Virosaurus A Reference to Explore and Capture Virus Genetic Diversity

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection and Cleaning Up of Viral Sequences
2.2. Generation of Complete Virus Sequence Dataset
2.3. Clustering
2.4. Generation of Virus Genes Dataset
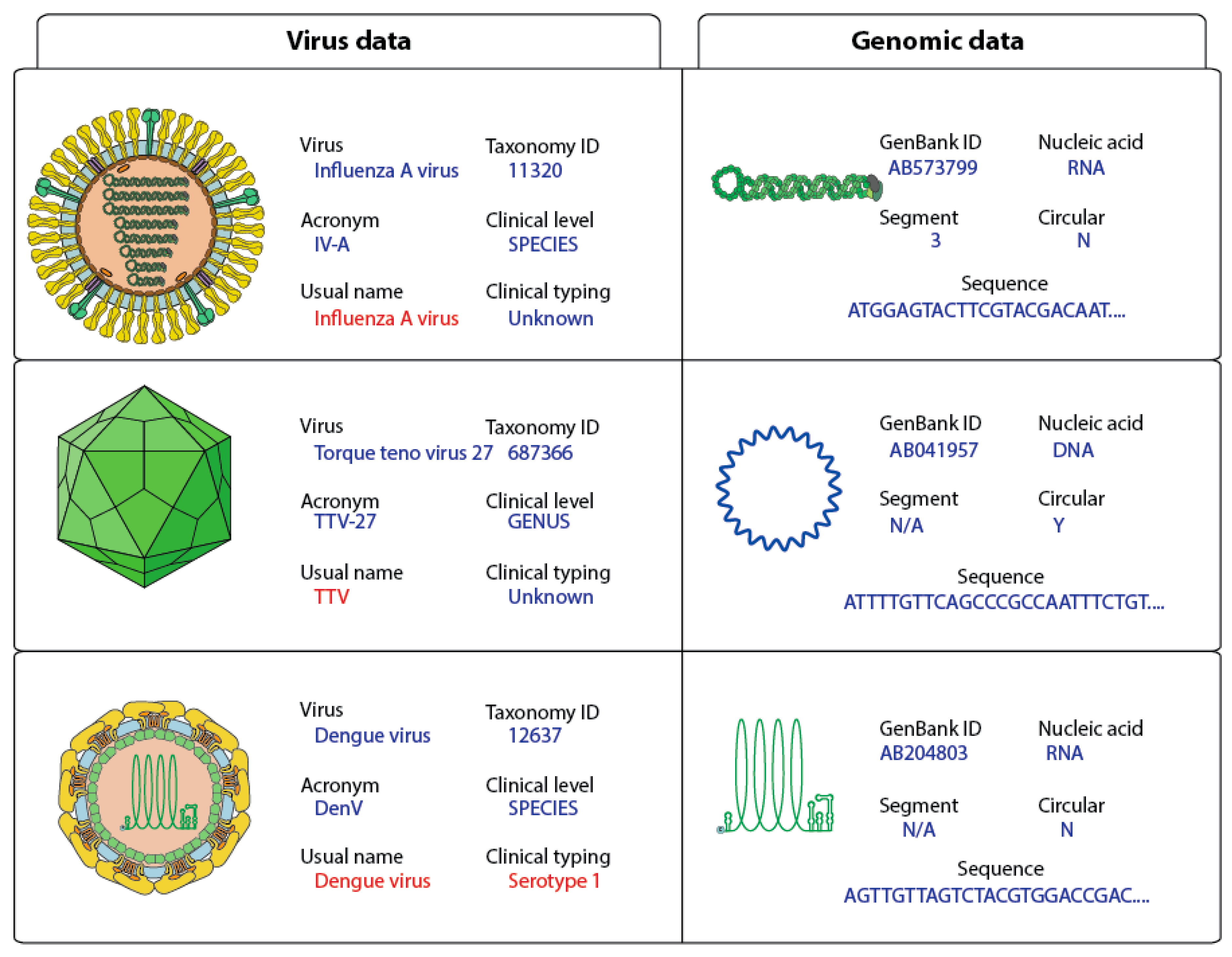
2.5. Clinical Annotation
2.6. FASTA Format
2.7. Evaluation on Clinical Samples
3. Results
