Methamphetamine Increases the Proportion of SIV-Infected Microglia/Macrophages, Alters Metabolic Pathways, and Elevates Cell Death Pathways: A Single-Cell Analysis

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Samples, SIV Infection, and Methamphetamine Treatment
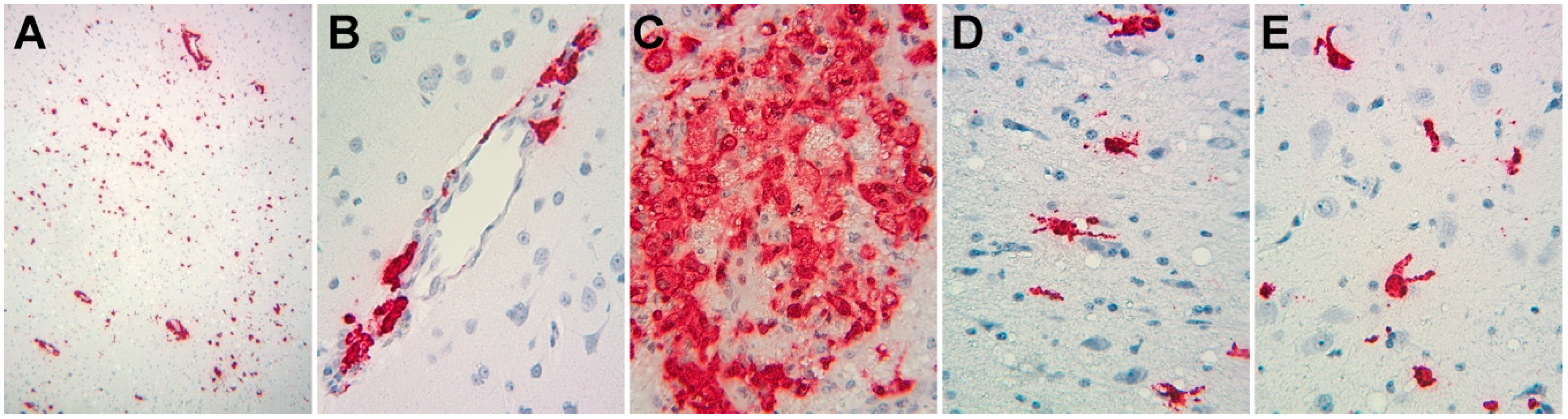
2.2. In Situ Hybridization
2.3. Single Cell Preparation and RNA Sequencing
2.4. Bioinformatic Analyses
2.5. Statistics
2.6. Data Availability
3. Results
3.1. Characterization of Samples
3.2. Single-Cell Cluster Analysis and SIV Infection
3.3. Effect of Meth on SIVE
3.3.1. Analysis of Macrophage/Microglia Marker Genes
3.3.2. Pathway Analysis
3.3.3. Differential Gene Expression
3.3.4. Upstream Regulators
3.4. Analysis of Microglial Clusters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vollm, B.A.; de Araujo, I.E.; Cowen, P.J.; Rolls, E.T.; Kringelbach, M.L.; Smith, K.A.; Jezzard, P.; Heal, R.J.; Matthews, P.M. Methamphetamine activates reward circuitry in drug naive human subjects. Neuropsychopharmacology 2004, 29, 1715–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbruscato, T.J.; Trippier, P.C. DARK Classics in Chemical Neuroscience: Methamphetamine. ACS Chem. Neurosci. 2018, 9, 2373–2378. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.L.; Bisagno, V.; Milroy, C.M. Neuropathology of substance use disorders. Acta Neuropathol. 2014, 127, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chen, T.; Xue, L.; Wang, J.; Jia, Y.; Li, G.; Ren, H.; Wu, F.; Wu, M.; Chen, Y. Methamphetamine exacerbates neuroinflammatory response to lipopolysaccharide by activating dopamine D1-like receptors. Int. Immunopharmacol. 2019, 73, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Adhikary, S.; Sobesky, J.L.; Weber, M.D.; Watkins, L.R.; Maier, S.F. The danger-associated molecular pattern HMGB1 mediates the neuroinflammatory effects of methamphetamine. Brain Behav. Immun. 2016, 51, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, J.; Leitao, R.A.; Higuera-Matas, A.; Assis, M.A.; Coria, S.M.; Fontes-Ribeiro, C.; Ambrosio, E.; Silva, A.P. Extended-access methamphetamine self-administration elicits neuroinflammatory response along with blood-brain barrier breakdown. Brain Behav. Immun. 2017, 62, 306–317. [Google Scholar] [CrossRef]
- Clark, K.H.; Wiley, C.A.; Bradberry, C.W. Psychostimulant abuse and neuroinflammation: Emerging evidence of their interconnection. Neurotox. Res. 2013, 23, 174–188. [Google Scholar] [CrossRef]
- Kohno, M.; Morales, A.M.; Ghahremani, D.G.; Hellemann, G.; London, E.D. Risky decision making, prefrontal cortex, and mesocorticolimbic functional connectivity in methamphetamine dependence. JAMA Psychiatry 2014, 71, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Hoenigl, M.; Chaillon, A.; Moore, D.J.; Morris, S.R.; Smith, D.M.; Little, S.J. Clear Links between Starting Methamphetamine and Increasing Sexual Risk Behavior: A Cohort Study among Men Who Have Sex with Men. J. Acquir. Immune Defic. Syndr. 2016, 71, 551–557. [Google Scholar] [CrossRef] [Green Version]
- Finlayson, T.J.; Le, B.; Smith, A.; Bowles, K.; Cribbin, M.; Miles, I.; Oster, A.M.; Martin, T.; Edwards, A.; Dinenno, E.; et al. HIV risk, prevention, and testing behaviors among men who have sex with men--National HIV Behavioral Surveillance System, 21 U.S. cities, United States, 2008. MMWR Surveill. Summ. 2011, 60, 1–34. [Google Scholar]
