Next-Generation Sequencing Analysis of Cellular Response to Influenza B Virus Infection



Abstract
1. Introduction
2. Materials and Methods
2.1. Virus and Cell Culture
2.2. IBV Infection of A549 Cells and Next-Generation Sequencing
2.3. Transcriptome Read Processing
2.4. Differential Gene Expression Analysis
2.5. Gene Ontology and Pathway Enrichment Analysis
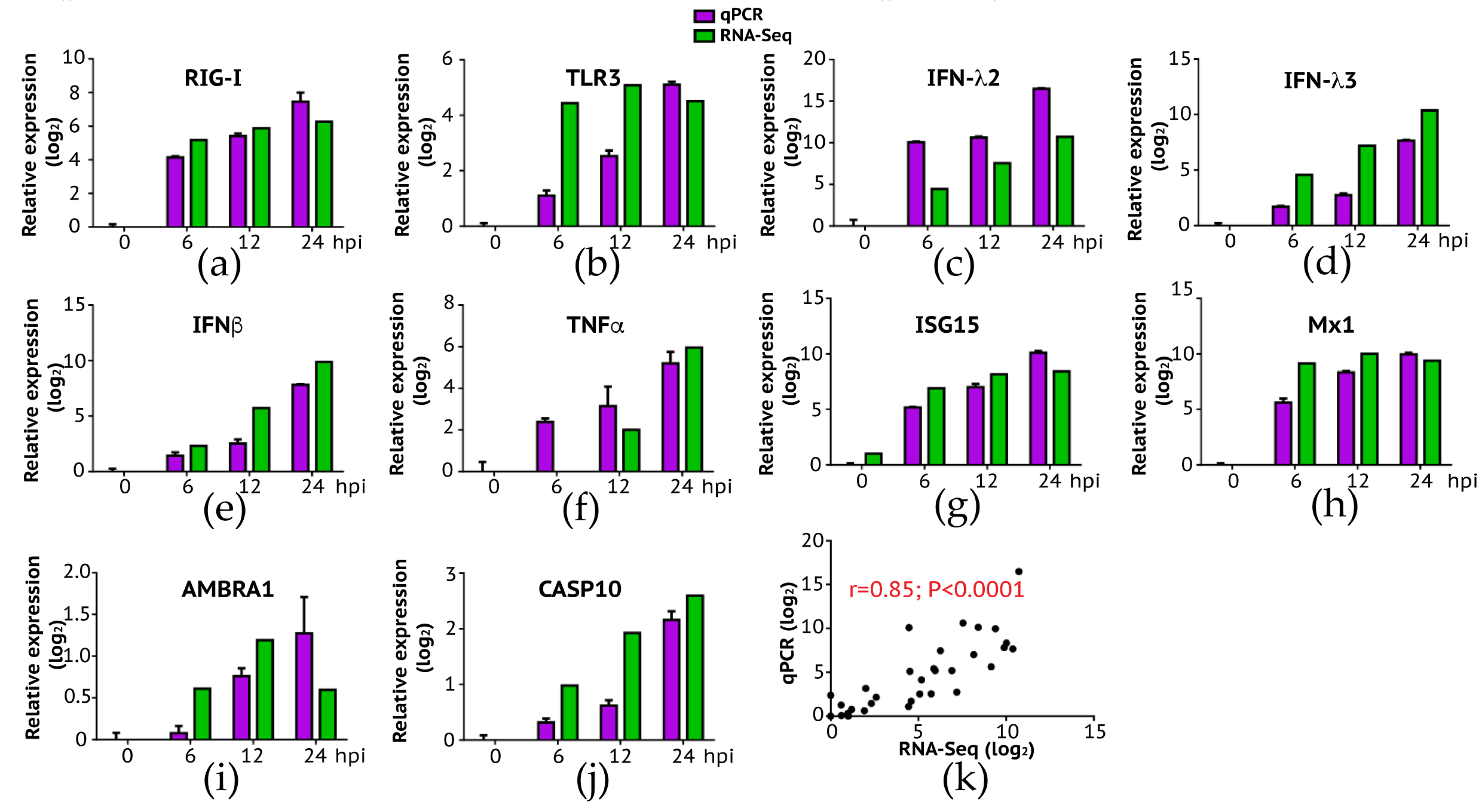
2.6. Validation of RNA-Seq Results by RT-qPCR
3. Results
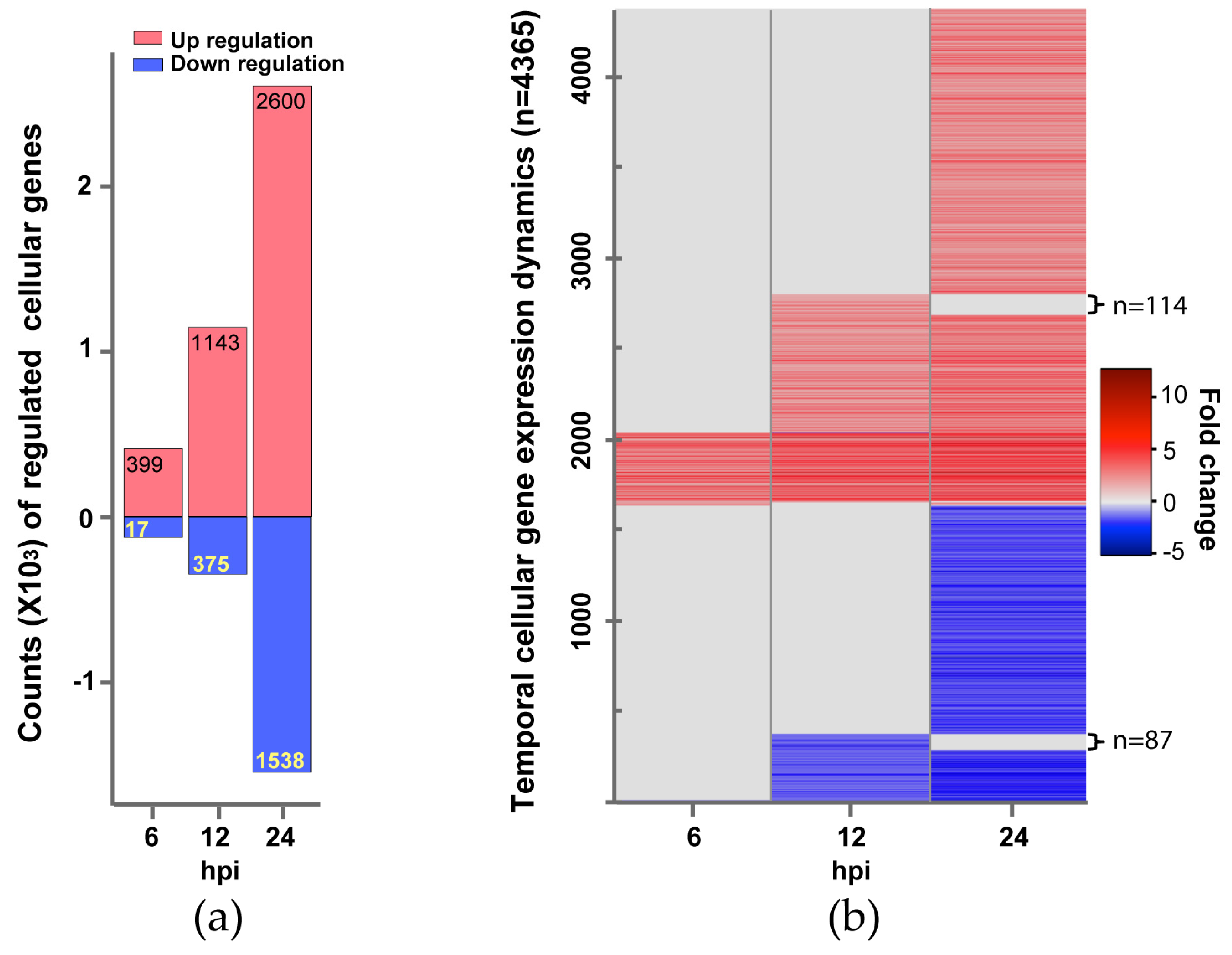
3.1. Next-Generation Sequencing of IBV Infected A549 Cells
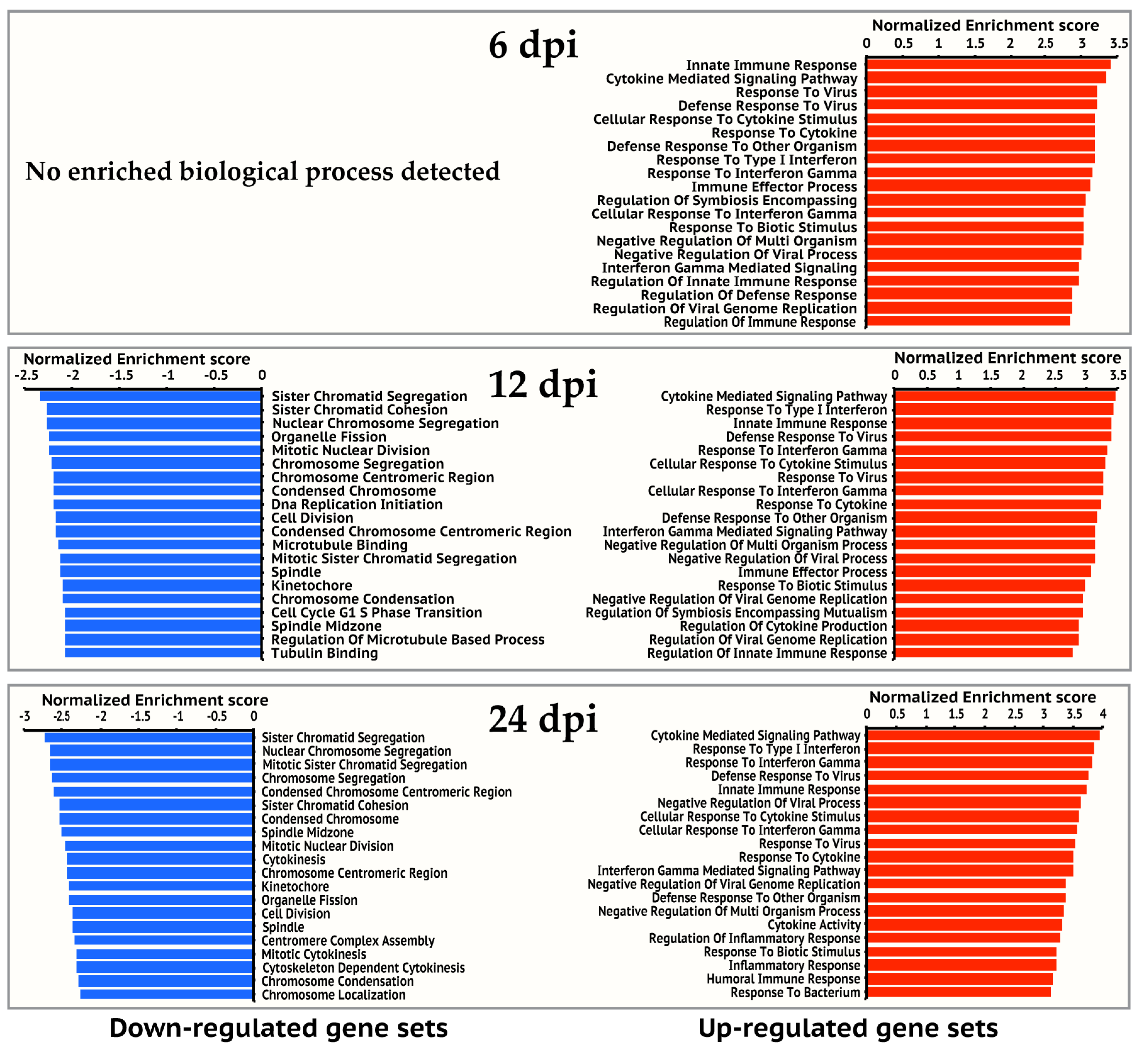
3.2. Cellular Processes and Pathways Regulated by IBV Infection
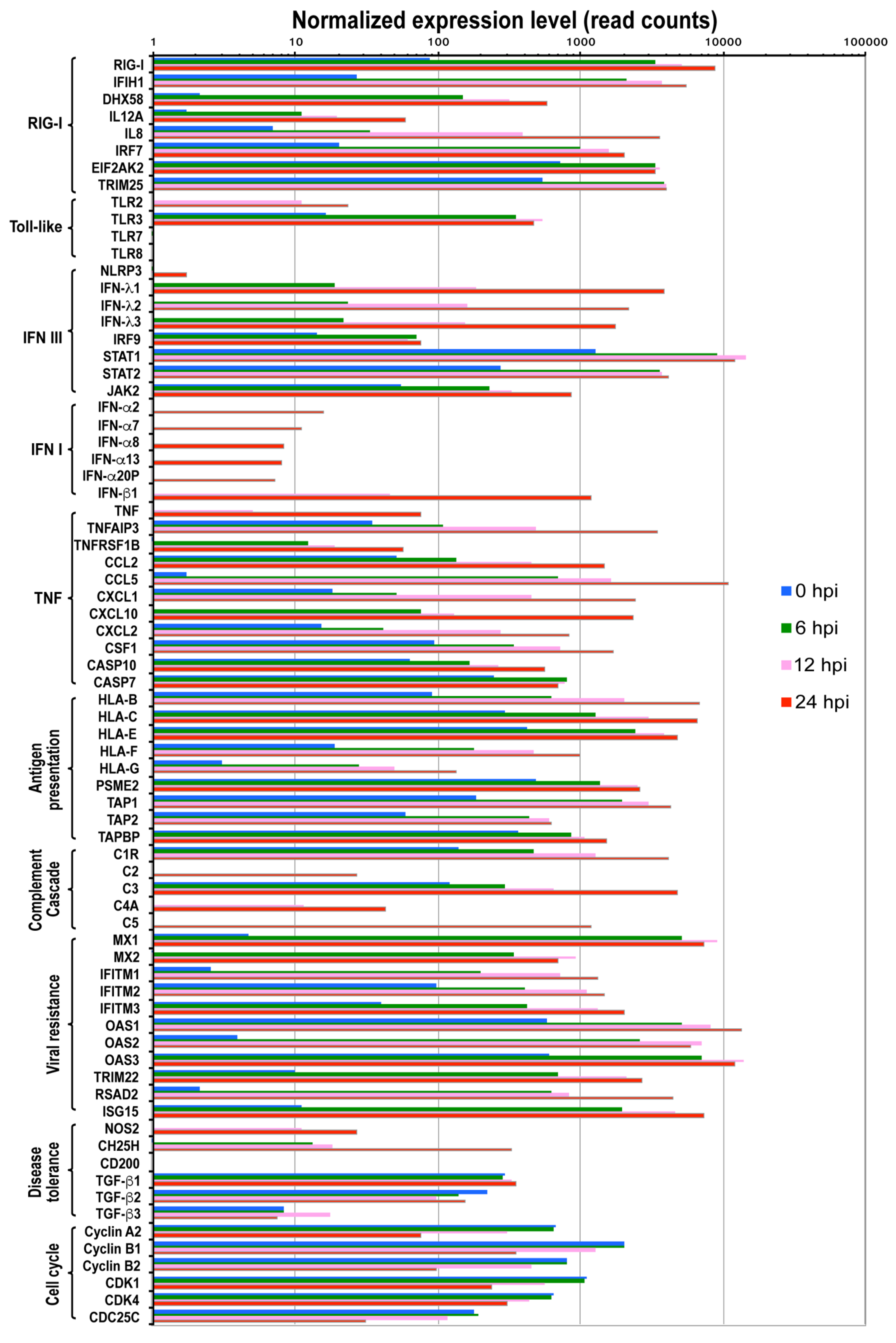
3.3. Sensing IBV Infection
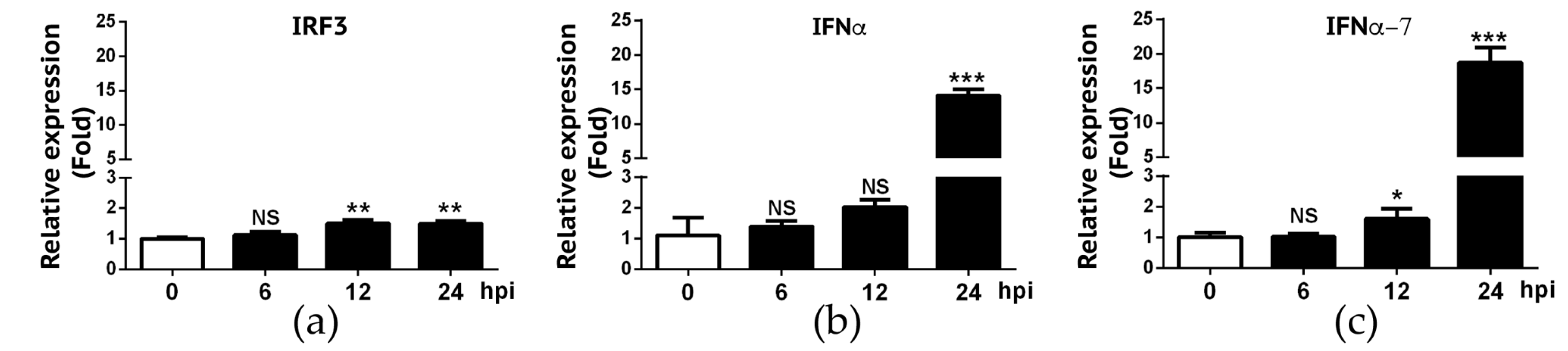
3.4. Interferon Response
3.5. Cytokine and Chemokine Cascades
3.6. Complement Cascade
3.7. Antigen Processing and Presentation
3.8. Host Resistance and Tolerance to IBV Infection
3.9. Metabolic Processes Were Up Regulated in IBV-infected Cells at 12 and 24 hpi
3.10. Cell Proliferation Pathway Was Repressed at 12 and 24 hpi
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Palese, P.; Shaw, M. Orthomyxoviridae: The viruses and their replication. In Field’s Virology, 5th ed.; Fields, B.N., Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 2, pp. 1647–1689. [Google Scholar]
- Hause, B.M.; Collin, E.A.; Liu, R.; Huang, B.; Sheng, Z.; Lu, W.; Wang, D.; Nelson, E.A.; Li, F. Characterization of a novel influenza virus in cattle and Swine: Proposal for a new genus in the Orthomyxoviridae family. MBio 2014, 5, e00031-14. [Google Scholar] [CrossRef]
- Hause, B.M.; Ducatez, M.; Collin, E.A.; Ran, Z.; Liu, R.; Sheng, Z.; Armien, A.; Kaplan, B.; Chakravarty, S.; Hoppe, A.D.; et al. Isolation of a novel swine influenza virus from Oklahoma in 2011 which is distantly related to human influenza C viruses. PLoS Pathog. 2013, 9, e1003176. [Google Scholar] [CrossRef] [PubMed]