3.1. Data Collection
3.2. Clinical Annotation
3.3. Clustering
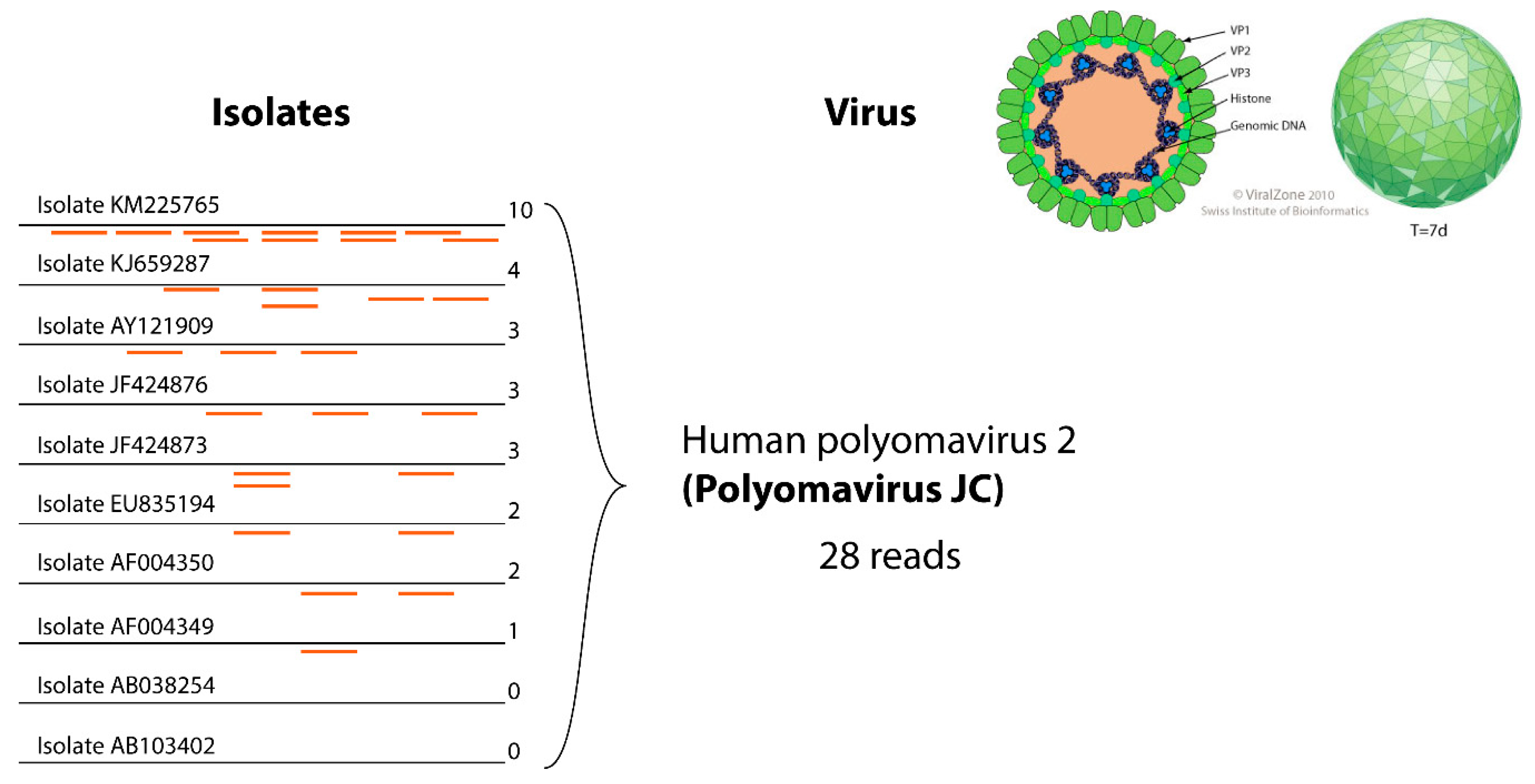
3.4. Reducing False Negatives in Virus Detection
3.5. Validation on Clinical Samples
3.6. Virosaurus Availability and Updates
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Human Viruses Table—ViralZone Page. Available online: https://swissprot.sib.swiss/prime/678 (accessed on 9 July 2020).
