1. Introduction
The human immunodeficiency virus type 1 (HIV-1) is a retrovirus that contains two copies of its genomic single-stranded RNA embedded into the nucleocapsid of mature particles. During the assembly of the virus, some tRNAs from the host cell are selectively packaged into the budding particles [
1,
2]. This includes tRNA
Lys,3 that serves as a primer for initiation of reverse transcription, an essential step of the life cycle of the virus that is believed to take place in the virus shortly after budding, before infection of host cells [
3]. In virio analyses reveal that annealing of tRNA
Lys,3 to the primer binding site (PBS) of viral RNA occurs in the viruses [
4], consistent with the finding that the level of viral infectivity is correlated with the level of tRNA
Lys,3 encapsidation into the virions [
5]. Lysyl-tRNA synthetase, an enzyme of the translation machinery that catalyzes aminoacylation of tRNA
Lys,3 with lysine, is also detected in HIV-1 virions [
6,
7] and its interactions with Gag and GagPol polyproteins is suggested to be essential for tRNA
Lys,3 encapsidation into newly formed viral particles [
8,
9]. The early model suggested that cytoplasmic LysRS, after dissociation from the multisynthetase complex, following phosphorylation on Ser207 by MAPK, associates with Gag [
6,
10]. Monospecific antibodies directed to human mitochondrial lysyl-tRNA synthetase (mLysRS) identified a cross-reactive protein in extracts of HIV-1 particles [
7], corresponding to the mature form of mLysRS produced after cleavage of its N-terminal mitochondria-targeting sequence [
11]. The tRNA
Lys,3 packaging complex is formed by the association of GagPol with the LysRS:tRNA
Lys,3 complex. The catalytic domain of mLysRS was shown to interact with the transframe and integrase domains of the GagPol polyprotein precursor [
8]. The interaction between mLysRS and the integrase subunit (IN) from the Pol domain of the GagPol precursor is the major contributor to the stability of the GagPol:mLysRS complex [
12].
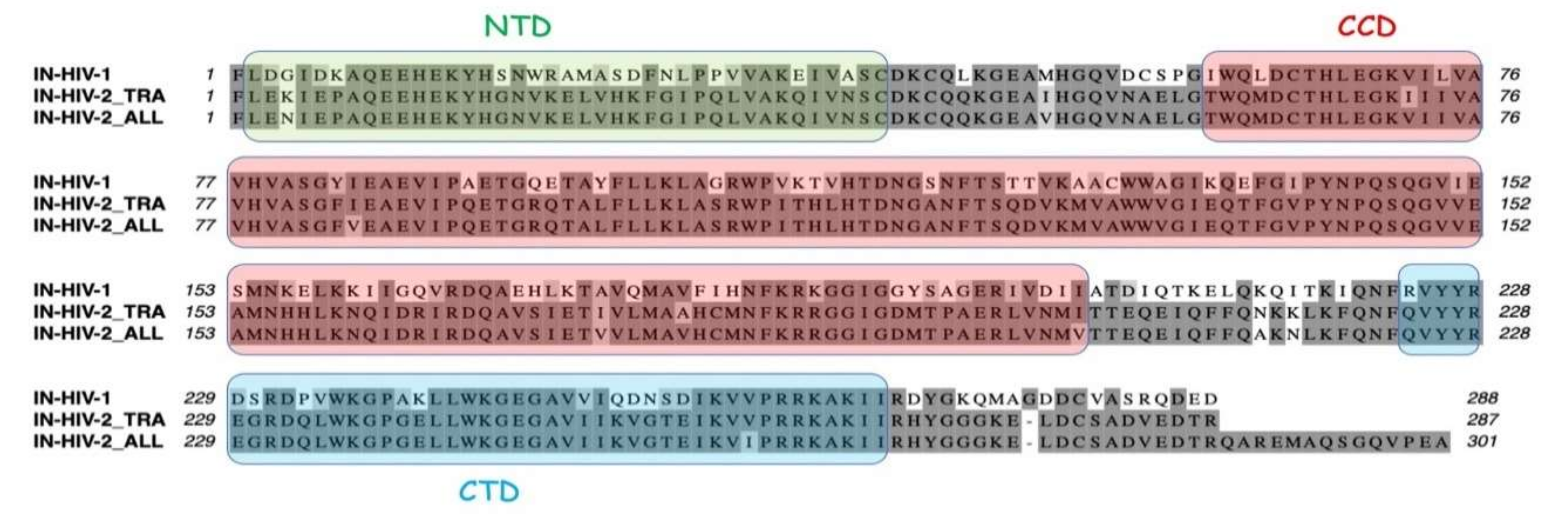
In human, cytoplasmic and mitochondrial LysRSs are encoded by the
KARS1 gene by means of alternative splicing [
13]. They share 576 amino acid residues in common. The cytoplasmic and the mature mitochondrial enzymes possess specific N-terminal sequences of 21 and 19 amino acid residues, respectively [
11]. They are homodimers composed of a C-terminal catalytic domain, a central tRNA anticodon-binding domain, and a eukaryote-specific N-terminal domain that stabilizes the LysRS:tRNA
Lys complex [
11,
14]. In the crystal-structure of LysRS, only the catalytic and tRNA anticodon-binding domains are visible [
15]. These two domains are shared by the cytoplasmic and mitochondrial LysRS.