- Cox, N.J.; Subbarao, K. Global epidemiology of influenza: Past and present. Annu. Rev. Med. 2000, 51, 407–421. [Google Scholar] [CrossRef] [PubMed]
- McCullers, J.A.; Facchini, S.; Chesney, P.J.; Webster, R.G. Influenza B virus encephalitis. Clin. Infect. Dis 1999, 28, 898–900. [Google Scholar] [CrossRef] [PubMed]
- McCullers, J.A.; Saito, T.; Iverson, A.R. Multiple genotypes of influenza B virus circulated between 1979 and 2003. J. Virol. 2004, 78, 12817–12828. [Google Scholar] [CrossRef]
- Monto, A.S. Epidemiology of influenza. Vaccine 2006, 26 (Suppl. 4), D45–D48. [Google Scholar] [CrossRef] [PubMed]
- Osterhaus, A.D.; Rimmelzwaan, G.F.; Martina, B.E.; Bestebroer, T.M.; Fouchier, R.A. Influenza B virus in seals. Science 2000, 288, 1051–1053. [Google Scholar] [CrossRef] [PubMed]
- Ran, Z.; Shen, H.; Lang, Y.; Kolb, E.A.; Turan, N.; Zhu, L.; Ma, J.; Bawa, B.; Liu, Q.; Liu, H.; et al. Domestic pigs are susceptible to infection with influenza B viruses. J. Virol. 2015, 89, 4818–4826. [Google Scholar] [CrossRef] [PubMed]
- Paul Glezen, W.; Schmier, J.K.; Kuehn, C.M.; Ryan, K.J.; Oxford, J. The burden of influenza B: A structured literature review. Am. J. Public Health 2013, 103, e43–e51. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Pizarraya, A.; Perez-Romero, P.; Alvarez, R.; Aydillo, T.A.; Osorio-Gomez, G.; Milara-Ibanez, C.; Sanchez, M.; Pachon, J.; Cordero, E. Unexpected severity of cases of influenza B infection in patients that required hospitalization during the first postpandemic wave. J. Infect. 2012, 65, 423–430. [Google Scholar] [CrossRef]
- Wu, P.; Goldstein, E.; Ho, L.M.; Yang, L.; Nishiura, H.; Wu, J.T.; Ip, D.K.; Chuang, S.K.; Tsang, T.; Cowling, B.J. Excess mortality associated with influenza A and B virus in Hong Kong, 1998–2009. J. Infect. Dis 2012, 206, 1862–1871. [Google Scholar] [CrossRef]
