Characterization of Potato Virus Y Isolates and Assessment of Nanopore Sequencing to Detect and Genotype Potato Viruses


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. PVY Serological Tests
2.3. Nucleic Acids Extraction
2.4. RT-PCR
2.5. Illumina and ONT Sequencing
2.5.1. Illumina
2.5.2. Oxford Nanopore Technologies (ONT)
2.6. Analysis of Sequencing Reads
2.6.1. Illumina Datasets
2.6.2. ONT Datasets
2.6.3. Accuracy of ONT Sequencing
2.7. PVY Recombination and Phylogenetic Analyses
3. Results
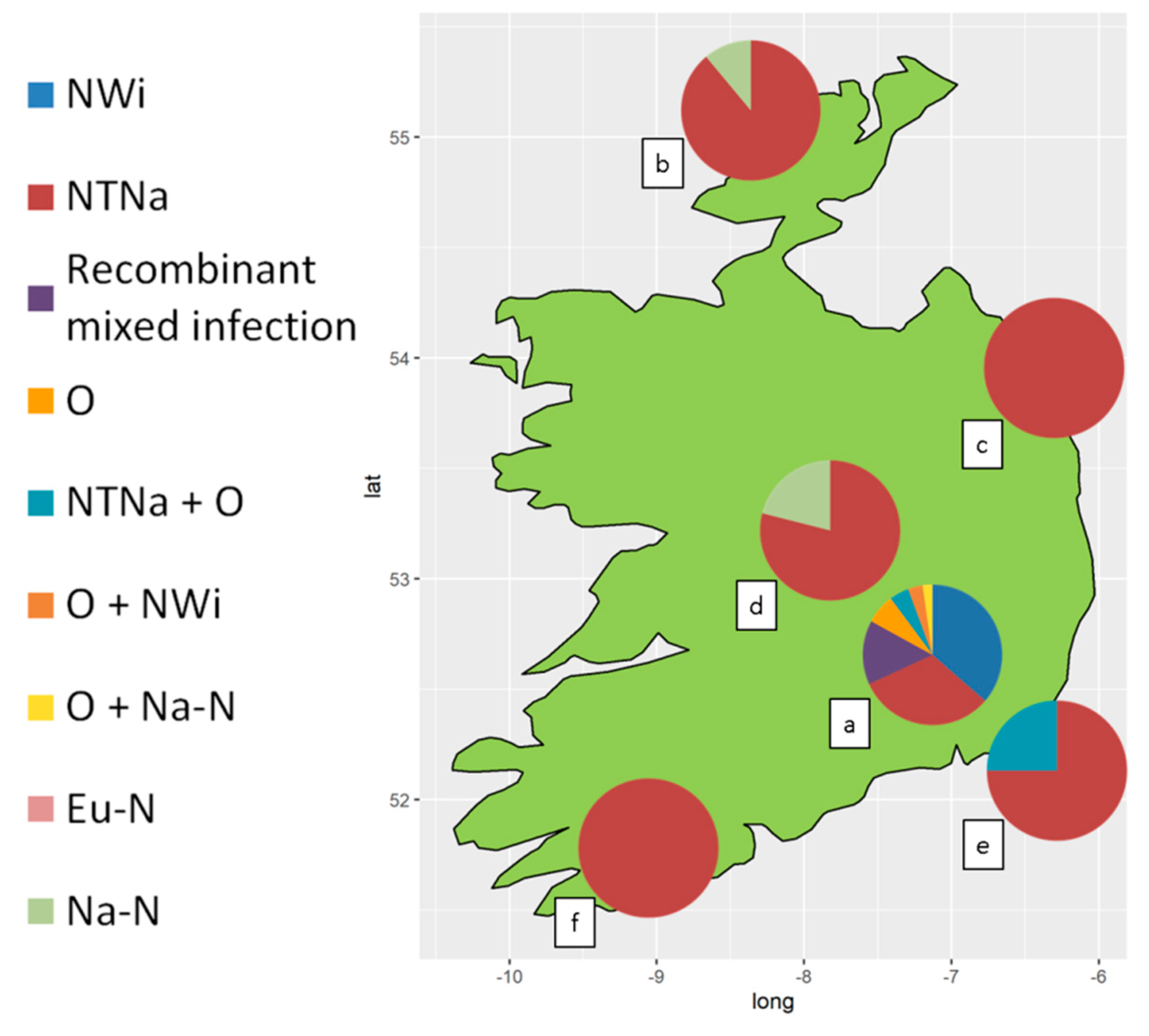
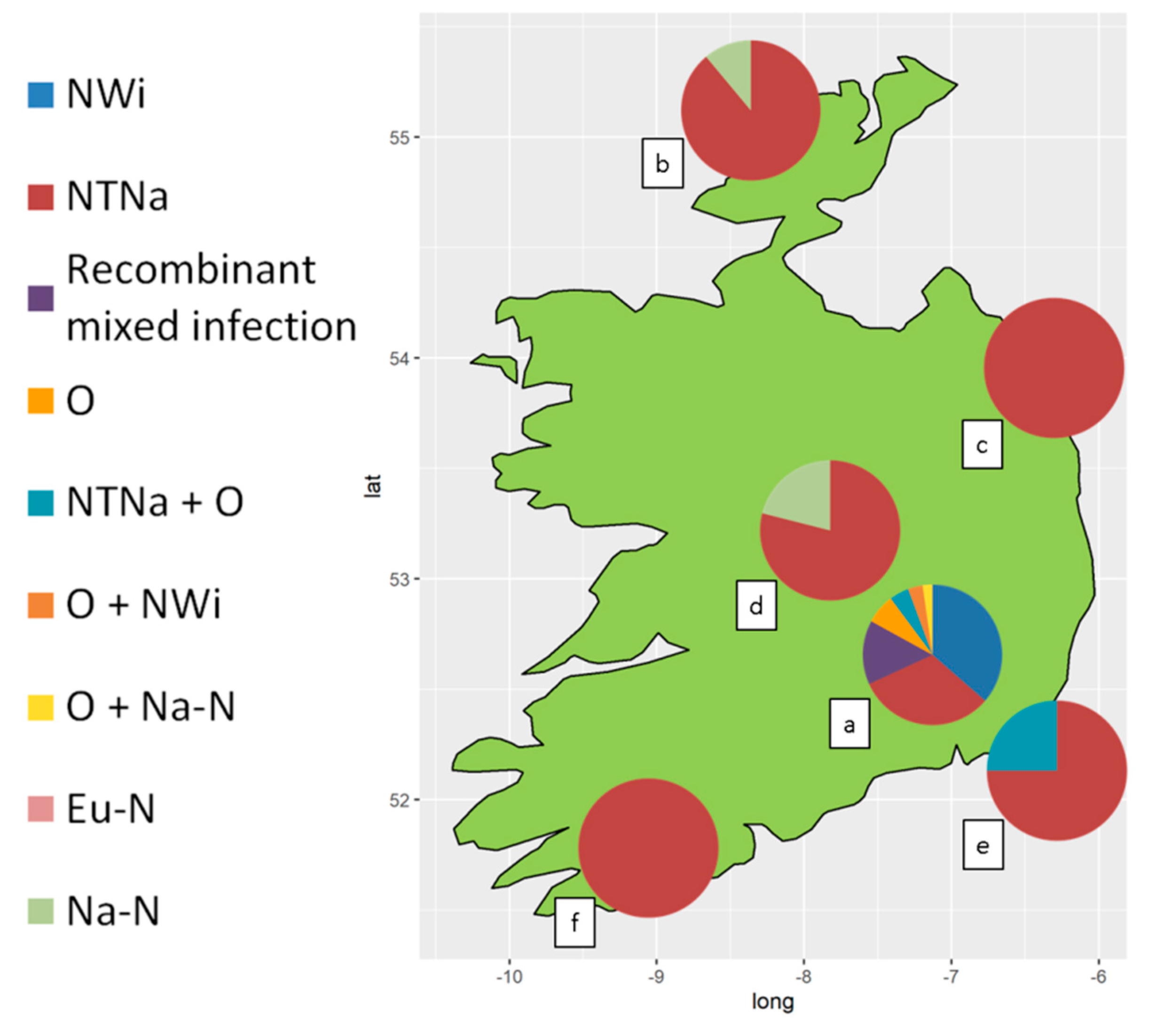
3.1. PVY Serological Detection and Characterization
3.2. RT-PCR Assays for Virus Detection and PVY Strain-Typing
3.3. Illumina Sequencing
3.4. ONT Sequencing
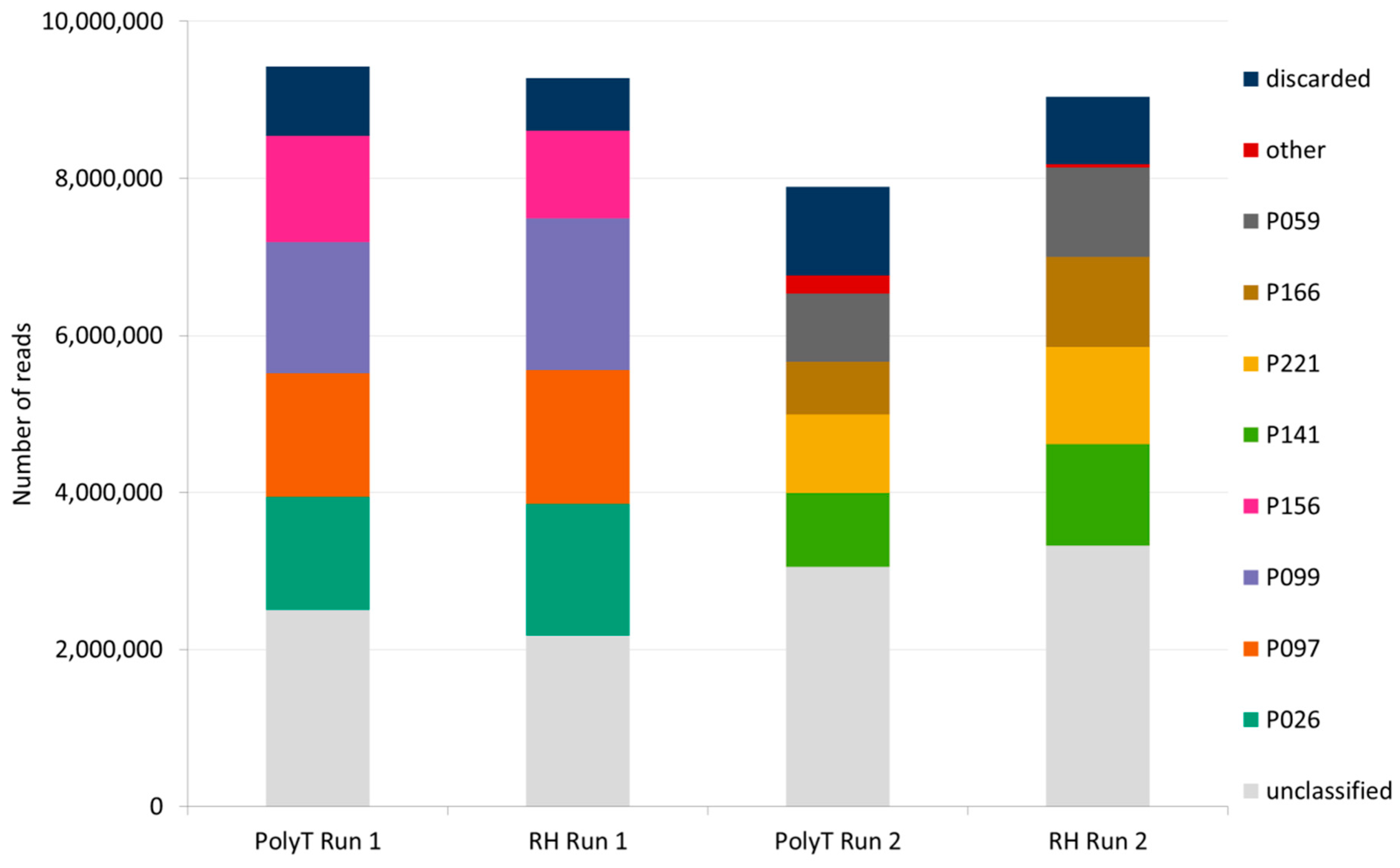
3.4.1. Nanopore Sequencing Throughput
3.4.2. De Novo Assembly and Virus Detection
3.4.3. Map to Reference
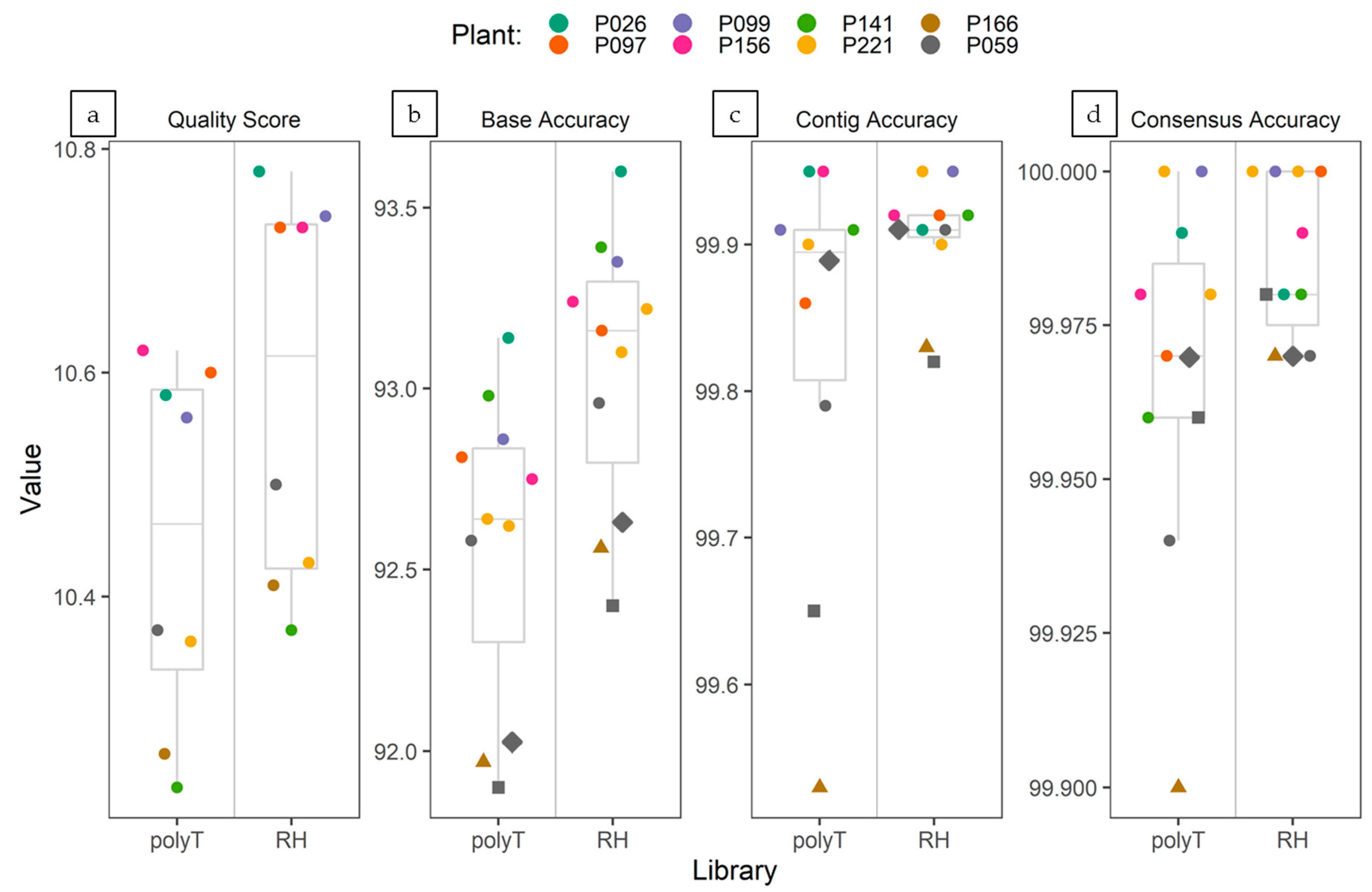
3.4.4. Accuracy of ONT Sequencing
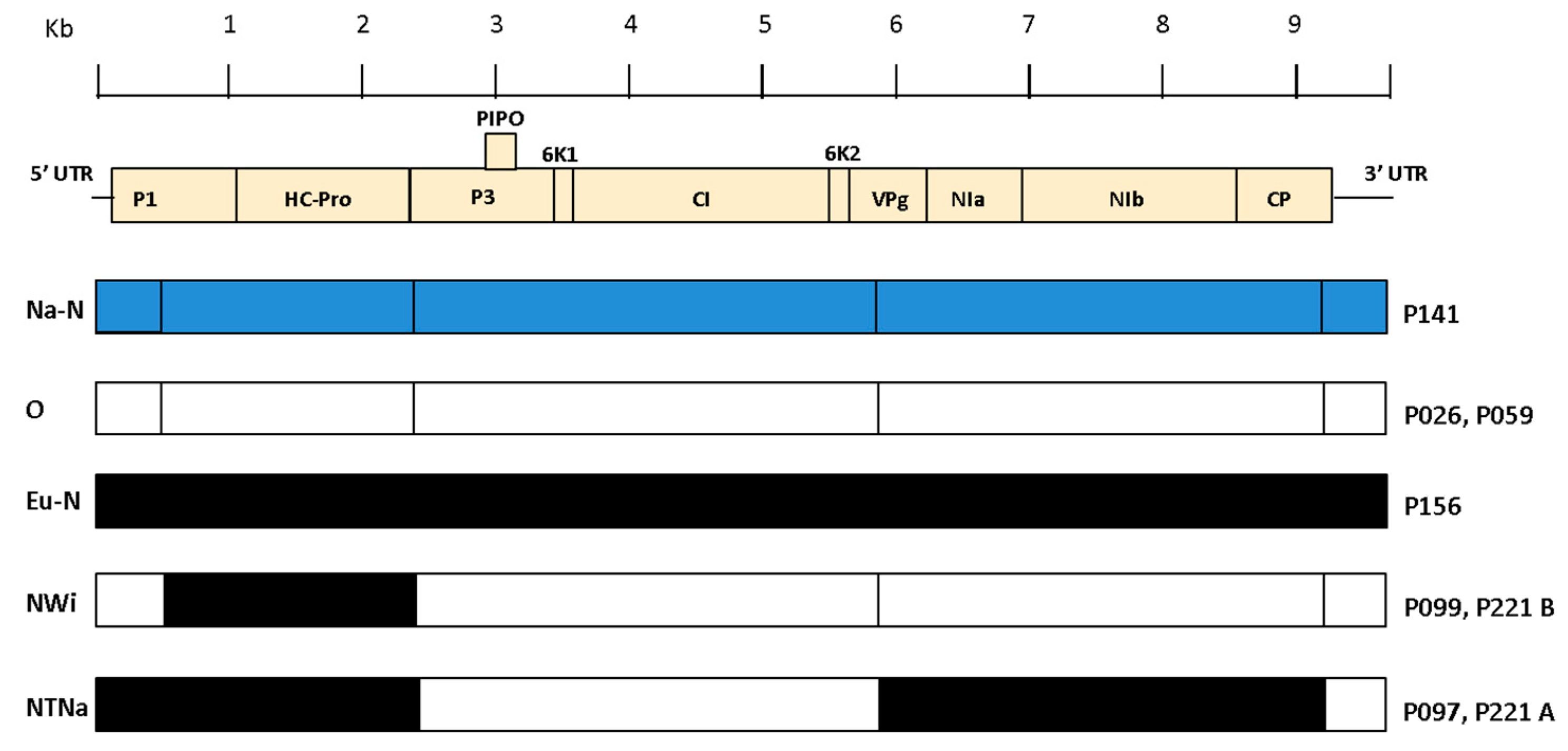
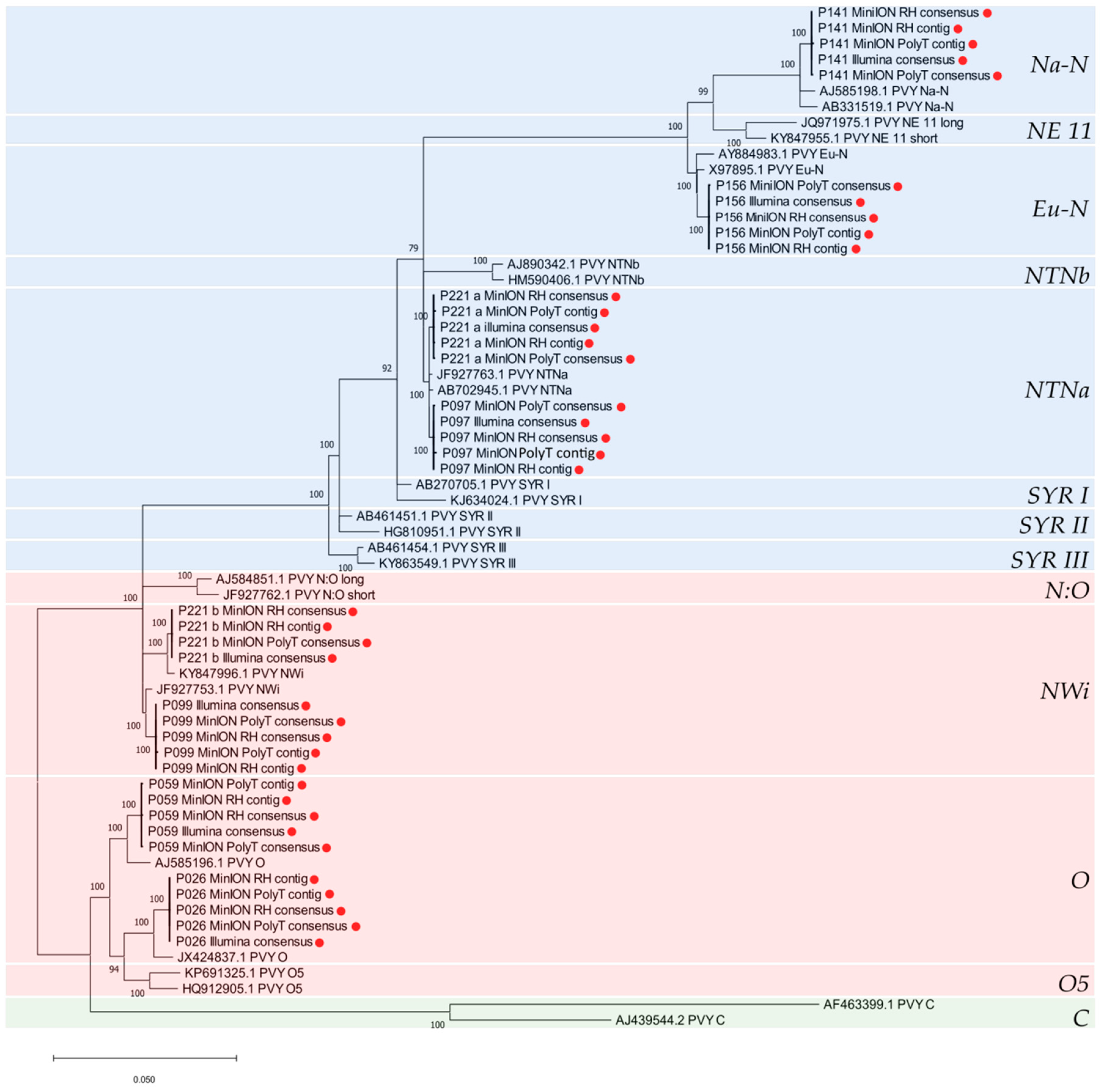
3.5. PVY Recombination and Phylogenetic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bond, J.K. Potato utilization and markets. In The Potato: Botany, Production and Uses; CABI: Wallingford, UK, 2014; pp. 29–44. [Google Scholar]
- Jones, R.A.C. Virus disease problems facing potato industries worldwide: Viruses found, climate change implications, rationalizing virus strain nomenclature, and addressing the Potato virus Y issue. In The Potato: Botany, Production and Uses; CABI: Wallingford, UK, 2014; pp. 202–224. [Google Scholar]