After maturation of the GagPol precursor by viral protease, the integrase (IN) from HIV-1 is released as a dimer formed of two identical subunits made of 288 amino acid residues, which also forms a dimer of dimers at high concentration, or when it binds RNA [
16,
17]. The primary function of integrase is integration of viral DNA into the host genome. Integrase strand-transfer inhibitors (INSTIs) that target this function have been developed and are used to treat HIV-1 infections [
18]. Recent studies showed that IN is also essential during morphogenesis of the particles [
17]. It binds viral RNA and is necessary for proper localization of the viral genome inside the capsid. Allosteric IN inhibitors (ALLINIs) define a new class of antiretroviral agents that target IN oligomerization and compromise viral replication [
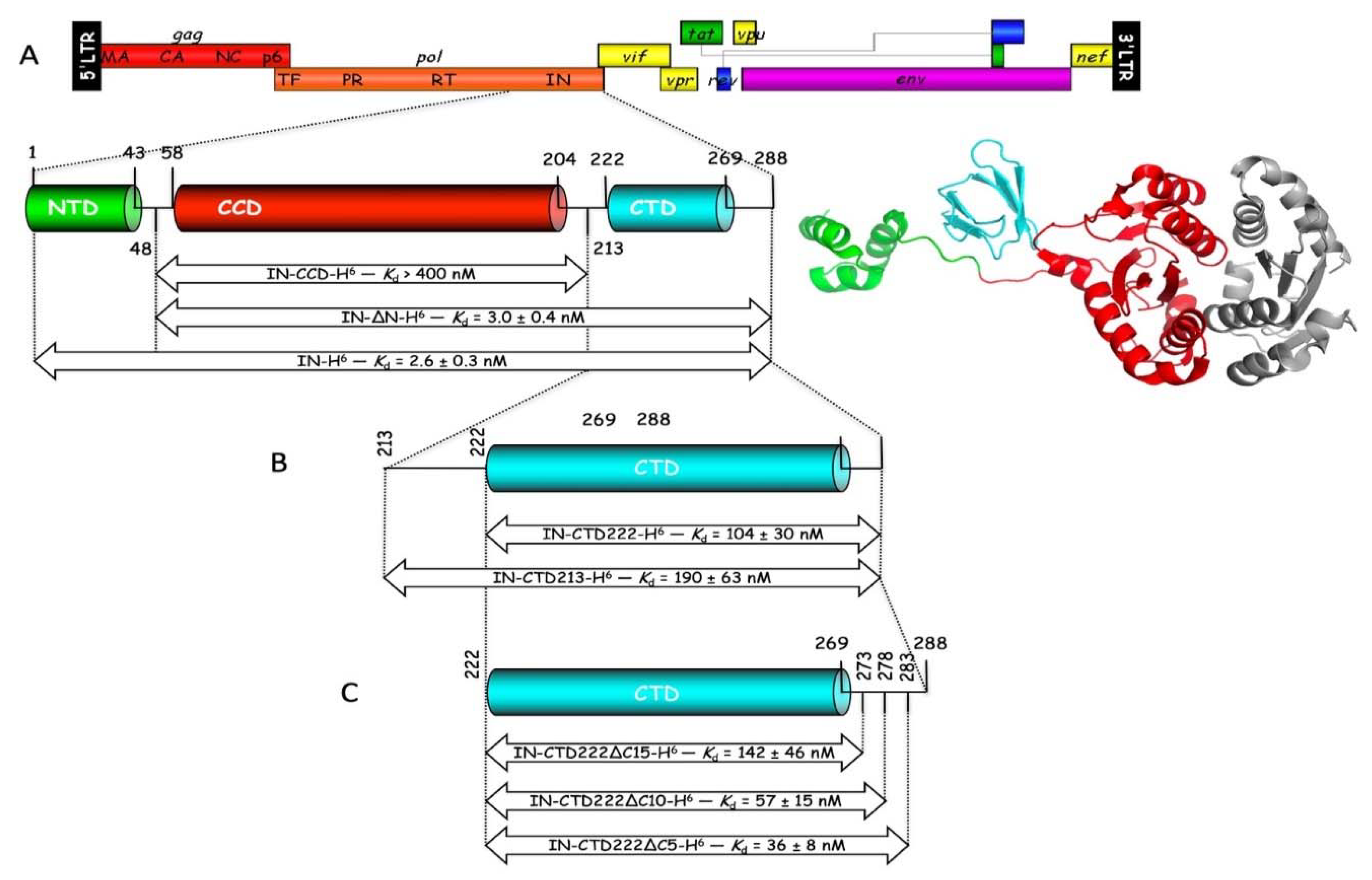
19]. HIV-1 integrase is constituted of three distinct structural domains, a α-helical N-terminal domain (IN-NTD), the catalytic core domain (IN-CCD), and a β-barrel C-terminal domain (IN-CTD). Crystal structures of IN-CCD alone, or of IN-CCD with either the NTD or the CTD have been reported [
20,
21]. The structural domains are linked by long spacer polypeptides which are not always visible in the crystal structures. The three domains of integrase are clearly visible in the cryo-EM structure of the HIV-1 strand transfer complex intasome [
22]. Interactions of integrase with viral and target DNA within the intasome involve the CCD and CTD of integrase, and the NTD-CCD linker.
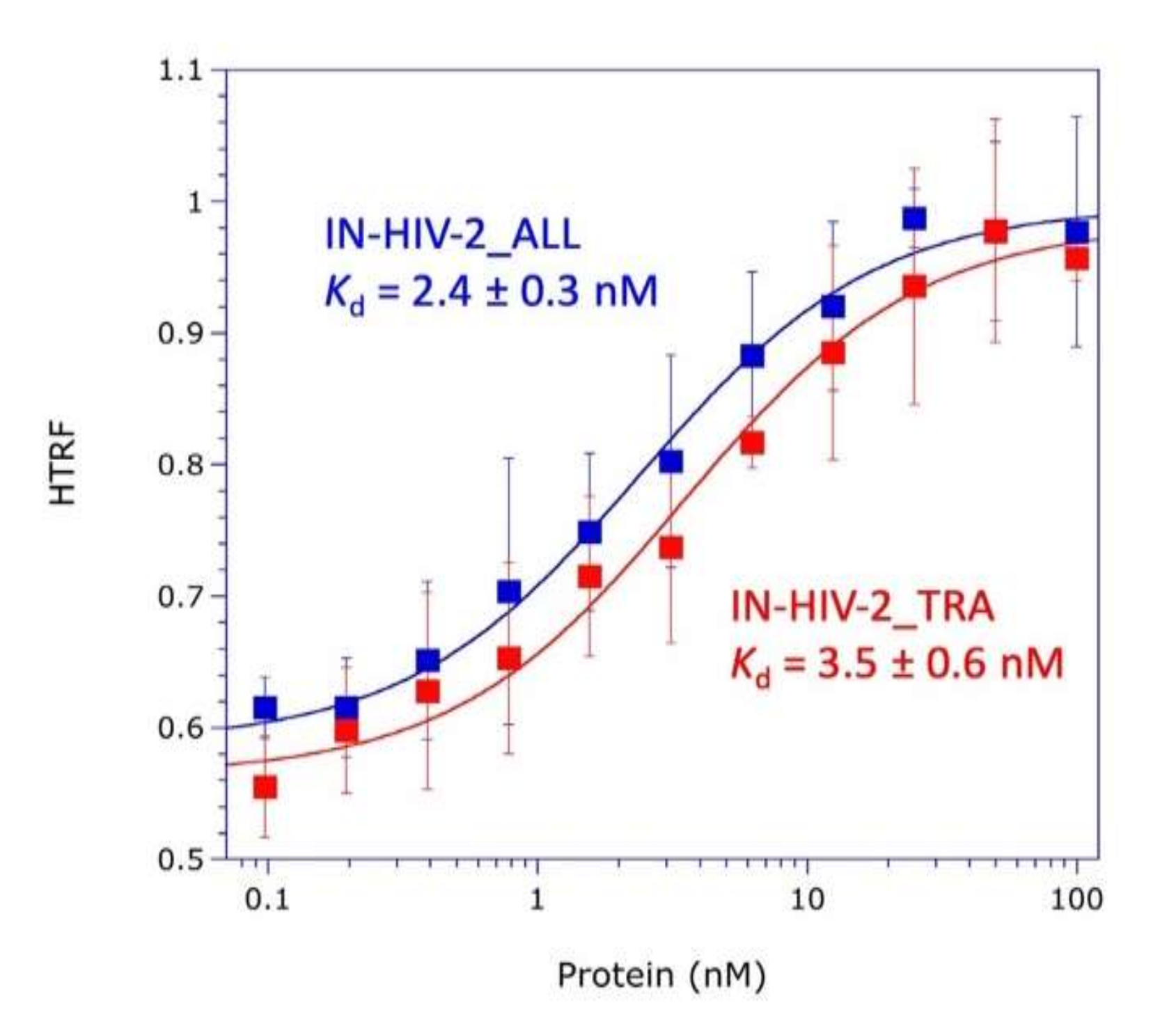
The knowledge of the three-dimensional (3D) structure of the three structural domains of integrase and of the catalytic and anticodon-binding domains of LysRS opens the way to explore the mechanism of mLysRS:IN association. In this work, we conducted a detailed analysis of the complex of mLysRS with the integrase from HIV-1. We first determined that the CTD of IN from HIV-1 is responsible for its association with mLysRS. HIV-2 is a retrovirus that also uses tRNALys,3 as a primer for reverse transcription but displays significant sequence variations in its genome, including the integrase coding sequence. We determined that IN from HIV-2 binds mLysRS with an efficiency similar to IN from HIV-1. Then, we developed experimental and computational approaches in order to propose a structural model of the mLysRS:IN complex. Finally, this structural model was subjected to mutational probing. In particular, five compensatory mutants were constructed on the basis of the structural model, which eventually led to the validation of a 3D model of mLysRS:IN interaction. The combination of experimental and in silico approaches used in this study allowed to determine the mode of interaction of mLysRS with IN, an interaction believed to be essential for the replication of the virus. Targeting the mLysRS:IN-CTD complex with inhibitors of its assembly may prove useful to develop new antiviral drugs with original resistance profiles.
