Transcription with dCas9-KRAB
3.2. dCas9-KRAB Inhibits Reactivation of Latent HIV
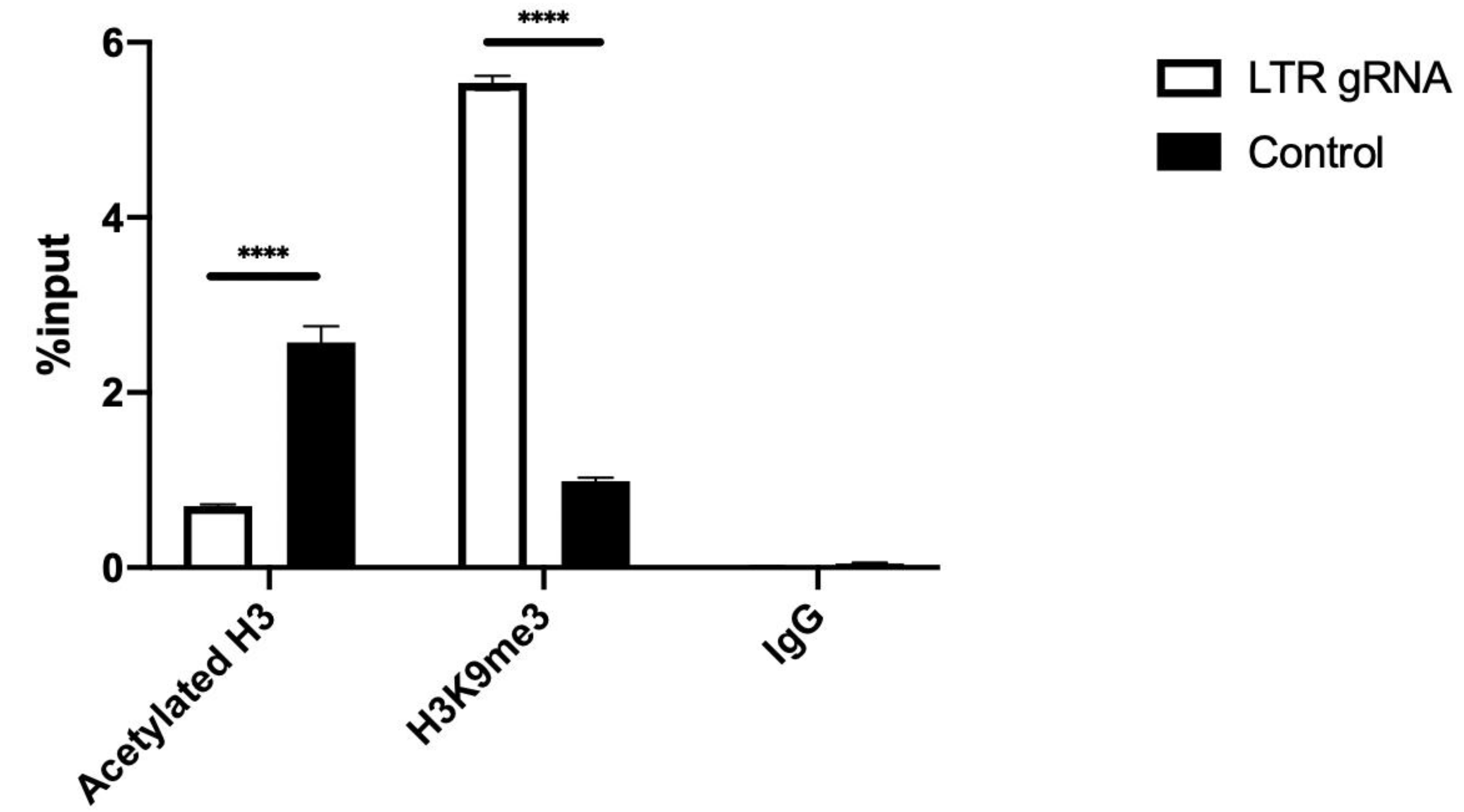
3.3. dCas9-KRAB Repression of HIV-1 Transcription Correlates with Epigenetic Modifications
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, M.R.; Joseph, S.B.; Garrett, N.; Tyers, L.; Moeser, M.; Archin, N.; Council, O.D.; Matten, D.; Zhou, S.; Doolabh, D.; et al. The replication-competent HIV-1 latent reservoir is primarily established near the time of therapy initiation. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nat. Cell Biol. 1997, 387, 183–188. [Google Scholar] [CrossRef]
- Wong, J.K.; Hezareh, M.; Günthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of Replication-Competent HIV Despite Prolonged Suppression of Plasma Viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef]
- Chavez, L.; Calvanese, V.; Verdin, E. HIV Latency Is Established Directly and Early in Both Resting and Activated Primary CD4 T Cells. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [Green Version]
- Agosto, L.M.; Herring, M.B.; Mothes, W.; Henderson, A.J. HIV-1-Infected CD4+ T Cells Facilitate Latent Infection of Resting CD4+ T Cells through Cell-Cell Contact. Cell Rep. 2018, 24, 2088–2100. [Google Scholar] [CrossRef] [Green Version]
- Spina, C.A.; Guatelli, J.C.; Richman, D.D. Establishment of a stable, inducible form of human immunodeficiency virus type 1 DNA in quiescent CD4 lymphocytes in vitro. J. Virol. 1995, 69, 2977–2988. [Google Scholar] [CrossRef] [Green Version]
- Gagne, M.; Michaels, D.; Lester, G.M.S.; Gummuluru, S.; Wong, W.W.; Henderson, A.J. Strength of T cell signaling regulates HIV-1 replication and establishment of latency. PLoS Pathog. 2019, 15. [Google Scholar] [CrossRef] [Green Version]
- Oswald-Richter, K.; Grill, S.M.; Leelawong, M.; Unutmaz, D. HIV infection of primary human T cells is determined by tunable thresholds of T cell activation. Eur. J. Immunol. 2004, 34, 1705–1714. [Google Scholar] [CrossRef]
- Shan, L.; Deng, K.; Gao, H.; Xing, S.; Capoferri, A.A.; Durand, C.M.; Rabi, S.A.; Laird, G.M.; Kim, M.; Hosmane, N.N.; et al. Transcriptional Reprogramming during Effector-to-Memory Transition Renders CD4+ T Cells Permissive for Latent HIV-1 Infection. Immunity 2017, 47, 766–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzon, M.J.; Sun, H.; Li, C.; Shaw, A.; Seiss, K.; Ouyang, Z.; Martin-Gayo, E.; Leng, J.; Henrich, T.J.; Li, J.Z.; et al. HIV-1 persistence in CD4+ T cells with stem cell–like properties. Nat. Med. 2014, 20, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Couturier, J.; Orozco, A.F.; Liu, H.; Budhiraja, S.; Siwak, E.B.; Nehete, P.N.; Sastry, K.J.; Rice, A.S.C.; Lewis, D.E. Regulation of cyclin T1 during HIV replication and latency establishment in human memory CD4 T cells. Virol. J. 2019, 16, 22. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolski, C.; Valadkhan, S.; Graham, A.C.; Shukla, M.; Ciuffi, A.; Telenti, A.; Karn, J.; Ott, M.; Henderson, A.; Spina, C.A. Entry of Polarized Effector Cells into Quiescence Forces HIV Latency. mBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, M.; Pearson, R.J.; Karn, J. Establishment of HIV Latency in Primary CD4+ Cells Is due to Epigenetic Transcriptional Silencing and P-TEFb Restriction. J. Virol. 2010, 84, 6425–6437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolis, D.M.; Archin, N.M.; Cohen, M.S.; Eron, J.J.; Ferrari, G.; Garcia, J.V.; Gay, C.L.; Goonetilleke, N.; Joseph, S.B.; Swanstrom, R.; et al. Curing HIV: Seeking to Target and Clear Persistent Infection. Cell 2020, 181, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.R.; Siliciano, R.F. Progress Toward HIV Eradication: Case Reports, Current Efforts, and the Challenges Associated with Cure. Annu. Rev. Med. 2016, 67, 215–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzer, R.; Gramatica, A.; Greene, W.C. Reduce and Control: A Combinatorial Strategy for Achieving Sustained HIV Remissions in the Absence of Antiretroviral Therapy. Viruses 2020, 12, 188. [Google Scholar] [CrossRef] [Green Version]
- Spivak, A.M.; Planelles, V. HIV-1 Eradication: Early Trials (and Tribulations). Trends Mol. Med. 2016, 22, 10–27. [Google Scholar] [CrossRef] [Green Version]
- Nixon, C.C.; Mavigner, M.; Sampey, G.C.; Brooks, A.D.; Spagnuolo, R.A.; Irlbeck, D.M.; Mattingly, C.; Ho, P.T.; Schoof, N.; Cammon, C.G.; et al. Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nat. Cell Biol. 2020, 578, 160–165. [Google Scholar] [CrossRef]
- Abner, E.; Jordan, A. HIV “shock and kill” therapy: In need of revision. Antivir. Res. 2019, 166, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Olson, A.; Basukala, B.; Wong, W.W.; Henderson, A.J. Targeting HIV-1 proviral transcription. Curr. Opin. Virol. 2019, 38, 89–96. [Google Scholar] [CrossRef] [PubMed]
- VanSant, G.; Bruggemans, A.; Janssens, J.; Debyser, Z. Block-And-Lock Strategies to Cure HIV Infection. Viruses 2020, 12, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, R.; Berges, B.K.; Solis-Leal, A.; Igbinedion, O.; Strong, C.L.; Schiller, M.R. TALEN gene editing takes aim on HIV. Qual. Life Res. 2016, 135, 1059–1070. [Google Scholar] [CrossRef] [Green Version]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Kwarteng, A.; Ahuno, S.; Kwakye-Nuako, G. The therapeutic landscape of HIV-1 via genome editing. AIDS Res. Ther. 2017, 14, 32. [Google Scholar] [CrossRef]
- Kang, H.; Minder, P.; Park, M.A.; Mesquitta, W.T.; Torbett, B.; Slukvin, I.I. CCR5 Disruption in Induced Pluripotent Stem Cells Using CRISPR/Cas9 Provides Selective Resistance of Immune Cells to CCR5-tropic HIV-1 Virus. Mol. Ther. Nucleic Acids 2015, 4. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, S.; Jin, X.; Wang, Q.; Yang, K.; Li, C.; Xiao, Q.; Hou, P.; Liu, S.; Wu, S.; et al. Genome editing of the HIV co-receptors CCR5 and CXCR4 by CRISPR-Cas9 protects CD4+ T cells from HIV-1 infection. Cell Biosci. 2017, 7, 47. [Google Scholar] [CrossRef]