- Scholthof, K.B.G.; Adkins, S.; Czosnek, H.; Palukaitis, P.; Jacquot, E.; Hohn, T.; Hohn, B.; Saunders, K.; Candresse, T.; Ahlquist, P.; et al. Top 10 plant viruses in molecular plant pathology. Mol. Plant Pathol. 2011, 12, 938–954. [Google Scholar] [CrossRef]
- Valkonen, J.P.T. Viruses: Economical losses and biotechnological potential. In Potato Biology and Biotechnology: Advances and Perspectives; Elsevier: Amsterdam, The Netherlands, 2007; pp. 619–641. ISBN 9780444510181. [Google Scholar]
- Kerlan, C.; Moury, B. Potato Virus Y. In Encyclopedia of Virology; Association of Applied Biologists: Weisborne, UK, 2008; pp. 287–296. ISBN 9780123744104. [Google Scholar]
- Lacomme, C.; Pickup, J.; Fox, A.; Glais, L.; Dupuis, B.; Steinger, T.; Rolot, J.L.; Valkonen, J.P.T.; Kruger, K.; Nie, X.; et al. Transmission and epidemiology of Potato virus Y. In Potato Virus Y: Biodiversity, Pathogenicity, Epidemiology and Management; Springer International Publishing: Basel, Switzerland, 2017; pp. 141–176. ISBN 9783319588605. [Google Scholar]
- Wylie, S.J.; Adams, M.; Chalam, C.; Kreuze, J.; López-Moya, J.J.; Ohshima, K.; Praveen, S.; Rabenstein, F.; Stenger, D.; Wang, A.; et al. ICTV virus taxonomy profile: Potyviridae. J. Gen. Virol. 2017, 98, 352–354. [Google Scholar] [CrossRef]