- Diagnostic Methods in Virology, Virological Methods, Virus Culture, Virus Isolation. Available online: http://virology-online.com/general/Test1.htm (accessed on 9 July 2020).
- Happi, A.N.; Happi, C.T.; Schoepp, R.J. Lassa fever diagnostics: Past, present, and future. Curr. Opin. Virol. 2019, 37, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Lipkin, W.I.; Firth, C. Viral surveillance and discovery. Curr. Opin. Virol. 2013, 3, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipkin, W.I.; Anthony, S.J. Virus hunting. Virology 2015, 479–480, 194–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, C.Y.; Miller, S.A. Clinical metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, D.W.; Zagordi, O.; Zbinden, A.; Schuurmans, M.M.; Schreiber, P.; Geissberger, F.-D.; Huder, J.B.; Böni, J.; Benden, C.; Mueller, N.J.; et al. Unbiased metagenomic sequencing complements specific routine diagnostic methods and increases chances to detect rare viral strains. Diagn. Microbiol. Infect. Dis. 2015, 83, 133–138. [Google Scholar] [CrossRef]
- Nooij, S.; Schmitz, D.; Vennema, H.; Kroneman, A.; Koopmans, M.P.G. Overview of Virus Metagenomic Classification Methods and Their Biological Applications. Front. Microbiol 2018, 9, 749. [Google Scholar] [CrossRef]
- Petty, T.J.; Cordey, S.; Padioleau, I.; Docquier, M.; Turin, L.; Preynat-Seauve, O.; Zdobnov, E.M.; Kaiser, L. Comprehensive human virus screening using high-throughput sequencing with a user-friendly representation of bioinformatics analysis: A pilot study. J. Clin. Microbiol. 2014, 52, 3351–3361. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Ling, Y.; Xi, Y.; Ma, H.; Wang, H.; Hu, H.-M.; Liu, Q.; Li, Y.-M.; Deng, X.-T.; Yang, S.-X.; et al. Viral metagenomics updated the prevalence of human papillomavirus types in anogenital warts. Emerg. Microbes Infect. 2019, 8, 1291–1299. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, S.R.; Wang, D. Origins and challenges of viral dark matter. Virus Res. 2017, 239, 136–142. [Google Scholar] [CrossRef]
- Rose, R.; Constantinides, B.; Tapinos, A.; Robertson, D.L.; Prosperi, M. Challenges in the analysis of viral metagenomes. Virus Evol. 2016, 2, vew022. [Google Scholar] [CrossRef] [Green Version]
- Kiselev, D.; Matsvay, A.; Abramov, I.; Dedkov, V.; Shipulin, G.; Khafizov, K. Current Trends in Diagnostics of Viral Infections of Unknown Etiology. Viruses 2020, 12, 211. [Google Scholar] [CrossRef] [Green Version]
- Stano, M.; Beke, G.; Klucar, L. viruSITE-integrated database for viral genomics. Database (Oxford) 2016, 2016. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D7337–D7345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodacre, N.; Aljanahi, A.; Nandakumar, S.; Mikailov, M.; Khan, A.S. A Reference Viral Database (RVDB) To Enhance Bioinformatics Analysis of High-Throughput Sequencing for Novel Virus Detection. mSphere 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Cochrane, G.; Karsch-Mizrachi, I. International Nucleotide Sequence Database Collaboration the International Nucleotide Sequence Database Collaboration. Nucleic Acids Res. 2013, 41, D212–D214. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Cordey, S.; Hartley, M.-A.; Keitel, K.; Laubscher, F.; Brito, F.; Junier, T.; Kagoro, F.; Samaka, J.; Masimba, J.; Said, Z.; et al. Detection of novel astroviruses MLB1 and MLB2 in the sera of febrile Tanzanian children. Emerg. Microbes Infect. 2018, 7, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.H.; Cordey, S.; Bhuva, N.; Laubscher, F.; Hartley, M.