2. Materials and Methods
2.1. Expression of HIV-1 Integrase in E. coli and Purification
The HIV-1 integrase coding region from pNL4-3 (nucleotides 4230 to 5093) was amplified by PCR with oligonucleotides GP119 (5′-GTTTAACTTTAAGAAGGAGATATACCATGGCGTTTTTAG ATGGAATAGATAA) and GP159 (5′-ATCTCAGTGGTGGTGGTGGTGGTGCTCGAGATCCTCATC CTGTCTACTTG), and introduced by the SLIC method [
23] into pET28b digested with NcoI and XhoI. For cloning of IN-∆N, PCR was performed with primers GP120 (5′-GTTTAACTTT AAGAAGGAGATATACCATGGCGATGCATGGACAAGTAGACTG) and GP159, and for cloning IN-CCD, primers were GP120 and GP160 (5′-ATCTCAGTGGTGGTGGTGGTGGTGCTCGAGTTCTT TAGTTTGTATGTCTGTTG). For cloning of IN-CTD213 and IN-CTD222, the 5′-primers were IN003 (5′-GTTTAACTTTAAGAAGGAGATATACCATGGCGTTACAAAAACAAATTACAAAAATTC) and IN005 (5′-GTTTAACTTTAAGAAGGAGATATACCATGGCGAATTTTCGGGTTTATTACA GG), respectively, and the 3′-primer was GP159. For cloning of IN-CTD222∆C5, IN-CTD222∆C10 or IN-CTD222∆C15, the 5′-primer was IN005, and the 3′-primers were IN104 (5′-ATCTCAGTGGTGGTGGTGGTGGTGCTCGAGACTTGCCACACAATCATCACCTGC), IN105 (5′-ATCTCAGTGGTGGTGGTGGTGGTGCTCGAGATCACCTGCCATCTGTTTTCCA), or IN106 (5′-ATCTCAGTGGTGGTGGTGGTGGTGCTCGAGTTTTCCATAATCCCTGATGATCT), respectively. The sequence of the recombinant plasmids was verified by DNA sequencing. A C-terminal His-Tag is appended to all these constructs.
The proteins (IN-H6, IN-∆N-H6, IN-CCD-H6) were expressed in E. coli BL21(DE3) grown in LB medium supplemented with kanamycin (50 µg/mL). Cultures (6 liters) were grown at 37 °C to an A600 = 0.20, transferred to 15 °C, and grown to an A600 = 0.5, and expression was induced by addition of 1 mM IPTG for 16 h. Cells were collected by centrifugation (3000× g, 10 min, 4 °C), washed twice with ice-cold buffer 150/10 (20 mM K-phosphate pH 7.5, 150 mM NaCl, 10 mM imidazole, 5% glycerol, 5 mM 2-mercaptoethanol), resuspended in the same buffer (1 mL per g of cell pellet) and lysed in an Eaton Press after freezing in dry ice. All subsequent steps were conducted at 4 °C. After addition of 2 vol. of buffer 150/10 supplemented with protease inhibitors (1 mM Pefabloc, 10 mM benzamidine, and 10 mM PMSF), extracts were clarified by sonication and by centrifugation at 70,000× g for 30 min, and incubated for 1 h at 4 °C with 1 mL of Ni-NTA Superflow matrix (Qiagen, Hilden, Germany). Beads were extensively washed with buffer 500/50 (20 mM K-phosphate pH 7.5, 500 mM NaCl, 50 mM imidazole, 5% glycerol, 5 mM 2-mercaptoethanol), and elution was performed in Bio-Spin columns (BioRad, Hercules, CA, USA) by adding 6 × 1 mLof buffer 500/400 (20 mM K-phosphate pH 7.5, 500 mM NaCl, 400 mM imidazole, 5% glycerol, 5 mM 2-mercaptoethanol) and centrifugation 1 min at 1500× g.
Eluted proteins were either dialyzed against buffer ASU (20 mM Tris-HCl pH 7.5, 100 mM NaCl, 500 mM Urea, 10% glycerol, 1 mM EDTA, 10 mM 2-mercaptoethanol) for IN-H6 and IN-∆N-H6 (pI = 8.74 and 9.46, respectively), or against buffer AQU (20 mM Tris-HCl pH 7.5, 500 mM Urea, 10% glycerol, 1 mM EDTA, 10 mM 2-mercaptoethanol) for IN-CCD-H6 (pI = 7.8), and applied either to a Mono S HR 5/5 column (IN-H6 and IN-∆N-H6) or to a Mono Q HR 5/5 column (IN-CCD-H6) equilibrated in the same buffers. Proteins were eluted by linear gradients (40 column vol.) of NaCl from 100 to 300 mM (Mono S) or from 0 to 300 mM (Mono Q). Purified proteins were concentrated by ultrafiltration (Vivaspin 6, 10 kDa), dialyzed against storage buffer (20 mM K-phosphate pH 7.5, 100 mM NaCl, 0.02% Triton X-100, 2 mM DTT), and stored at −80 °C. Protein concentrations were determined by using calculated absorption coefficients of 1.578, 1.632, or 1.552 A280 units mg−1 cm2, respectively for IN-H6, IN-∆N-H6, or IN-CCD-H6.
The various derivatives of the CTD of IN (IN-CTD213-H6, IN-CTD222-H6, IN-CTD222∆C5-H6, IN-CTD222∆C10-H6, and IN-CTD222∆C15-H6) were expressed as described above, except that cells were grown at 37 °C to an A600 = 0.25, transferred to 28 °C and grown to an A600 = 0.5, and expression was induced by addition of 1 mM IPTG for 6 h. Cell lysis and Ni-NTA chromatography were conducted as described above. Purification on Mono S HR 5/5 columns was performed as described above, except that elution was achieved by gradients (50 column vol.) of NaCl from 100 to 500 mM. Purified proteins were concentrated by ultrafiltration (Vivaspin 6, 3 kDa), dialyzed against storage buffer (20 mM K-phosphate pH 7.5, 100 mM NaCl, 2 mM DTT), and stored at −80 °C. Protein concentrations were determined by using calculated absorption coefficients of 1.536, 1.72, 1.853, 1.965, or 2.099 A280 units mg−1 cm2, respectively for IN-CTD213-H6, IN-CTD222-H6, IN-CTD222∆C5-H6, IN-CTD222∆C10-H6, and IN-CTD222∆C15-H6.