- Steinhauer, D.A.; Domingo, E.; Holland, J.J. Lack of evidence for proofreading mechanisms associated with an RNA virus polymerase. Gene 1992, 122, 281–288. [Google Scholar] [CrossRef]
- Gibbs, A.; Ohshima, K. Potyviruses and the Digital Revolution. Annu. Rev. Phytopathol. 2010, 48, 205–223. [Google Scholar] [CrossRef]
- Revers, F.; Le Gall, O.; Candresse, T.; Le Romancer, M.; Dunez, J. Frequent occurrence of recombinant potyvirus isolates. J. Gen. Virol. 1996, 77, 1953–1965. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Valkonen, J.P.T.; Gray, S.M.; Boonham, N.; Jones, R.A.C.; Kerlan, C.; Schubert, J. Discussion paper: The naming of Potato virus Y strains infecting potato. Arch. Virol. 2008, 153, 1–13. [Google Scholar] [CrossRef]
- Kehoe, M.A.; Jones, R.A.C. Improving Potato virus Y strain nomenclature: Lessons from comparing isolates obtained over a 73-year period. Plant Pathol. 2016, 65, 322–333. [Google Scholar] [CrossRef]
- Glais, L.; Tribodet, M.; Kerlan, C. Genomic variability in Potato potyvirus Y (PVY): Evidence that PVYNW and PVYNTN variants are single to multiple recombinants between PVYO and PVYN isolates. Arch. Virol. 2002, 147, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Green, K.J.; Brown, C.J.; Gray, S.M.; Karasev, A.V. Phylogenetic study of recombinant strains of Potato virus Y. Virology 2017, 507, 40–52. [Google Scholar] [CrossRef]
- Gibbs, A.J.; Ohshima, K.; Yasaka, R.; Mohammadi, M.; Gibbs, M.J.; Jones, R.A.C. The phylogenetics of the global population of potato virus Y and its necrogenic recombinants. Virus Evol. 2017, 3, vex002. [Google Scholar] [CrossRef] [Green Version]