-A.; Boillat-Blanco, N.; Mbarack, Z.; Samaka, J.; Mlaganile, T.; Jain, K.; et al. Investigation of the Plasma Virome from Cases of Unexplained Febrile Illness in Tanzania from 2013 to 2014: A Comparative Analysis between Unbiased and VirCapSeq-VERT High-Throughput Sequencing Approaches. mSphere 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Cotmore, S.F.; Agbandje-McKenna, M.; Chiorini, J.A.; Mukha, D.V.; Pintel, D.J.; Qiu, J.; Soderlund-Venermo, M.; Tattersall, P.; Tijssen, P.; Gatherer, D.; et al. The family Parvoviridae. Arch. Virol. 2014, 159, 1239–1247. [Google Scholar] [CrossRef]
- Ball, J.K.; Curran, R.; Berridge, S.; Grabowska, A.M.; Jameson, C.L.; Thomson, B.J.; Irving, W.L.; Sharp, P.M. TT virus sequence heterogeneity in vivo: Evidence for co-infection with multiple genetic types. J. Gen. Virol. 1999, 80, 1759–1768. [Google Scholar] [CrossRef] [Green Version]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuilleumier, S.; Bonhoeffer, S. Contribution of recombination to the evolutionary history of HIV. Curr. Opin. HIV AIDS 2015, 10, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Cordey, S.; Laubscher, F.; Hartley, M.-A.; Junier, T.; Pérez-Rodriguez, F.J.; Keitel, K.; Vieille, G.; Samaka, J.; Mlaganile, T.; Kagoro, F.; et al. Detection of dicistroviruses RNA in blood of febrile Tanzanian children. Emerg. Microbes Infect. 2019, 8, 613–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, J.F.; Laubscher, F.; Held, J.; Eckerle, I.; Docquier, M.; Grobusch, M.P.; Mordmüller, B.; Kaiser, L.; Cordey, S. Unbiased metagenomic next-generation sequencing of blood from hospitalized febrile children in Gabon. Emerg. Microbes Infect. 2020, 9, 1242–1244. [Google Scholar] [CrossRef]
- Fernandes, J.F.; Held, J.; Dorn, M.; Lalremruata, A.; Schaumburg, F.; Alabi, A.; Agbanrin, M.D.; Kokou, C.; Ben Adande, A.; Esen, M.; et al. Causes of fever in Gabonese children: A cross-sectional hospital-based study. Sci Rep. 2020, 10, 2080. [Google Scholar] [CrossRef]
- Forum on Microbial Threats; Board on Global Health; Institute of Medicine. Emerging Viral Diseases: The One Health Connection: Workshop Summary; The National Academies Collection: Reports funded by National Institutes of Health; National Academies Press (US): Washington, DC, USA, 2015; ISBN 978-0-309-31397-1. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gleizes, A.; Laubscher, F.; Guex, N.; Iseli, C.; Junier, T.; Cordey, S.; Fellay, J.; Xenarios, I.; Kaiser, L.; Mercier, P.L. Virosaurus A Reference to Explore and Capture Virus Genetic Diversity. Viruses 2020, 12, 1248. https://doi.org/10.3390/v12111248
Gleizes A, Laubscher F, Guex N, Iseli C, Junier T, Cordey S, Fellay J, Xenarios I, Kaiser L, Mercier PL. Virosaurus A Reference to Explore and Capture Virus Genetic Diversity. Viruses. 2020; 12(11):1248. https://doi.org/10.3390/v12111248
Chicago/Turabian StyleGleizes, Anne, Florian Laubscher, Nicolas Guex, Christian Iseli, Thomas Junier, Samuel Cordey, Jacques Fellay, Ioannis Xenarios, Laurent Kaiser, and Philippe Le Mercier. 2020. "Virosaurus A Reference to Explore and Capture Virus Genetic Diversity" Viruses 12, no. 11: 1248. https://doi.org/10.3390/v12111248
APA StyleGleizes, A., Laubscher, F., Guex, N., Iseli, C., Junier, T., Cordey, S., Fellay, J., Xenarios, I., Kaiser, L., & Mercier, P. L. (2020). Virosaurus A Reference to Explore and Capture Virus Genetic Diversity. Viruses, 12(11), 1248. https://doi.org/10.3390/v12111248