- Su, S.; Chaves, S.S.; Perez, A.; D’Mello, T.; Kirley, P.D.; Yousey-Hindes, K.; Farley, M.M.; Harris, M.; Sharangpani, R.; Lynfield, R.; et al. Comparing Clinical Characteristics Between Hospitalized Adults With Laboratory-Confirmed Influenza A and B Virus Infection. Clin. Infect. Dis. 2014, 59, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, N.; Higashi, N.; Nakagawa, T. Cocirculation of antigenic variants and the vaccine-type virus during the 2004–2005 influenza B virus epidemics in Japan. J. Clin. Microbiol. 2009, 47, 352–357. [Google Scholar] [CrossRef][Green Version]
- Nerome, R.; Hiromoto, Y.; Sugita, S.; Tanabe, N.; Ishida, M.; Matsumoto, M.; Lindstrom, S.E.; Takahashi, T.; Nerome, K. Evolutionary characteristics of influenza B virus since its first isolation in 1940: Dynamic circulation of deletion and insertion mechanism. Arch. Virol. 1998, 143, 1569–1583. [Google Scholar] [CrossRef]
- Shen, J.; Kirk, B.D.; Ma, J.; Wang, Q. Diversifying selective pressure on influenza B virus hemagglutinin. J. Med. Virol. 2009, 81, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Chiu, S.C.; Shaw, M.W.; Lin, Y.C.; Lee, C.H.; Chen, H.Y.; Klimov, A. Characterization of the epidemic influenza B viruses isolated during 2004–2005 season in Taiwan. Virus Res. 2007, 124, 204–211. [Google Scholar] [CrossRef]
- McCullers, J.A.; Wang, G.C.; He, S.; Webster, R.G. Reassortment and insertion-deletion are strategies for the evolution of influenza B viruses in nature. J. Virol. 1999, 73, 7343–7348. [Google Scholar] [CrossRef] [PubMed]
- Glezen, W.P. Editorial commentary: Changing epidemiology of influenza B virus. Clin. Infect. Dis 2014, 59, 1525–1526. [Google Scholar] [CrossRef]
- Vijaykrishna, D.; Holmes, E.C.; Joseph, U.; Fourment, M.; Su, Y.C.; Halpin, R.; Lee, R.T.; Deng, Y.M.; Gunalan, V.; Lin, X.; et al. The contrasting phylodynamics of human influenza B viruses. eLife 2015, 4, e05055. [Google Scholar] [CrossRef]
- McCullers, J.A.; Hayden, F.G. Fatal influenza B infections: Time to reexamine influenza research priorities. J. Infect. Dis. 2012, 205, 870–872. [Google Scholar] [CrossRef]