- Green, K.J.; Brown, C.J.; Karasev, A.V. Genetic diversity of potato virus Y (PVY): Sequence analyses reveal ten novel PVY recombinant structures. Arch. Virol. 2018, 163, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Karasev, A.V.; Hu, X.; Brown, C.J.; Kerlan, C.; Nikolaeva, O.V.; Crosslin, J.M.; Gray, S.M. Genetic diversity of the ordinary strain of Potato virus y (pvy) and origin of recombinant PVY strains. Phytopathology 2011, 101, 778–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.A.C.; Kehoe, M.A. A proposal to rationalize within-species plant virus nomenclature: Benefits and implications of inaction. Arch. Virol. 2016, 161, 2051–2057. [Google Scholar] [CrossRef] [Green Version]
- Elwan, E.A.; Aleem, E.E.A.; Fattouh, F.A.; Green, K.J.; Tran, L.T.; Karasev, A.V. Occurrence of diverse recombinant strains of potato virus y circulating in potato fields in Egypt. Plant Dis. 2017, 101, 1463–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanigliulo, A.; Comes, S.; Pacella, R.; Harrach, B.; Martin, D.P.; Crescenzi, A. Characterisation of Potato virus Y nnp strain inducing veinal necrosis in pepper: A naturally occurring recombinant strain of PVY. Arch. Virol. 2005, 150, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Galvino-Costa, S.B.F.; dos Reis Figueira, A.; de Assis Câmara Rabelo-Filho, F.; Moraes, F.H.R.; Nikolaeva, O.V.; Karasev, A.V. Molecular and serological typing of Potato virus Y isolates from Brazil reveals a diverse set of recombinant strains. Plant Dis. 2012, 96, 1451–1458. [Google Scholar] [CrossRef] [Green Version]