- Freeman, P.; Walker, B.C.; Harris, D.R.; Garofalo, R.; Willard, N.; Ellen, J.M.; Adolescent Trials Network for HIV/AIDS Interventions 016b Team. Methamphetamine use and risk for HIV among young men who have sex with men in 8 US cities. Arch. Pediatr. Adolesc. Med. 2011, 165, 736–740. [Google Scholar] [CrossRef] [Green Version]
- Mayer, K.H.; O’Cleirigh, C.; Skeer, M.; Covahey, C.; Leidolf, E.; Vanderwarker, R.; Safren, S.A. Which HIV-infected men who have sex with men in care are engaging in risky sex and acquiring sexually transmitted infections: Findings from a Boston community health centre. Sex. Transm. Infect. 2010, 86, 66–70. [Google Scholar] [CrossRef] [Green Version]
- Shoptaw, S.; Reback, C.J. Associations between methamphetamine use and HIV among men who have sex with men: A model for guiding public policy. J. Urban Health 2006, 83, 1151–1157. [Google Scholar] [CrossRef] [Green Version]
- Buchacz, K.; McFarland, W.; Kellogg, T.A.; Loeb, L.; Holmberg, S.D.; Dilley, J.; Klausner, J.D. Amphetamine use is associated with increased HIV incidence among men who have sex with men in San Francisco. Aids 2005, 19, 1423–1424. [Google Scholar] [CrossRef]
- Koblin, B.A.; Husnik, M.J.; Colfax, G.; Huang, Y.; Madison, M.; Mayer, K.; Barresi, P.J.; Coates, T.J.; Chesney, M.A.; Buchbinder, S. Risk factors for HIV infection among men who have sex with men. Aids 2006, 20, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Lambert, E.; Normand, J.; Stall, R.; Aral, S.; Vlahov, D. Introduction: New dynamics of HIV risk among drug-using men who have sex with men. J. Urban Health 2005, 82, i1–i8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plankey, M.W.; Ostrow, D.G.; Stall, R.; Cox, C.; Li, X.; Peck, J.A.; Jacobson, L.P. The relationship between methamphetamine and popper use and risk of HIV seroconversion in the multicenter AIDS cohort study. J. Acquir. Immune Defic. Syndr. 2007, 45, 85–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurt, C.B.; Torrone, E.; Green, K.; Foust, E.; Leone, P.; Hightow-Weidman, L. Methamphetamine use among newly diagnosed HIV-positive young men in North Carolina, United States, from 2000 to 2005. PLoS ONE 2010, 5, e11314. [Google Scholar] [CrossRef]
- Kaul, M.; Garden, G.A.; Lipton, S.A. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 2001, 410, 988–994. [Google Scholar]
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R., Jr.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010, 75, 2087–2096. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, F.; Amieva, H.; Marquant, F.; Bernard, C.; Bruyand, M.; Dauchy, F.A.; Mercie, P.; Greib, C.; Richert, L.; Neau, D.; et al. Cognitive disorders in HIV-infected patients: Are they HIV-related? Aids 2013, 27, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Simioni, S.; Cavassini, M.; Annoni, J.M.; Abraham, A.R.; Bourquin, I.; Schiffer, V.; Calmy, A.; Chave, J.P.; Giacobini, E.; Hirschel, B.; et al. Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. Aids 2010, 24, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, N.; Rosen, E.P.; Gilliland, W.M., Jr.; Kovarova, M.; Remling-Mulder, L.; De La Cruz, G.; White, N.; Adamson, L.; Schauer, A.P.; Sykes, C.; et al. Antiretroviral concentrations and surrogate measures of efficacy in the brain tissue and CSF of preclinical species. Xenobiotica 2019, 49, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Letendre, S.; Marquie-Beck, J.; Capparelli, E.; Best, B.; Clifford, D.; Collier, A.C.; Gelman, B.B.; McArthur, J.C.; McCutchan, J.A.; Morgello, S.; et al. Validation of the CNS Penetration-Effectiveness rank for quantifying antiretroviral penetration into the central nervous system. Arch. Neurol. 2008, 65, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, M.; Bumpus, N.N.; Ma, Q.; Ellis, R.J.; Soontornniyomkij, V.; Fields, J.A.; Bharti, A.; Achim, C.L.; Moore, D.J.; Letendre, S.L. Antiretroviral drug concentrations in brain tissue of adult decedents. Aids 2020. [Google Scholar] [CrossRef]
- Gill, A.J.; Kolson, D.L. Chronic inflammation and the role for cofactors (hepatitis C, drug abuse, antiretroviral drug toxicity, aging) in HAND persistence. Curr. HIV AIDS Rep. 2014, 11, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.L.; Connaghan, K.P.; Wei, Y.; Li, M.D. NeuroHIV and use of addictive substances. Int. Rev. Neurobiol. 2014, 118, 403–440. [Google Scholar] [CrossRef]
- Nath, A. Human immunodeficiency virus-associated neurocognitive disorder: Pathophysiology in relation to drug addiction. Ann. N. Y. Acad. Sci. 2010, 1187, 122–128. [Google Scholar] [CrossRef]
- Loftis, J.M.; Janowsky, A. Neuroimmune basis of methamphetamine toxicity. Int. Rev. Neurobiol. 2014, 118, 165–197. [Google Scholar] [CrossRef] [Green Version]
- Salamanca, S.A.; Sorrentino, E.E.; Nosanchuk, J.D.; Martinez, L.R. Impact of methamphetamine on infection and immunity. Front. Neurosci. 2014, 8, 445. [Google Scholar] [CrossRef] [Green Version]
- Durvasula, R.; Miller, T.R. Substance abuse treatment in persons with HIV/AIDS: Challenges in managing triple diagnosis. Behav. Med. 2014, 40, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, M.B.; Thomas, J.A.; Alexy, E.R.; Irvine, M.K. Crystal methamphetamine use and HIV medical outcomes among HIV-infected men who have sex with men accessing support services in New York. Drug Alcohol Depend. 2015, 147, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Chana, G.; Everall, I.P.; Crews, L.; Langford, D.; Adame, A.; Grant, I.; Cherner, M.; Lazzaretto, D.; Heaton, R.; Ellis, R.; et al. Cognitive deficits and degeneration of interneurons in HIV+ methamphetamine users. Neurology 2006, 67, 1486–1489. [Google Scholar] [CrossRef] [PubMed]
- Langford, D.; Adame, A.; Grigorian, A.; Grant, I.; McCutchan, J.A.; Ellis, R.J.; Marcotte, T.D.; Masliah, E. Patterns of selective neuronal damage in methamphetamine-user AIDS patients. J. Acquir. Immune Defic. Syndr. 2003, 34, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.P.; Schoenbaum, E.E.; Gourevitch, M.N.; Buono, D.; Klein, R.S. A prospective study of HIV disease progression in female and male drug users. Aids 1999, 13, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Rippeth, J.D.; Heaton, R.K.; Carey, C.L.; Marcotte, T.D.; Moore, D.J.; Gonzalez, R.; Wolfson, T.; Grant, I.; Group, H. Methamphetamine dependence increases risk of neuropsychological impairment in HIV infected persons. J. Int. Neuropsychol. Soc. 2004, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, M.; Atluri, V.S.; Nair, M. Drugs of Abuse in HIV infection and neurotoxicity. Front. Microbiol. 2015, 6, 217. [Google Scholar] [CrossRef] [Green Version]