- Paddock, C.D.; Liu, L.; Denison, A.M.; Bartlett, J.H.; Holman, R.C.; Deleon-Carnes, M.; Emery, S.L.; Drew, C.P.; Shieh, W.J.; Uyeki, T.M.; et al. Myocardial injury and bacterial pneumonia contribute to the pathogenesis of fatal influenza B virus infection. J. Infect. Dis. 2012, 205, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A Simple Method of Estimating Fifty Per Cent Endpoints12. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Wu, T.D.; Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010, 26, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆Ct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Sheng, Z.; Liu, R.; Yu, J.; Ran, Z.; Newkirk, S.J.; An, W.; Li, F.; Wang, D. Identification and characterization of viral defective RNA genomes in influenza B virus. J. Gen. Virol. 2018, 99, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Tissari, J.; Siren, J.; Meri, S.; Julkunen, I.; Matikainen, S. IFN-alpha enhances TLR3-mediated antiviral cytokine expression in human endothelial and epithelial cells by up-regulating TLR3 expression. J. Immunol. 2005, 174, 4289–4294. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, M.; Suryawanshi, A.; Tundup, S.; Perez, J.T.; Schmolke, M.; Manicassamy, S.; Manicassamy, B. RIG-I Signaling Is Critical for Efficient Polyfunctional T Cell Responses during Influenza Virus Infection. PLoS Pathog. 2016, 12, e1005754. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Verhelst, J.; Spitaels, J.; Nurnberger, C.; De Vlieger, D.; Ysenbaert, T.; Staeheli, P.; Fiers, W.; Saelens, X. Functional Comparison of Mx1 from Two Different Mouse Species Reveals the Involvement of Loop L4 in the Antiviral Activity against Influenza A Viruses. J. Virol. 2015, 89, 10879–10890. [Google Scholar] [CrossRef]
- Verhelst, J.; Parthoens, E.; Schepens, B.; Fiers, W.; Saelens, X. Interferon-inducible protein Mx1 inhibits influenza virus by interfering with functional viral ribonucleoprotein complex assembly. J. Virol. 2012, 86, 13445–13455. [Google Scholar] [CrossRef]