- Galvino-Costa, S.B.F.; dos Reis Figueira, A.; Camargos, V.V.; Geraldino, P.S.; Hu, X.J.; Nikolaeva, O.V.; Kerlan, C.; Karasev, A.V. A novel type of Potato virus Y recombinant genome, determined for the genetic strain PVY E. Plant Pathol. 2012, 61, 388–398. [Google Scholar] [CrossRef]
- Hataya, T.; Inoue, A.K.; Ohshima, K.; Shikata, E. Characterization and strain identification of a potato virus Y isolate non-reactive with monoclonal antibodies specific to the ordinary and necrotic strains. Intervirology 1994, 37, 12–19. [Google Scholar] [CrossRef]
- Karasev, A.V.; Gray, S.M. Continuous and Emerging Challenges of Potato virus Y in Potato. Annu. Rev. Phytopathol. 2013, 51, 571–586. [Google Scholar] [CrossRef]
- Lorenzen, J.; Nolte, P.; Martin, D.; Pasche, J.S.; Gudmestad, N.C. NE-11 represents a new strain variant class of Potato virus Y. Arch. Virol. 2008, 153, 517–525. [Google Scholar] [CrossRef]
- Moodley, V.; Ibaba, J.D.; Naidoo, R.; Gubba, A. Full-genome analyses of a Potato Virus Y (PVY) isolate infecting pepper (Capsicum annuum L.) in the Republic of South Africa. Virus Genes 2014, 49, 466–476. [Google Scholar] [CrossRef]
- Ogawa, T.; Nakagawa, A.; Hataya, T.; Ohshima, K. The Genetic Structure of Populations of Potato virus Y in Japan; Based on the Analysis of 20 Full Genomic Sequences. J. Phytopathol. 2012, 160, 661–673. [Google Scholar] [CrossRef]