2.2. Expression of HIV-2 Integrase in E. coli and Purification
The coding regions of integrase from the two strains HIV-2_ALL and HIV-2_TRA, were amplified by PCR with oligonucleotides IN001 (5′-GTTTAACTTTAAGAAGGAGATATACCATGGC GTTTTTAGAGAACATAGAACC) and IN101 (5′-ATCTCAGTGGTGGTGGTGGTGGTGCTCGAGT GCTTCAGGTACTTGACCAGAC) or IN002 (5′-GTTTAACTTTAAGAAGGAGATATACCATGGCG TTCCTAGAAAAAATAGAACC) and IN102 (5′-ATCTCAGTGGTGGTGGTGGTGGTGCTCGAGC CTGGTATCCTCCACGTCGGCA), respectively, and introduced by the SLIC method into pET28b digested with NcoI and XhoI.
The two integrase species from HIV-2_ALL and HIV-2_TRA were expressed in E. coli BL21(DE3) grown at 37 °C to an A600 = 0.25, transferred to 28 °C and grown to an A600 = 0.5, and expression was induced by addition of 1 mM IPTG for 6 h. Purification was conducted exactly as described above for IN-H6 from HIV-1. Protein concentrations were determined by using calculated absorption coefficients of 1.156 and 1.203 A280 units mg−1 cm2, respectively, for IN from HIV-2_ALL and HIV-2_TRA.
2.3. HTRF Assay
Homogeneous time-resolved fluorescence (HTRF) assays were performed in black, flat bottom, half-area, non-binding surface, 96-well microplates (Corning #3993). Human mitochondrial LysRS (mLysRS) with a C-terminal HA-tag (mLysRS-HA) was incubated at a dimer concentration of 1.5 nM with the various integrase derivatives carrying a C-terminal His-tag at concentrations indicated in the legends of the figures, in 10 mM Tris-HCl pH 7.5, 50 mM NaCl, 10 mM 2-mercaptoethanol, and BSA at 1 mg/mL, for 1 h on ice. Antibodies (Cisbio) directed to the His-tag and conjugated with Eu3+ cryptate (Cisbio #61HISKLB), and to the HA-tag conjugated with XL665 (Cisbio #610HAXLB), were added and incubation was continued for 30 min. After addition of 50 mM KF, fluorescence of Eu3+ cryptate and of XL665 was recorded at 620 nm (I620) and 665 nm (I665), respectively, after excitation of Eu3+ cryptate at 317 nm, in an Infinite M1000 PRO microplate reader (TECAN). Results are expressed as the ratio of I665/I620.
2.4. Protein Photo-Cross-Linking
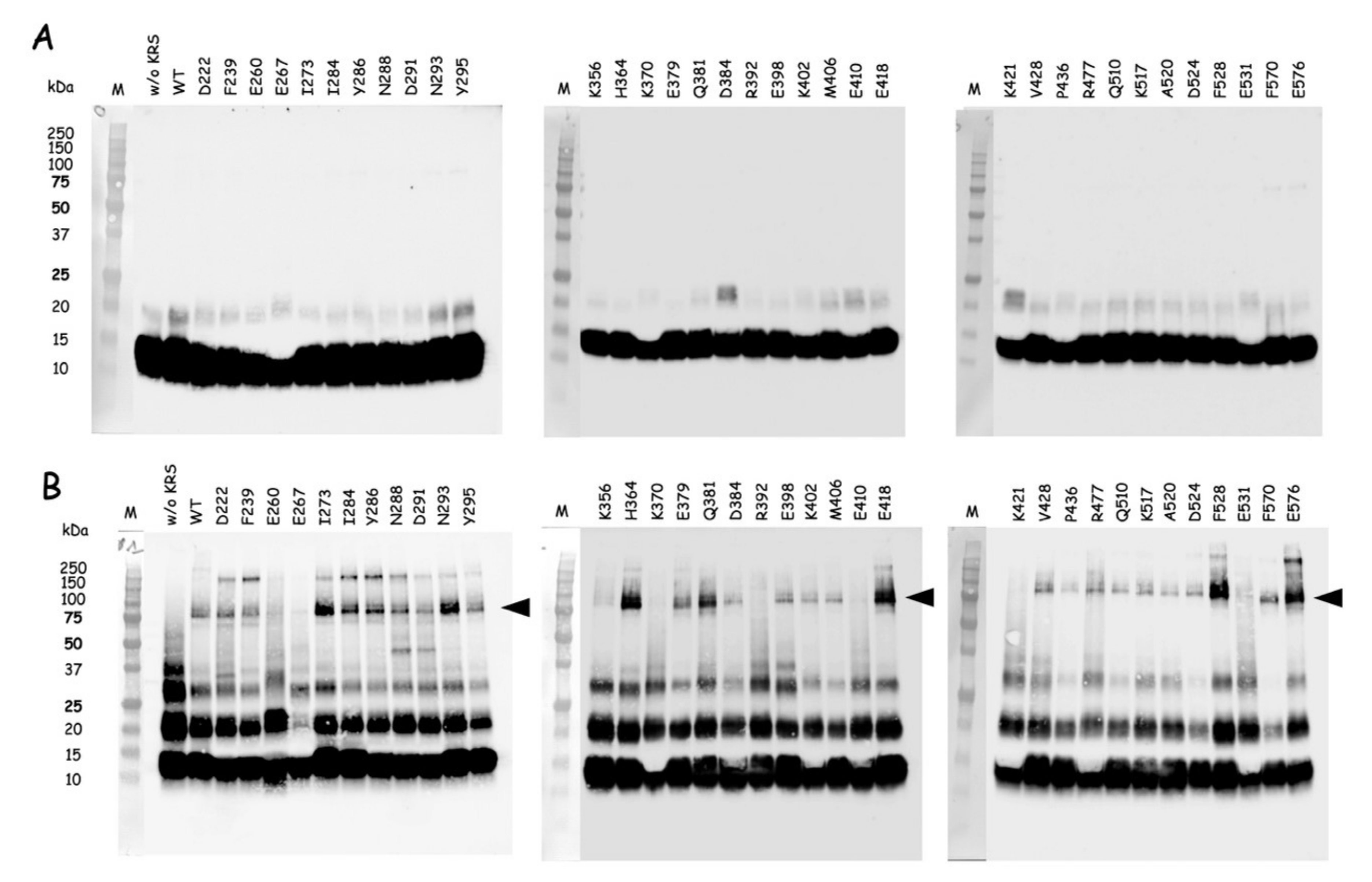
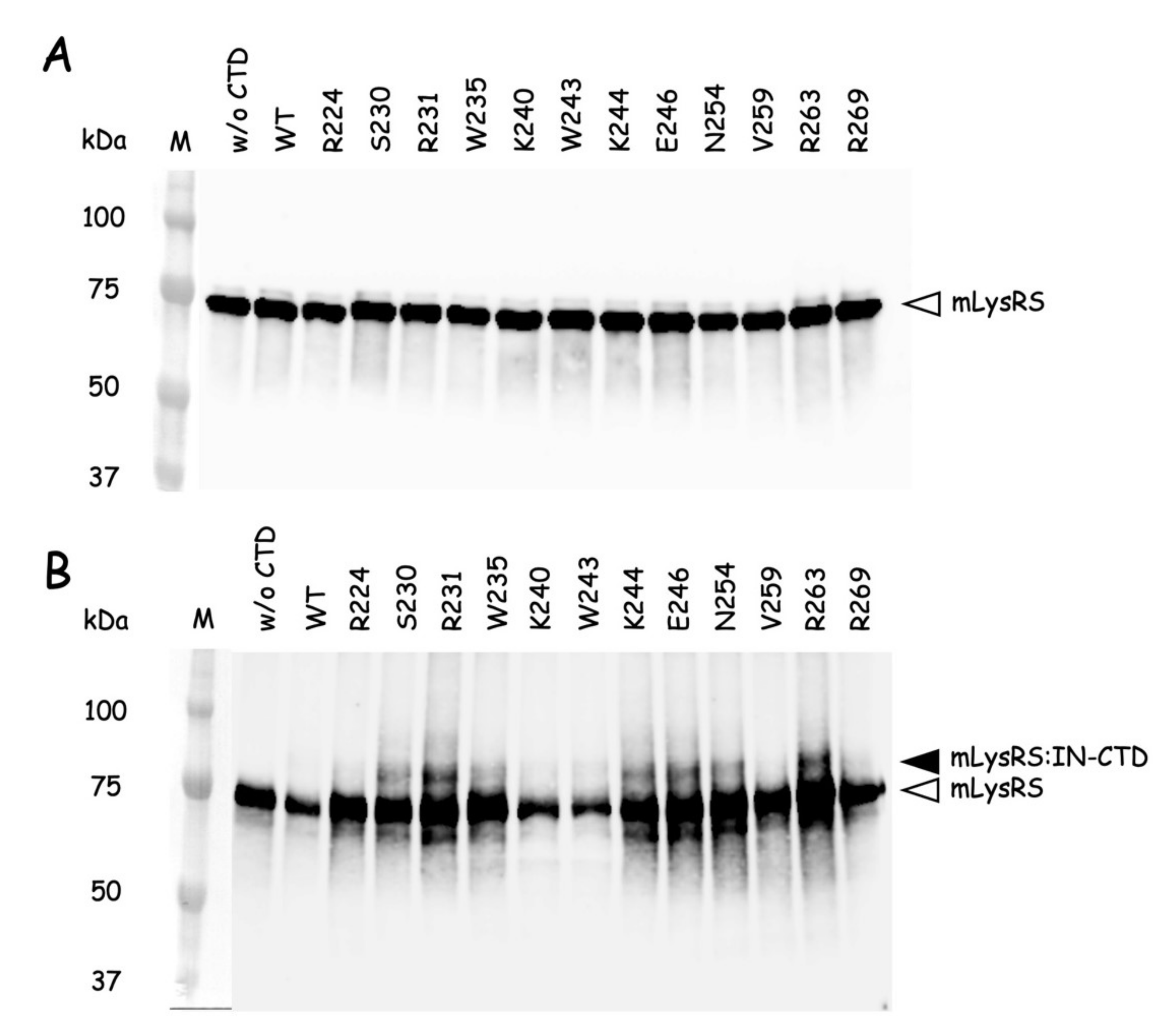
The QuickChange Lightning Site-directed Mutagenesis Kit from Agilent Technologies was used to introduce amber (TAG) stop codons at discrete sites within the nucleotide sequence encoding the catalytic domain of mLysRS or the CTD222 domain of integrase from HIV-1 as described previously [
24]. Incorporation of
p-benzoyl-L-phenylalanine (
pBpa) into mutant proteins was conducted in
E. coli BL21(DE3) transformed with pEVOL-Bpa that expresses the orthologous supression system [
25]. The
E. coli strains containing pET28b expressing the various mLysRS or IN-CTD222 mutants carrying TAG stop codons were grown at 37 °C in 1L of LB medium supplemented with kanamycin (50 µg/mL) and chloramphenicol (34 µg/mL), and containing 0.2% arabinose and 1 mM