- Kroncke, K.D.; Fehsel, K.; Kolb-Bachofen, V. Inducible nitric oxide synthase in human diseases. Clin. Exp. Immunol. 1998, 113, 147–156. [Google Scholar] [CrossRef]
- Bosworth, A.; Dowall, S.D.; Garcia-Dorival, I.; Rickett, N.Y.; Bruce, C.B.; Matthews, D.A.; Fang, Y.; Aljabr, W.; Kenny, J.; Nelson, C.; et al. A comparison of host gene expression signatures associated with infection in vitro by the Makona and Ecran (Mayinga) variants of Ebola virus. Sci. Rep. 2017, 7, 43144. [Google Scholar] [CrossRef]
- Ramos, I.; Smith, G.; Ruf-Zamojski, F.; Martinez-Romero, C.; Fribourg, M.; Carbajal, E.A.; Hartmann, B.M.; Nair, V.D.; Marjanovic, N.; Monteagudo, P.L.; et al. Innate Immune Response to Influenza Virus at Single-Cell Resolution in Human Epithelial Cells Revealed Paracrine Induction of Interferon Lambda 1. J. Virol. 2019, 93, e00559-19. [Google Scholar] [CrossRef]
- Crotta, S.; Davidson, S.; Mahlakoiv, T.; Desmet, C.J.; Buckwalter, M.R.; Albert, M.L.; Staeheli, P.; Wack, A. Type I and Type III Interferons Drive Redundant Amplification Loops to Induce a Transcriptional Signature in Influenza-Infected Airway Epithelia. PLoS Pathog. 2013, 9, e1003773. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.B.; Elshina, E.; Kowalsky, J.R.; te Velthuis, A.J.W.; Bloom, J.D. Single-Cell Virus Sequencing of Influenza Infections That Trigger Innate Immunity. J. Virol. 2019, 93, e00500-19. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhang, C.; Jia, X.; Wang, S.; Wang, J.; Chen, Y.; Zhao, J.; Tian, S.; Han, X.; Han, L. Transcriptome Profiles of Human Lung Epithelial Cells A549 Interacting with Aspergillus fumigatus by RNA-Seq. PLoS ONE 2015, 10, e0135720. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Qiu, X.; Song, C.; Sun, Y.; Meng, C.; Liao, Y.; Tan, L.; Ding, Z.; Liu, X.; Ding, C. Deep Sequencing-Based Transcriptome Profiling Reveals Avian Interferon-Stimulated Genes and Provides Comprehensive Insight into Newcastle Disease Virus-Induced Host Responses. Viruses 2018, 10, 162. [Google Scholar] [CrossRef]