- Teque, F.; Ye, L.; Xie, F.; Wang, J.; Morvan, M.G.; Kan, Y.W.; Levy, J.A. Genetically-edited induced pluripotent stem cells derived from HIV-1-infected patients on therapy can give rise to immune cells resistant to HIV-1 infection. AIDS 2020, 34, 1141–1149. [Google Scholar] [CrossRef]
- Yu, S.; Yao, Y.; Xiao, H.; Li, J.; Liu, Q.; Yang, Y.; Adah, D.; Lu, J.; Zhao, S.; Qin, L.; et al. Simultaneous Knockout of CXCR4 and CCR5 Genes in CD4+ T Cells via CRISPR/Cas9 Confers Resistance to Both X4- and R5-Tropic Human Immunodeficiency Virus Type 1 Infection. Hum. Gene Ther. 2018, 29, 51–67. [Google Scholar] [CrossRef]
- Dash, P.K.; Kaminski, R.; Bella, R.; Su, H.; Mathews, S.; Ahooyi, T.M.; Chen, C.; Mancuso, P.; Sariyer, R.; Ferrante, P.; et al. Sequential LASER ART and CRISPR Treatments Eliminate HIV-1 in a Subset of Infected Humanized Mice. Nat. Commun. 2019, 10, 2753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Kaminski, R.; Yang, F.; Zhang, Y.; Cosentino, L.; Li, F.; Luo, B.; Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Karn, J.; et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 11461–11466. [Google Scholar] [CrossRef] [Green Version]
- Park, R.J.; Wang, T.; Koundakjian, D.; Hultquist, J.F.; Lamothe-Molina, P.; Monel, B.; Schumann, K.; Yu, H.; Krupzcak, K.M.; Garcia-Beltran, W.; et al. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 2016, 49, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Zhang, T.; Qu, X.; Zhang, Y.; Putatunda, R.; Xiao, X.; Li, F.; Xiao, W.; Zhao, H.; Dai, S.; et al. In Vivo Excision of HIV-1 Provirus by saCas9 and Multiplex Single-Guide RNAs in Animal Models. Mol. Ther. 2017, 25, 1168–1186. [Google Scholar] [CrossRef] [Green Version]
- Bialek, J.K.; Dunay, G.A.; Voges, M.; Schäfer, C.; Spohn, M.; Stucka, R.; Hauber, J.; Lange, U.C. Targeted HIV-1 Latency Reversal Using CRISPR/Cas9-Derived Transcriptional Activator Systems. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [Green Version]
- Limsirichai, P.; Gaj, T.; Schaffer, D. CRISPR-mediated Activation of Latent HIV-1 Expression. Mol. Ther. 2016, 24, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Azzaz, A.M.; Vitalini, M.W.; Thomas, A.S.; Price, J.P.; Blacketer, M.J.; Cryderman, D.E.; Zirbel, L.N.; Woodcock, C.L.; Elcock, A.H.; Wallrath, L.L.; et al. Human Heterochromatin Protein 1α Promotes Nucleosome Associations That Drive Chromatin Condensation. J. Biol. Chem. 2014, 289, 6850–6861. [Google Scholar] [CrossRef] [Green Version]
- Iyengar, S.; Farnham, P.J. KAP1 Protein: An Enigmatic Master Regulator of the Genome. J. Biol. Chem. 2011, 286, 26267–26276. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Yang, T.; Luo, Y.; Wu, L.; Jiang, Y.; Song, Z.; Pan, T.; Liu, B.; Liu, G.; Liu, J.; et al. TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb. eLife 2019, 8. [Google Scholar] [CrossRef]