pBpa (Bachem, Bubendorf, Switzerland). When the culture reached an
A600 = 1.0, expression was induced by addition of 1 mM IPTG for 5 h. The mLysRS
pBpa variants with
pBpa inserted at the 35 distinct positions listed in
Table S2 were purified as described previously [
24], and the IN-CTD222
pBpa variants with
pBpa inserted at the 12 distinct positions listed in
Table S3 were purified as described above for wild-type IN-CTD222-H
6. All the proteins were dialyzed against PBS and stored at −80 °C. During their purification, all the mutants of mLysRS
pBpa or IN-CTD222
pBpa were eluted similarly to their wild-type counterparts, mLysRS
WT or IN-CTD222
WT, suggesting that their oligomeric structure was not affected by insertion of
pBpa, as expected for mutations of residues accessible to the solvent.
Photo-cross-linking was conducted essentially as described in [
26]. The different mLysRS
pBpa species and IN-CTD222
pBpa species were mixed with IN-CTD222
WT and mLysRS
WT, respectively, in a final volume of 80 µL into the wells of a 96-well plate cooled on ice, at protein concentrations indicated in the legends of the figures. Plates were covered with their polystyrene lids and with 3 mm-thick glass plates to filter short-wavelength UV light, and incubated on ice into a CL-1000 Ultraviolet Crosslinker (UVP) equipped with a 365 nm UV lamp. Control samples were withdrawn before starting irradiation, and cross-linked products were analyzed by SDS-PAGE and western blotting after exposure to UV light.
2.5. Docking Simulation for the mLysRS and IN Domains
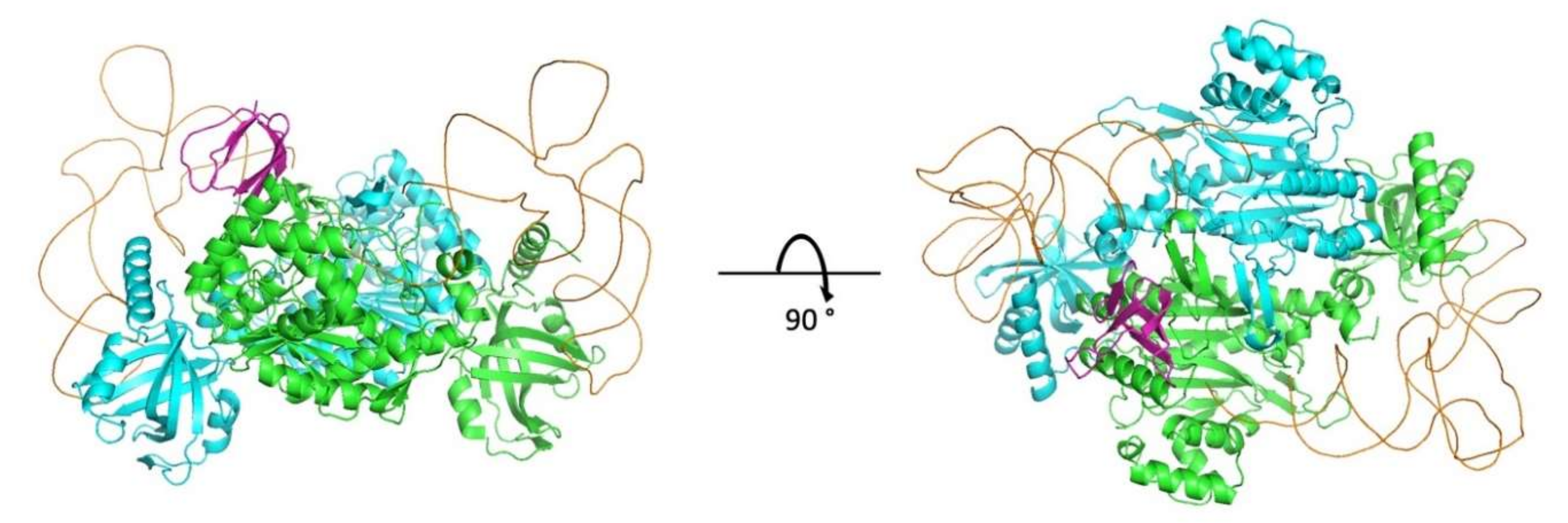
Rigid-body docking was performed between a dimeric structure of lysine-tRNA synthetase and a crystal structure of the C-terminal CTD domain of HIV-1 integrase. The input structure used for lysyl-tRNA synthetase (PDB: 3BJU) was crystallized without a tRNA molecule. The catalytic and tRNA anticodon-binding domains visible in the crystal structure are shared by cytoplasmic and mitochondrial LysRS. We modeled the structure of the bound tRNA using as template the yeast tRNA:aspartyl-tRNA synthetase complex structure (PDB: 1ASY) and superimposed the synthetase chains to deduce the conformation of the bound tRNA. An input structure containing the bound tRNA was preferred so as to prevent docking models to accumulate in unlikely regions where tRNA binds. As for the HIV-1 integrase partner, the input structure was a homodimeric structure containing both the catalytic core and the CTD (PDB: 1EX4). For the docking step, we did not isolate the CTD domain of the HIV-1 integrase, and rather kept all the domains present in the 1EX4 homodimeric structure. In that way, the helical linker connecting the catalytic core and the CTD prevented docking models to accumulate in unlikely regions.