- Makela, S.M.; Osterlund, P.; Westenius, V.; Latvala, S.; Diamond, M.S.; Gale, M., Jr.; Julkunen, I. RIG-I Signaling Is Essential for Influenza B Virus-Induced Rapid Interferon Gene Expression. J. Virol. 2015, 89, 12014–12025. [Google Scholar] [CrossRef]
- Osterlund, P.; Strengell, M.; Sarin, L.P.; Poranen, M.M.; Fagerlund, R.; Melen, K.; Julkunen, I. Incoming influenza A virus evades early host recognition, while influenza B virus induces interferon expression directly upon entry. J. Virol. 2012, 86, 11183–11193. [Google Scholar] [CrossRef]
- Schoenborn, J.R.; Wilson, C.B. Regulation of Interferon-γ During Innate and Adaptive Immune Responses. In Advances in Immunology; Academic Press: Cambridge, MA, USA, 2007; Volume 96, pp. 41–101. [Google Scholar]
- Bagga, S.; Bouchard, M.J. Cell cycle regulation during viral infection. Methods Mol. Biol. 2014, 1170, 165–227. [Google Scholar]
- Li, L.; Gu, B.; Zhou, F.; Chi, J.; Feng, D.; Xie, F.; Wang, F.; Ma, C.; Li, M.; Wang, J.; et al. Cell cycle perturbations induced by human herpesvirus 6 infection and their effect on virus replication. Arch. Virol. 2014, 159, 365–370. [Google Scholar] [CrossRef]
- Dove, B.; Brooks, G.; Bicknell, K.; Wurm, T.; Hiscox, J.A. Cell cycle perturbations induced by infection with the coronavirus infectious bronchitis virus and their effect on virus replication. J. Virol. 2006, 80, 4147–4156. [Google Scholar] [CrossRef]
- Jiang, W.; Wang, Q.; Chen, S.; Gao, S.; Song, L.; Liu, P.; Huang, W. Influenza A virus NS1 induces G0/G1 cell cycle arrest by inhibiting the expression and activity of RhoA protein. J. Virol. 2013, 87, 3039–3052. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KEGG ID | Pathway Description | Num. of Up-Regulated Genes | False Discovery Rate |
|---|---|---|---|
| 4060 | Cytokine–cytokine receptor interaction | 81 | 2.72 × 10−20 |
| 4062 | Chemokine signaling pathway | 33 | 0.00116 |
| 4064 | NF-kappa B signaling pathway | 36 | 3.12 × 10−13 |