- Robaglia, C.; Durand-Tardif, M.; Tronchet, M.; Boudazin, G.; Astier-Manifacier, S.; Casse-Delbart, F. Nucleotide sequence of potato virus Y (N Strain) genomic RNA. J. Gen. Virol. 1989, 70 (Pt 4), 935–947. [Google Scholar] [CrossRef]
- Schubert, J.; Fomitcheva, V.; Sztangret-Wiśniewska, J. Differentiation of Potato virus Y strains using improved sets of diagnostic PCR-primers. J. Virol. Methods 2007, 140, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Schubert, J.; Thieme, T.; Thieme, R.; Ha, C.V.; Hoang, G.T. Molecular and Biological Characterization of Potato virus Y Isolates from Vietnam. J. Phytopathol. 2015, 163, 620–631. [Google Scholar] [CrossRef]
- Hu, X.; He, C.; Xiao, Y.; Xiong, X.; Nie, X. Molecular characterization and detection of recombinant isolates of potato virus Y from China. Arch. Virol. 2009, 154, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Romancer, M.L.; Kerlan, C.; Nedellec, M. Biological characterisation of various geographical isolates of potato virus Y inducing superficial necrosis on potato tubers. Plant Pathol. 1994, 43, 138–144. [Google Scholar] [CrossRef]
- Gray, S.; De Boer, S.; Lorenzen, J.; Karasev, A.; Whitworth, J.; Nolte, P.; Singh, R.; Boucher, A.; Xu, H. Potato virus Y: An evolving concern for potato crops in the United States and Canada. Plant Dis. 2010, 94, 1384–1397. [Google Scholar] [CrossRef] [Green Version]