The rigid-body docking step was performed using the InterEvDock2 server [
27] that takes into account both the physicochemical nature of protein surfaces and co-evolutionary information and uses three complementary scores Frodock [
28], SOAP-PP [
29] and InterEvScore [
30] to identify the most likely interfaces (
http://bioserv.rpbs.univ-paris-diderot.fr/services/InterEvDock2/). In the context of this particular complex, no co-alignments between both partners could be generated since they belong to different species. The docking protocol was performed as described previously following the standard protocol of the server [
31,
32], using as input the two dimeric structures described above. In the result archive, the 50 best decoys of every three scores (Frodock, SOAP-PP, InterEvScore) used in the consensus selection of the docking models were considered. Among those 150 models, 119 solutions involved the IN-CTD in the interface with LysRS. For the next refinement steps, only the CTD and not the catalytic core domain was considered in the structural models of the complex with LysRS. Models were clustered using fcc [
33] with a cutoff threshold of 0.5 and removing similarities between symmetrical structures. Forty-nine non-redundant representative models of complexes were retrieved and were refined using Rosetta [
34] through a standard relax protocol under native coordinate constraints and the scoring of the resulting interface energy between IN-CTD and LysRS using the beta_nov15 scoring function. The model with the lowest interface energy reached −45.9 rosetta units, significantly lower than any of the alternative configurations (second best model at −41.3) and was first selected for in-depth structural analysis and design of disruptive compensatory mutants. The coordinates of the refined structural model were deposited on the ModelArchive database (DOI: 10.1016/j.str.2008.12.014) and can be downloaded at (
https://modelarchive.org/doi/10.5452/ma-bxirn).
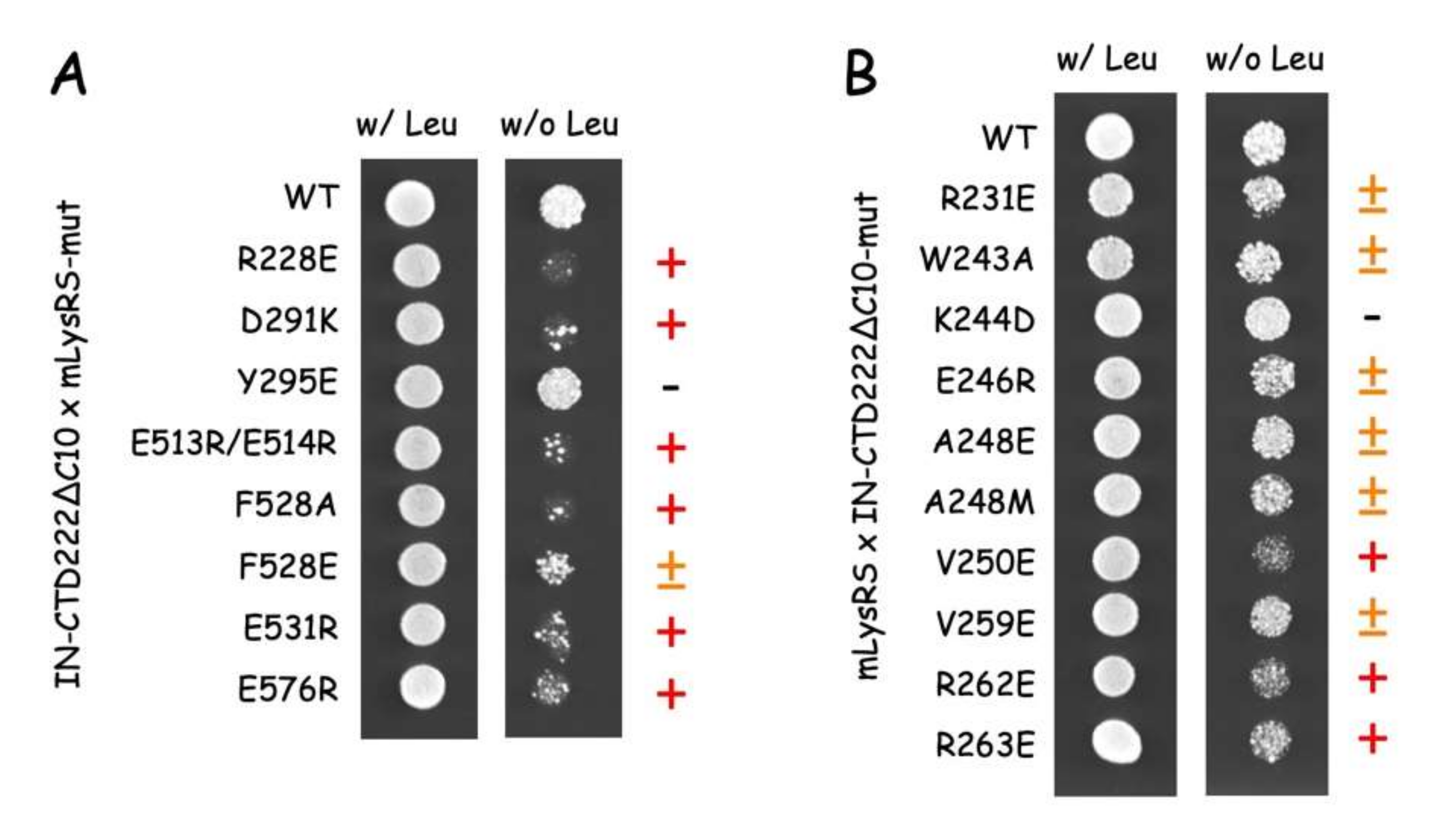
2.6. Yeast Two-Hybrid Analysis
The yeast two-hybrid system developed by Brent et al. was used [
35]. The mLysRS and IN-CTD222∆C10 coding sequences were inserted into the plasmids pJG4-5 (fused to the B42-activator domain placed under the control of a galactose-inducible promoter) and pEG202 (fused to the LexA DNA binding domain), respectively. Mutations in mLysRS or IN-CTD222∆C10 coding sequences listed in
Table S3 were generated using the QuickChange Lightning Site-directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA). The yeast strain SKY54 (
Matα
his3 leu2::
3LexAop-LEU2 ura3 trp1 lys2::λcI-op-LYS2) [
36], which contains a chromosomal
LEU2 gene placed under the control of LexA operators was transformed to his
+ with pEG202-derivatives and to trp
+ with pJG4-5 derivatives. At least four independent colonies were analyzed for their ability to grow in the absence of leucine (expression of
LexAop-LEU2). SKY54 expressing a pair of interactive proteins grew on galactose medium (YNBGal) lacking leucine (expression of B42-fusions that interacted with LexA-fusions) but did not grow on glucose medium (YNB) lacking leucine (no expression of B42-fusions).