- Toussi, S.S.; Joseph, A.; Zheng, J.H.; Dutta, M.; Santambrogio, L.; Goldstein, H. Short communication: Methamphetamine treatment increases in vitro and in vivo HIV replication. AIDS Res. Hum. Retrovir. 2009, 25, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Wang, X.; Chen, H.; Song, L.; Ye, L.; Wang, S.H.; Wang, Y.J.; Zhou, L.; Ho, W.Z. Methamphetamine enhances HIV infection of macrophages. Am. J. Pathol. 2008, 172, 1617–1624. [Google Scholar] [CrossRef]
- Marcondes, M.C.; Flynn, C.; Watry, D.D.; Zandonatti, M.; Fox, H.S. Methamphetamine increases brain viral load and activates natural killer cells in simian immunodeficiency virus-infected monkeys. Am. J. Pathol. 2010, 177, 355–361. [Google Scholar] [CrossRef]
- National Institute on Drug Abuse. Overdose Death Rates (from the Centers for Disease Control and Prevention, National Center for Health Statistics). Available online: https://www.drugabuse.gov/drug-topics/trends-statistics/overdose-death-rates (accessed on 15 October 2020).
- Twillman, R.K.; Dawson, E.; LaRue, L.; Guevara, M.G.; Whitley, P.; Huskey, A. Evaluation of Trends of Near-Real-Time Urine Drug Test Results for Methamphetamine, Cocaine, Heroin, and Fentanyl. JAMA Netw. Open 2020, 3, e1918514. [Google Scholar] [CrossRef] [Green Version]
- Strickland, J.C.; Havens, J.R.; Stoops, W.W. A nationally representative analysis of “twin epidemics”: Rising rates of methamphetamine use among persons who use opioids. Drug Alcohol Depend. 2019, 204, 107592. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.S.; Kasper, Z.A.; Cicero, T.J. Twin epidemics: The surging rise of methamphetamine use in chronic opioid users. Drug Alcohol Depend. 2018, 193, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2019, 101, 207–223.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Bottcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Sousa, C.; Golebiewska, A.; Poovathingal, S.K.; Kaoma, T.; Pires-Afonso, Y.; Martina, S.; Coowar, D.; Azuaje, F.; Skupin, A.; Balling, R.; et al. Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Aguilar, S.V.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; Zelada Gonzalez, F.; Perrin, P.; et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016, 353, aad8670. [Google Scholar] [CrossRef]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [Green Version]
- Jordao, M.J.C.; Sankowski, R.; Brendecke, S.M.; Sagar; Locatelli, G.; Tai, Y.H.; Tay, T.L.; Schramm, E.; Armbruster, S.; Hagemeyer, N.; et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 2019, 363. [Google Scholar] [CrossRef] [PubMed]
- Sankowski, R.; Bottcher, C.; Masuda, T.; Geirsdottir, L.; Sagar; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Marcondes, M.C.; Burudi, E.M.; Huitron-Resendiz, S.; Sanchez-Alavez, M.; Watry, D.; Zandonatti, M.; Henriksen, S.J.; Fox, H.S. Highly activated CD8(+) T cells in the brain correlate with early central nervous system dysfunction in simian immunodeficiency virus infection. J. Immunol. 2001, 167, 5429–5438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leon-Rivera, R.; Morsey, B.; Niu, M.; Fox, H.S.; Berman, J.W. Interactions of Monocytes, HIV, and ART Identified by an Innovative scRNAseq Pipeline: Pathways to Reservoirs and HIV-Associated Comorbidities. mBio 2020, 11. [Google Scholar] [CrossRef]