| 4145 | Phagosome | 23 | 0.0367 |
| 4210 | Apoptosis | 23 | 2.37 × 10−05 |
| 4610 | Complement and coagulation cascades | 23 | 4.29 × 10−07 |
| 4612 | Antigen processing and presentation | 20 | 1.98 × 10−05 |
| 4620 | Toll-like receptor signaling pathway | 37 | 4.58 × 10−12 |
| 4621 | NOD-like receptor signaling pathway | 23 | 8.86 × 10−09 |
| 4622 | RIG-I-like receptor signaling pathway | 29 | 3.00 × 10−11 |
| 4623 | Cytosolic DNA-sensing pathway | 23 | 5.02 × 10−08 |
| 4630 | Jak-STAT signaling pathway | 49 | 5.03 × 10−13 |
| 4650 | Natural killer cell mediated cytotoxicity | 28 | 0.0001 |
| 4668 | TNF signaling pathway | 47 | 6.41 × 10−19 |
| 4917 | Prolactin signaling pathway | 16 | 0.00386 |
| 4940 | Type I diabetes mellitus | 12 | 0.0017 |
| 5133 | Pertussis | 30 | 4.58 × 10−12 |
| 5134 | Legionellosis | 23 | 2.73 × 10−09 |
| 5142 | Chagas disease (American trypanosomiasis) | 30 | 4.32 × 10−08 |
| 5143 | African trypanosomiasis | 11 | 0.00117 |
| 5144 | Malaria | 12 | 0.00552 |
| 5145 | Toxoplasmosis | 29 | 4.72 × 10−06 |
| 5150 | Staphylococcus aureus infection | 16 | 0.000115 |
| 5160 | Hepatitis C | 36 | 3.12 × 10−08 |
| 5161 | Hepatitis B | 37 | 8.17 × 10−08 |
| 5162 | Measles | 44 | 5.90 × 10−13 |
| 5164 | Influenza A | 58 | 1.79 × 10−17 |
| 5166 | HTLV-I infection | 43 | 0.000802 |
| 5168 | Herpes simplex infection | 62 | 6.41 × 10−19 |
| 5169 | Epstein–Barr virus infection | 33 | 0.00296 |
| 5203 | Viral carcinogenesis | 45 | 2.64 × 10−08 |
| 5320 | Autoimmune thyroid disease | 13 | 0.0032 |
| 5323 | Rheumatoid arthritis | 25 | 2.28 × 10−06 |
| 5330 | Allograft rejection | 10 | 0.00542 |
| 5332 | Graft-versus-host disease | 11 | 0.00245 |
| 5416 | Viral myocarditis | 13 | 0.00893 |
| Time Point | KEGG ID | Pathway Description | Num. of Down-Regulated Genes | False Discovery Rate |
|---|---|---|---|---|
| 12 hpi | 4110 | Cell cycle | 11 | 0.00193 |
| 24 hpi | 3030 | DNA replication | 19 | 3.00 × 10−11 |