2.7. Antibodies and Western Blot Analysis
Rabbit anti-IN-CTD antibodies were generated against a synthetic peptide (NFRVYYRDSRDPV WKGPAKLLWKGEGAVVIQDNSDIKVVPRRKAKIIRDYGK) corresponding to residues 222-273 of HIV-1 integrase and affinity purified (GeneCust, Boynes, France). The specificity of these antibodies was controlled by western blotting (
Figure S1). Polyclonal antibodies to LysRS have been described previously [
37]. Western blot analyses were conducted with goat anti-rabbit secondary antibodies conjugated to peroxidase (Chemicon) and the SuperSignal West Pico chemiluminescent substrates (Thermo Scientific, Waltham, MA, USA). Chemiluminescence was detected with a LAS-3000 Imaging System (Fuji, Tokyo, Japan).
4. Discussion
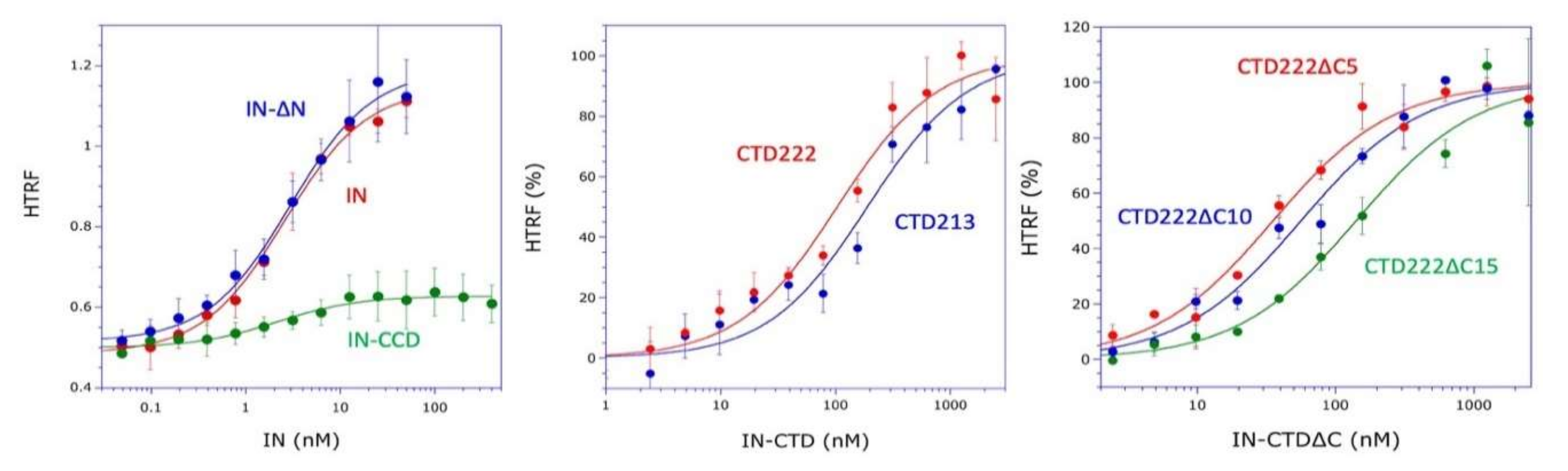
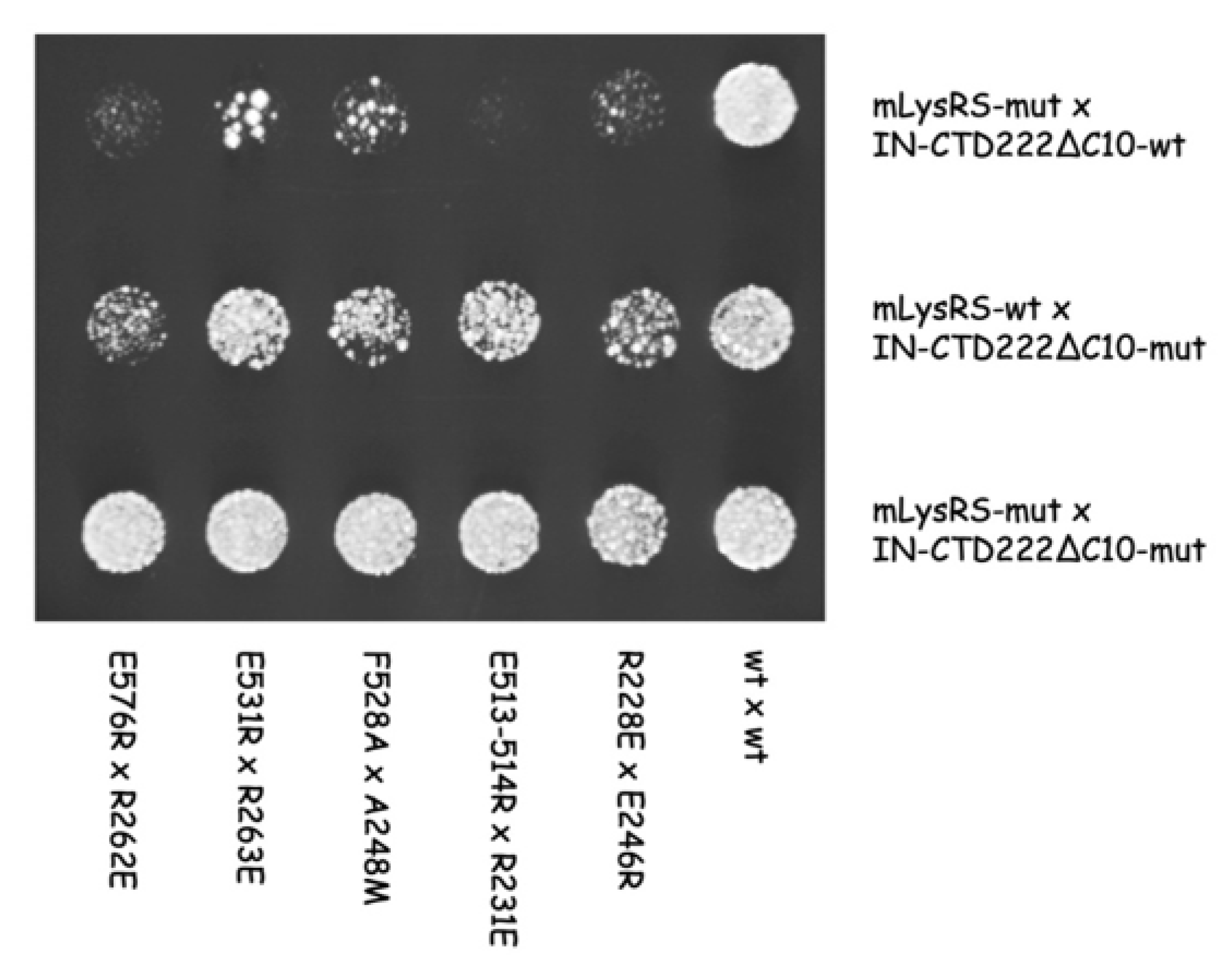
In this work, we obtained a structural model of the complex between mLysRS and IN-CTD strongly supported by three sets of experimental constraints, six cross-links with
pBpa-containing mLysRS mutants, five cross-links with
pBpa-containing IN-CTD proteins, and five pairs of compensatory mutations. This structural model brings several key insights. First, association of IN-CTD requires that mLysRS is a dimer. Indeed, among the residues clearly involved in the interaction, as assessed by compensatory mutant experiments (
Figure 9), two belong to one subunit of the dimer (R228 and E576), and four to the other subunit (E513, E514, F528 and E531) (
Figure S5). The two symmetrical binding sites on mLysRS are 30 Å apart, and thus, could bind two IN-CTDs. The polypeptide linkers between IN-CCD and IN-CTD of a dimer of integrase are made of 18 amino acid residues, and were described as non-structured polypeptides in the intasome structure [
22] or as long α-helices in the crystal structure of a dimer of IN-CCD-CTD [
21]. Therefore, this linker is very flexible and the two CTDs of a dimer of integrase are likely to be able to bind to a dimer of LysRS. This is also consistent with our finding that monomeric IN-CTD binds mLysRS with a
Kd of about 40 to 140 nM (
Figure 1C), whereas oligomeric IN binds to a dimer of mLysRS with a 20-fold higher affinity (
Kd of about 3.0 nM,
Figure 1A). However, in the context of the GagPol polyprotein, it is not known if a monomeric domain of IN located at the very C-terminus of GagPol interacts with mLysRS to form the GagPol:mLysRS:tRNA
Lys,3 packaging complex, or if the IN domain of GagPol is able to form a dimer in the context of the polyprotein precursor. The second noticeable feature of the mLysRS-IN complex is the proximity of the tRNA molecules with the CTD domains of integrase (
Figure 10). The acceptor stem of the tRNA molecule is close to the IN-CTD, which suggests a possible protein-tRNA interaction that could stabilize the association of the tRNA within the mLysRS:IN complex. In particular, Lys266 and Arg269 are directed towards the TΨC stem-loop of the tRNA molecule and could establish salt bridges. This is consistent with our previous report showing that the affinity of tRNA
Lys,3 for mLysRS is increased by about two-fold in the presence of a derivative of integrase [
12].
The finding that the five compensatory mutants tested in this work (
Figure 9) successfully restored normal growth on yeast cells, strongly argue in favor of the proposed model of mLysRS:IN association. It is noteworthy that mutations introduced in IN-CTD have less pronounced growth phenotypes than those introduced in mLysRS (
Figure 8). IN-CTD is a small domain made of 57 amino acid residues that form a β-barrel structure (
Figure S3). Residues of IN-CTD that build the site of interaction with mLysRS are mostly located in loops that join the β-strands of this small structural domain, which suggests that they may be more mobile than the residues of mLysRS engaged in this assembly platform, which are part of a more compact structure, and are generally located in α-helices. Thus, mutations in IN-CTD could be more easily tolerated due to the flexibility of this domain that could accommodate some variations.