- Hunt, S.E.; McLaren, W.; Gil, L.; Thormann, A.; Schuilenburg, H.; Sheppard, D.; Parton, A.; Armean, I.M.; Trevanion, S.J.; Flicek, P.; et al. Ensembl variation resources. Database 2018, 2018. [Google Scholar] [CrossRef]
- Smedley, D.; Haider, S.; Durinck, S.; Pandini, L.; Provero, P.; Allen, J.; Arnaiz, O.; Awedh, M.H.; Baldock, R.; Barbiera, G.; et al. The BioMart community portal: An innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015, 43, W589–W598. [Google Scholar] [CrossRef] [Green Version]
- Westmoreland, S.V.; Halpern, E.; Lackner, A.A. Simian immunodeficiency virus encephalitis in rhesus macaques is associated with rapid disease progression. J. Neurovirol. 1998, 4, 260–268. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J.; Sisodia, S.S.; Ransohoff, R.M. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci. 2011, 14, 1227–1235. [Google Scholar] [CrossRef]
- Calef, C.; Mokili, J.; O’Connor, D.H.; Watkins, D.I.; Korber, B. Numbering Positions in SIV Relative to SIVMM239 (Revised*). Available online: https://www.hiv.lanl.gov/content/sequence/HIV/REVIEWS/SIV_NUMBERING2001/SivNumbering.html (accessed on 15 October 2020).
- Roberts, E.S.; Zandonatti, M.A.; Watry, D.D.; Madden, L.J.; Henriksen, S.J.; Taffe, M.A.; Fox, H.S. Induction of pathogenic sets of genes in macrophages and neurons in NeuroAIDS. Am. J. Pathol. 2003, 162, 2041–2057. [Google Scholar] [CrossRef] [Green Version]
- Roberts, E.S.; Masliah, E.; Fox, H.S. CD163 identifies a unique population of ramified microglia in HIV encephalitis (HIVE). J. Neuropathol. Exp. Neurol. 2004, 63, 1255–1264. [Google Scholar] [CrossRef] [Green Version]
- Nowlin, B.T.; Burdo, T.H.; Midkiff, C.C.; Salemi, M.; Alvarez, X.; Williams, K.C. SIV encephalitis lesions are composed of CD163(+) macrophages present in the central nervous system during early SIV infection and SIV-positive macrophages recruited terminally with AIDS. Am. J. Pathol. 2015, 185, 1649–1665. [Google Scholar] [CrossRef]
- Chitu, V.; Gokhan, S.; Nandi, S.; Mehler, M.F.; Stanley, E.R. Emerging Roles for CSF-1 Receptor and its Ligands in the Nervous System. Trends Neurosci. 2016, 39, 378–393. [Google Scholar] [CrossRef] [Green Version]
- Soulas, C.; Conerly, C.; Kim, W.K.; Burdo, T.H.; Alvarez, X.; Lackner, A.A.; Williams, K.C. Recently infiltrating MAC387(+) monocytes/macrophages a third macrophage population involved in SIV and HIV encephalitic lesion formation. Am. J. Pathol. 2011, 178, 2121–2135. [Google Scholar] [CrossRef] [Green Version]
- Chao, C.; Ghorpade, A. Production and Roles of Glial Tissue Inhibitor of Metalloproteinases-1 in Human Immunodeficiency Virus-1-Associated Dementia Neuroinflammation: A Review. Am. J. Infect. Dis. 2009, 5, 314–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdo, T.H.; Ellis, R.J.; Fox, H.S. Osteopontin is increased in HIV-associated dementia. J. Infect. Dis. 2008, 198, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, F.J.; Du, Y.; Greif, E.; Boucher, T.; Dannals, R.F.; Mathews, W.B.; Pomper, M.G.; Sysa-Shah, P.; Metcalf Pate, K.A.; Lyons, C.; et al. Osteopontin/secreted phosphoprotein-1 behaves as a molecular brake regulating the neuroinflammatory response to chronic viral infection. J. Neuroinflamm. 