- Meylan, S.; Groner, A.C.; Ambrosini, G.; Malani, N.; Quenneville, S.; Zangger, N.; Kapopoulou, A.; Kauzlaric, A.; Rougemont, J.; Ciuffi, A.; et al. A gene-rich, transcriptionally active environment and the pre-deposition of repressive marks are predictive of susceptibility to KRAB/KAP1-mediated silencing. BMC Genom. 2011, 12, 378. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.X.; El Farran, C.A.; Guo, H.C.; Yu, T.; Fang, H.T.; Wang, H.F.; Schlesinger, S.; Seah, Y.F.S.; Goh, G.Y.L.; Neo, S.P.; et al. Systematic identification of factors for provirus silencing in embryonic stem cells. Cell 2015, 163, 230–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef] [Green Version]
- Benchling. Available online: http://www.benchling.com (accessed on 27 November 2017).
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.D.; Scott, D.; Weinstein, J.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Kiani, S.; Chavez, A.; Tuttle, M.; Hall, R.N.; Chari, R.; Ter-Ovanesyan, D.; Qian, J.; Pruitt, B.W.; Beal, J.; Vora, S.; et al. Cas9 gRNA engineering for genome editing, activation and repression. Nat. Methods 2015, 12, 1051–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bintu, L.; Yong, J.; Antebi, Y.; McCue, K.; Kazuki, Y.; Uno, N.; Oshimura, M.; Elowitz, M.B. Dynamics of epigenetic regulation at the single-cell level. Science 2016, 351, 720–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Begg, G.; Schultz, D.C.; Friedman, J.R.; Jensen, D.; Speicher, D.W.; Rauscher, F.J. Reconstitution of the KRAB-KAP-1 repressor complex: a model system for defining the molecular anatomy of RING-B box-coiled-coil domain-mediated protein-protein interactions. J. Mol. Biol. 2000, 295, 1139–1162. [Google Scholar] [CrossRef]
- Urrutia, R. KRAB-containing zinc-finger repressor proteins. Genome Biol. 2003, 4, 231. [Google Scholar] [CrossRef] [Green Version]
- Schultz, D.C. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Sripathy, S.P.; Stevens, J.; Schultz, D.C. The KAP1 Corepressor Functions To Coordinate the Assembly of De Novo HP1-Demarcated Microenvironments of Heterochromatin Required for KRAB Zinc Finger Protein-Mediated Transcriptional Repression. Mol. Cell. Biol. 2006, 26, 8623–8638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agosto, L.M.; Gagne, M.; Henderson, A.J. Impact of Chromatin on HIV Replication. Genes 2015, 6, 957–976. [Google Scholar] [CrossRef] [PubMed]
- Saayman, S.M.; Lazar, D.C.; Scott, T.A.; Hart, J.R.; Takahashi, M.; Burnett, J.C.; Planelles, V.; Morris, K.V.; Weinberg, M.S. Potent and Targeted Activation of Latent HIV-1 Using the CRISPR/dCas9 Activator Complex. Mol. Ther. 2016, 24, 488–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Li, D.; Lin, M.H.; Li, L.; Harrich, D. Tat-Based Therapies as an Adjuvant for an HIV-1 Functional Cure. Viruses 2020, 12, 415. [Google Scholar] [CrossRef] [Green Version]
- Mousseau, G.; Valente, S.T. Role of Host Factors on the Regulation of Tat-Mediated HIV-1 Transcription. Curr. Pharm. Des. 2017, 23, 4079–4090. [Google Scholar] [CrossRef] [Green Version]