- Chikh-Ali, M.; Rodriguez-Rodriguez, M.; Green, K.J.; Kim, D.J.; Kuhl, J.C.; Chung, S.M.; Karasev, A.V. Identification and molecular characterization of recombinant potato virus Y (PVY) in potato from South Korea, PVY NTN strain. Plant Dis. 2019, 103, 137–142. [Google Scholar] [CrossRef] [Green Version]
- Chikh-Ali, M.; Bosque-Pérez, N.A.; Vanderpol, D.; Sembel, D.; Karasev, A.V. Occurrence and molecular characterization of recombinant Potato virus YNTN isolates from Sulawesi, Indonesia. Plant Dis. 2016, 100, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Chikh-Ali, M.; Alruwaili, H.; Vander Pol, D.; Karasev, A.V. Molecular characterization of recombinant strains of Potato virus Y from Saudi Arabia. Plant Dis. 2016, 100, 292–297. [Google Scholar] [CrossRef] [Green Version]
- Davie, K.; Holmes, R.; Pickup, J.; Lacomme, C. Dynamics of PVY strains in field grown potato: Impact of strain competition and ability to overcome host resistance mechanisms. Virus Res. 2017, 241, 95–104. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, T.D.B.; Nie, X.; Bisht, V.; Singh, M. Proliferation of Recombinant PVY Strains in Two Potato-Producing Regions of Canada, and Symptom Expression in 30 Important Potato Varieties with Different PVY Strains. Plant Dis. 2019, 103, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Chikh Ali, M.; Karasev, A.V.; Furutani, N.; Taniguchi, M.; Kano, Y.; Sato, M.; Natsuaki, T.; Maoka, T. Occurrence of potato virus Y strain PVYNTN in foundation seed potatoes in Japan, and screening for symptoms in Japanese potato cultivars. Plant Pathol. 2013, 62, 1157–1165. [Google Scholar] [CrossRef]