2020, 17, 273. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Pina-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for beta-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharif, O.; Knapp, S. From expression to signaling: Roles of TREM-1 and TREM-2 in innate immunity and bacterial infection. Immunobiology 2008, 213, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Ballarino, M.; Sorrentino, A.; Sthandier, O.; De Angelis, F.G.; Marchioni, M.; Masella, B.; Guarini, A.; Fatica, A.; Peschle, C.; et al. The interplay between the master transcription factor PU.1 and miR-424 regulates human monocyte/macrophage differentiation. Proc. Natl. Acad. Sci. USA 2007, 104, 19849–19854. [Google Scholar] [CrossRef] [Green Version]
- Buttgereit, A.; Lelios, I.; Yu, X.; Vrohlings, M.; Krakoski, N.R.; Gautier, E.L.; Nishinakamura, R.; Becher, B.; Greter, M. Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol. 2016, 17, 1397–1406. [Google Scholar] [CrossRef]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- McPhee, G.M.; Downey, L.A.; Stough, C. Neurotrophins as a reliable biomarker for brain function, structure and cognition: A systematic review and meta-analysis. Neurobiol. Learn. Mem. 2020, 175, 107298. [Google Scholar] [CrossRef] [PubMed]
- Balietti, M.; Giuli, C.; Conti, F. Peripheral Blood Brain-Derived Neurotrophic Factor as a Biomarker of Alzheimer’s Disease: Are There Methodological Biases? Mol. Neurobiol. 2018, 55, 6661–6672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattaneo, A.; Cattane, N.; Begni, V.; Pariante, C.M.; Riva, M.A. The human BDNF gene: Peripheral gene expression and protein levels as biomarkers for psychiatric disorders. Transl. Psychiatry 2016, 6, e958. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.Y.; Feng, J.C.; Cao, C.; Wu, H.T.; Loh, Y.P.; Cheng, Y. Association of Peripheral Blood Levels of Brain-Derived Neurotrophic Factor with Autism Spectrum Disorder in Children: A Systematic Review and Meta-analysis. JAMA Pediatr. 2016, 170, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Erjavec, G.N.; Sagud, M.; Perkovic, M.N.; Strac, D.S.; Konjevod, M.; Tudor, L.; Uzun, S.; Pivac, N. Depression: Biological markers and treatment. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 110139. [Google Scholar] [CrossRef]
- Pan, W.; Banks, W.A.; Fasold, M.B.; Bluth, J.; Kastin, A.J. Transport of brain-derived neurotrophic factor across the blood-brain barrier. Neuropharmacology 1998, 37, 1553–1561. [Google Scholar] [CrossRef]
- Klein, A.B.; Williamson, R.; Santini, M.A.; Clemmensen, C.; Ettrup, A.; Rios, M.; Knudsen, G.M.; Aznar, S. Blood BDNF concentrations reflect brain-tissue BDNF levels across species. Int. J. Neuropsychopharmacol. 2011, 14, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Poo, M.M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef]