| 24 hpi | 1100 | Metabolic pathways | 134 | 2.40 × 10−07 |
| 24 hpi | 3460 | Fanconi anemia pathway | 17 | 2.40 × 10−06 |
| 24 hpi | 4110 | Cell cycle | 26 | 1.17 × 10−05 |
| 24 hpi | 3430 | Mismatch repair | 9 | 0.000658 |
| 24 hpi | 4114 | Oocyte meiosis | 20 | 0.00135 |
| 24 hpi | 260 | Glycine, serine and threonine metabolism | 11 | 0.00181 |
| 24 hpi | 3440 | Homologous recombination | 9 | 0.00183 |
| 24 hpi | 4540 | Gap junction | 16 | 0.00477 |
| 24 hpi | 5032 | Morphine addiction | 16 | 0.00758 |
| 24 hpi | 230 | Purine metabolism | 24 | 0.00878 |
| 24 hpi | 3420 | Nucleotide excision repair | 10 | 0.0213 |
| 24 hpi | 4961 | Endocrine and other factor-regulated calcium reabsorption | 10 | 0.0213 |
| 24 hpi | 630 | Glyoxylate and dicarboxylate metabolism | 7 | 0.0237 |
| 24 hpi | 3410 | Base excision repair | 8 | 0.0279 |
| 24 hpi | 280 | Valine, leucine and isoleucine degradation | 9 | 0.0444 |
| 24 hpi | 1230 | Biosynthesis of amino acids | 12 | 0.0444 |
| 24 hpi | 4914 | Progesterone-mediated oocyte maturation | 13 | 0.0444 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheng, Z.; Huang, C.; Liu, R.; Guo, Y.; Ran, Z.; Li, F.; Wang, D. Next-Generation Sequencing Analysis of Cellular Response to Influenza B Virus Infection. Viruses 2020, 12, 383. https://doi.org/10.3390/v12040383
Sheng Z, Huang C, Liu R, Guo Y, Ran Z, Li F, Wang D. Next-Generation Sequencing Analysis of Cellular Response to Influenza B Virus Infection. Viruses. 2020; 12(4):383. https://doi.org/10.3390/v12040383
Chicago/Turabian StyleSheng, Zizhang, Chen Huang, Runxia Liu, Yicheng Guo, Zhiguang Ran, Feng Li, and Dan Wang. 2020. "Next-Generation Sequencing Analysis of Cellular Response to Influenza B Virus Infection" Viruses 12, no. 4: 383. https://doi.org/10.3390/v12040383
APA StyleSheng, Z., Huang, C., Liu, R., Guo, Y., Ran, Z., Li, F., & Wang, D. (2020). Next-Generation Sequencing Analysis of Cellular Response to Influenza B Virus Infection. Viruses, 12(4), 383. https://doi.org/10.3390/v12040383