Among 20,000 sequences of IN from HIV-1 (from non-redundant GenBank CDS translations+PDB+SwissProt+PIR+PRF), only 16 changes are observed for A248 (11S, 4T and 1V), 17 changes for V250 (15I, 2L and 1G), eight changes for R262 (7K and 1G), and 10 changes for R263 (7K, 2G and 1S). This very high level of conservation is also noted among 425 sequences of IN from HIV-2 (from non-redundant GenBank CDS translations+PDB+SwissProt+PIR+PRF), three changes are observed for R262 (1K and 2S), one change for R263 (1G), and A248 is strictly conserved. Concerning V250, this residue is mainly recovered as Ile (390) or Leu (27), corresponding to conservative changes. The high level of conservation of these residues in HIV-1 and HIV-2 suggests that they are important for the function of integrase. Nevertheless, conservation of functionally important residues is likely to be correlated to the many roles of integrase in the life cycle of the virus.
Integrase is a multifunctional protein involved in many aspects of HIV-1 biology. It catalyzes the strand-transfer reaction during the integration step of viral DNA into host genome, it fulfills an essential role in virus morphogenesis, and, as a component of the GagPol polyprotein precursor, it appears to be necessary for the packaging of tRNA
Lys,3 complexed with mLysRS, a crucial step for initiation of reverse transcription. The CTD of IN is involved in these three functional roles. Residues R228, R231, E246, A248, R263, and K266 are predicted to interact with viral DNA and residues R262, R263, and K266 with another monomer of IN, within the strand-transfer complex [
22]. Residues K264, K266, R269, and K273 interact with viral RNA, an essential step in virus morphogenesis [
17]. Mutation of these residues into Ala generates noninfectious particles that are unable to initiate reverse transcription. In the present study, mutation of residues R231, W243, E246, A248, V250, V259, R262, and R263 of IN-CTD compromised its interaction with mLysRS and are predicted to abolish tRNA
Lys,3 packaging into viral particles, and to inhibit viral replication. The conclusions of this work must now be validated by in cellulo approaches. The results obtained in this study offer the opportunity to test several mutants of mLysRS and of the IN domain of GagPol for mLysRS and tRNA
Lys,3 packaging, for the initiation of reverse transcription of viral RNA and for HIV-1 infectivity.
Although packaging of tRNA
Lys,3 into new virions is absolutely required to generate infectious particles, several of the conserved residues of IN-CTD identified as key residues in the interaction with mLysRS are also involved in electrostatic protein-DNA interactions within the strand transfer complex [
22]. Because residues R231, E246, A248, and R263 are predicted to be involved in these two functions, characterization of the effects of their mutation in a cellular system of HIV-1 replication requires a detailed analysis of the stages of the viral life cycle that are affected by these changes.
Two classes of inhibitors have been developed to inhibit IN functions. INSTIs, such as raltegravir or dolutegravir, inhibit the strand-transfer step catalyzed by IN and are used in antiviral therapies [
18]. ALLINIs induce abnormal IN multimerization, prevent interaction on IN with viral RNA, generating eccentric non-infectious particles defective in viral replication [
17,
19]. Because viral resistance to drugs frequently develops, there is a need to develop antiviral drugs with new resistance profiles. The structural model of the complex between IN-CTD and mLysRS reported in this study will provide a support for searching molecules likely to disrupt the interface between the two proteins. The best inhibitor candidates should be able to interfere with both the hydrophobic and electrostatic components of the assembly platform.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