- Zagrebelsky, M.; Korte, M. Form follows function: BDNF and its involvement in sculpting the function and structure of synapses. Neuropharmacology 2014, 76, 628–638. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.Y.; Pan, B.S.; Tsai, S.F.; Chiang, Y.T.; Huang, B.M.; Mo, F.E.; Kuo, Y.M. BDNF reverses aging-related microglial activation. J. Neuroinflamm. 2020, 17, 210. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, M.; Deng, D. KLF2 protects BV2 microglial cells against oxygen and glucose deprivation injury by modulating BDNF/TrkB pathway. Gene 2020, 735, 144277. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Gruber, S.H.; El Khoury, A.; Tonali, P.A.; Mathe, A.A. Chronic amphetamine treatment reduces NGF and BDNF in the rat brain. Eur. Neuropsychopharmacol. 2007, 17, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Le Foll, B.; Diaz, J.; Sokoloff, P. A single cocaine exposure increases BDNF and D3 receptor expression: Implications for drug-conditioning. Neuroreport 2005, 16, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Michael, H.; Mpofana, T.; Ramlall, S.; Oosthuizen, F. The Role of Brain Derived Neurotrophic Factor in HIV-Associated Neurocognitive Disorder: From the Bench-Top to the Bedside. Neuropsychiatr. Dis. Treat. 2020, 16, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.G.; Huang, L.; VanderVen, B.C. Immunometabolism at the interface between macrophages and pathogens. Nat. Rev. Immunol. 2019, 19, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.M.; O’Neill, L.A.J. Metabolic regulation of NLRP3. Immunol. Rev. 2018, 281, 88–98. [Google Scholar] [CrossRef]
- Prochnicki, T.; Latz, E. Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control. Cell Metab. 2017, 26, 71–93. [Google Scholar] [CrossRef] [Green Version]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef]
- Fleetwood, A.J.; Lee, M.K.S.; Singleton, W.; Achuthan, A.; Lee, M.C.; O’Brien-Simpson, N.M.; Cook, A.D.; Murphy, A.J.; Dashper, S.G.; Reynolds, E.C.; et al. Metabolic Remodeling, Inflammasome Activation, and Pyroptosis in Macrophages Stimulated by Porphyromonas gingivalis and Its Outer Membrane Vesicles. Front. Cell. Infect. Microbiol. 2017, 7, 351. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457.e13–470.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garaude, J.; Acin-Perez, R.; Martinez-Cano, S.; Enamorado, M.; Ugolini, M.; Nistal-Villan, E.; Hervas-Stubbs, S.; Pelegrin, P.; Sander, L.E.; Enriquez, J.A.; et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol. 2016, 17, 1037–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleeson, L.E.; Sheedy, F.J.; Palsson-McDermott, E.M.; Triglia, D.; O’Leary, S.M.; O’Sullivan, M.P.; O’Neill, L.A.; Keane, J. Cutting Edge: Mycobacterium tuberculosis Induces Aerobic Glycolysis in Human Alveolar Macrophages That Is Required for Control of Intracellular Bacillary Replication. J. Immunol. 2016, 196, 2444–2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekine, Y.; Ouchi, Y.; Sugihara, G.; Takei, N.; Yoshikawa, E.; Nakamura, K.; Iwata, Y.; Tsuchiya, K.J.; Suda, S.; Suzuki, K.; et al. Methamphetamine causes microglial activation in the brains of human abusers. J. Neurosci. 2008, 28, 5756–5761. [Google Scholar] [CrossRef]