- Mousseau, G.; Clementz, M.A.; Bakeman, W.N.; Nagarsheth, N.; Cameron, M.; Shi, J.; Baran, P.; Fromentin, R.; Chomont, N.; Valente, S.T. An Analog of the Natural Steroidal Alkaloid Cortistatin A Potently Suppresses Tat-Dependent HIV Transcription. Cell Host Microbe 2012, 12, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Friedli, M.; Trono, D. The Developmental Control of Transposable Elements and the Evolution of Higher Species. Annu. Rev. Cell Dev. Biol. 2015, 31, 429–451. [Google Scholar] [CrossRef]
- Wolf, G.; Greenberg, D.; Macfarlan, T.S. Spotting the enemy within: Targeted silencing of foreign DNA in mammalian genomes by the Krüppel-associated box zinc finger protein family. Mob. DNA 2015, 6, 17. [Google Scholar] [CrossRef] [Green Version]
- Morton, E.L.; Forst, C.V.; Zheng, Y.; DePaula-Silva, A.B.; Ramirez, N.G.P.; Planelles, V.; D’Orso, I. Transcriptional Circuit Fragility Influences HIV Proviral Fate. Cell Rep. 2019, 27, 154–171. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| gRNA | Gene | Strand | Sequence (5′-3′) | PAM | Specificity a | Activity b |
|---|---|---|---|---|---|---|
| 397 | 5′ LTR | + | GGTGTGGCCTGGGCGGGACTG | GGGAGT | 67.2 | 6.2 |
| 518 | 5′ LTR | − | AGCTTTATTGAGGCTTAAGCA | GTGGGT | 83.2 | 61.6 |
| 705 | NCR | − | TGCCGTGCGCGCTTCAGCAAG | CCGAGT | 89.6 | 27.4 |
| 818 | gag | + | GAGCGTCAGTATTAAGCGGGG | GAGAAT | 97.1 | 22.8 |
| 2285 | gag, pol | − | CCCCCTATCTTTACTTTGACG | AGGGGT | 91.0 | 27.5 |
| 2839 | pol | + | AATTAGGAATACCACATCCCG | CAGGGT | 96.4 | 66.4 |
| 5876 | tat | − | AAGCAGTTTTAGGCTGACTTC | CTGGAT | 79.5 | 3.1 |
| Construct | Type | Description |
|---|---|---|
| dCas9-KRAB | Plasmid | pCMV-dsaCas9-KRAB |
| dCas9-KRAB-BlasticidinR | Plasmid | pHR-SFFV-KRAB-dsaCas9-T2A-BlastR; stable J-Lat 6.3 |
| gRNA Control | Plasmid | phU6-gRNA (gRNA scaffold) |
| gRNA 397 | Plasmid | phU6-gRNA_Nl43-397 |
| gRNA 518 | Plasmid | phU6-gRNA_NL43-518 |
| gRNA 705 | Plasmid | phU6-gRNA_NL43-705 |
| gRNA 818 | Plasmid | phU6-gRNA_NL43-818 |
| gRNA 2285 | Plasmid | phU6-gRNA_NL43-2285 |
| gRNA 2839 | Plasmid | phU6-gRNA_NL43-2839 |
| gRNA 5876 | Plasmid | phU6-gRNA_NL43-5876 |
| gRNA 397 | Ultramer Oligo RNA | − |
| gRNA Scramble | Ultramer Oligo RNA | − |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olson, A.; Basukala, B.; Lee, S.; Gagne, M.; Wong, W.W.; Henderson, A.J. Targeted Chromatinization and Repression of HIV-1 Provirus Transcription with Repurposed CRISPR/Cas9. Viruses 2020, 12, 1154. https://doi.org/10.3390/v12101154
Olson A, Basukala B, Lee S, Gagne M, Wong WW, Henderson AJ. Targeted Chromatinization and Repression of HIV-1 Provirus Transcription with Repurposed CRISPR/Cas9. Viruses. 2020; 12(10):1154. https://doi.org/10.3390/v12101154
Chicago/Turabian StyleOlson, Alex, Binita Basukala, Seunghee Lee, Matthew Gagne, Wilson W. Wong, and Andrew J. Henderson. 2020. "Targeted Chromatinization and Repression of HIV-1 Provirus Transcription with Repurposed CRISPR/Cas9" Viruses 12, no. 10: 1154. https://doi.org/10.3390/v12101154
APA StyleOlson, A., Basukala, B., Lee, S., Gagne, M., Wong, W. W., & Henderson, A. J. (2020). Targeted Chromatinization and Repression of HIV-1 Provirus Transcription with Repurposed CRISPR/Cas9. Viruses, 12(10), 1154. https://doi.org/10.3390/v12101154