- Thomas, D.M.; Francescutti-Verbeem, D.M.; Kuhn, D.M. The newly synthesized pool of dopamine determines the severity of methamphetamine-induced neurotoxicity. J. Neurochem. 2008, 105, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.M.; Walker, P.D.; Benjamins, J.A.; Geddes, T.J.; Kuhn, D.M. Methamphetamine neurotoxicity in dopamine nerve endings of the striatum is associated with microglial activation. J. Pharmacol. Exp. Ther. 2004, 311, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Friend, D.M.; Keefe, K.A. A role for D1 dopamine receptors in striatal methamphetamine-induced neurotoxicity. Neurosci. Lett. 2013, 555, 243–247. [Google Scholar] [CrossRef] [Green Version]
- Ares-Santos, S.; Granado, N.; Oliva, I.; O’Shea, E.; Martin, E.D.; Colado, M.I.; Moratalla, R. Dopamine D(1) receptor deletion strongly reduces neurotoxic effects of methamphetamine. Neurobiol. Dis. 2012, 45, 810–820. [Google Scholar] [CrossRef]
- Cadet, J.L.; Brannock, C. Free radicals and the pathobiology of brain dopamine systems. Neurochem. Int. 1998, 32, 117–131. [Google Scholar] [CrossRef]
- Cadet, J.L.; Krasnova, I.N. Molecular bases of methamphetamine-induced neurodegeneration. Int. Rev. Neurobiol. 2009, 88, 101–119. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal | Age (Yrs) | Condition | Days p.i. | Terminal Plasma Viral Load (log10/mL) | Tissue Viral Load (log10/µg RNA) | |||
|---|---|---|---|---|---|---|---|---|
| Caudate | Frontal Cortex | Hippo-Campus | Spleen | |||||
| 17T | 5.1 | SIVE | 65 | 8.8 | 6.3 | 7.2 | 7.2 | 8.2 |
| 34T | 2.7 | SIVE | 92 | 7.8 | 7.8 | 8.1 | 7.7 | 7.8 |
| 21T | 5.4 | Meth-SIVE | 92 | 8.5 | 7.5 | 7.3 | 6.6 | 7.5 |
| 32T | 4.0 | Meth-SIVE | 78 | 7.7 | 7.3 | 7.7 | 7.6 | 8.0 |
| Region | Bases | SIVE | Meth-SIVE | Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 | Cluster 5 |
|---|---|---|---|---|---|---|---|---|
| SIV_A | 309 | 3.34% | 4.22% | 2.16% | 7.01% | 1.85% | 3.80% | 5.18% |
| SIV_B | 4225 | 4.20% | 5.85% | 2.92% | 6.95% | 3.15% | 7.44% | 5.93% |
| SIV_C | 1387 | 1.59% | 1.97% | 0.96% | 4.07% | 0.77% | 1.19% | 1.98% |
| SIV_D | 2208 | 1.81% | 2.23% | 1.12% | 4.17% | 0.86% | 1.61% | 3.07% |
| SIV_E | 1332 | 6.21% | 7.58% | 3.70% | 9.13% | 4.61% | 10.39% | 9.00% |
| SIV_All | 8.48% | 11.33% | 5.62% | 11.58% | 7.31% | 16.36% | 10.91% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, M.; Morsey, B.; Lamberty, B.G.; Emanuel, K.; Yu, F.; León-Rivera, R.; Berman, J.W.; Gaskill, P.J.; Matt, S.M.; Ciborowski, P.S.; et al. Methamphetamine Increases the Proportion of SIV-Infected Microglia/Macrophages, Alters Metabolic Pathways, and Elevates Cell Death Pathways: A Single-Cell Analysis. Viruses 2020, 12, 1297. https://doi.org/10.3390/v12111297
Niu M, Morsey B, Lamberty BG, Emanuel K, Yu F, León-Rivera R, Berman JW, Gaskill PJ, Matt SM, Ciborowski PS, et al. Methamphetamine Increases the Proportion of SIV-Infected Microglia/Macrophages, Alters Metabolic Pathways, and Elevates Cell Death Pathways: A Single-Cell Analysis. Viruses. 2020; 12(11):1297. https://doi.org/10.3390/v12111297
Chicago/Turabian StyleNiu, Meng, Brenda Morsey, Benjamin G. Lamberty, Katy Emanuel, Fang Yu, Rosiris León-Rivera, Joan W. Berman, Peter J. Gaskill, Stephanie M. Matt, Pawel S. Ciborowski, and et al. 2020. "Methamphetamine Increases the Proportion of SIV-Infected Microglia/Macrophages, Alters Metabolic Pathways, and Elevates Cell Death Pathways: A Single-Cell Analysis" Viruses 12, no. 11: 1297. https://doi.org/10.3390/v12111297