- Gray, S.M.; Power, A.G. Anthropogenic influences on emergence of vector-borne plant viruses: The persistent problem of Potato virus Y. Curr. Opin. Virol. 2018, 33, 177–183. [Google Scholar] [CrossRef]
- Karasev, A.V.; Gray, S.M. Genetic Diversity of Potato virus Y Complex. Am. J. Potato Res. 2013, 90, 7–13. [Google Scholar] [CrossRef]
- Yin, Z.; Chrzanowska, M.; Michalak, K.; Zagórska, H.; Zimnoch-Guzowska, E. Recombinants of PVY strains predominate among isolates from potato crop in Poland. J. Plant Prot. Res. 2012, 52, 214–219. [Google Scholar] [CrossRef]
- Avrahami-Moyal, L.; Tam, Y.; Sela, N.; Prakash, S.; Meller Harel, Y.; Bornstein, M.; Shulchani, R.; Dar, Z.; Gaba, V. Characterization of potato virus Y populations in potato in Israel. Arch. Virol. 2019, 164, 1691–1695. [Google Scholar] [CrossRef] [PubMed]
- Bahrami Kamangar, S.; Smagghe, G.; Maes, M.; De Jonghe, K. Potato virus Y (PVY) strains in Belgian seed potatoes and first molecular detection of the N-Wi strain. J. Plant Dis. Prot. 2014, 121, 10–19. [Google Scholar] [CrossRef]
- Bai, Y.; Han, S.; Gao, Y.; Zhang, W.; Fan, G.; Qiu, C.; Nie, X.; Wen, J.Z. Genetic diversity of potato virus y in potato production areas in Northeast China. Plant Dis. 2019, 103, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindner, K.; Trautwein, F.; Kellermann, A.; Bauch, G. Potato virus Y (PVY) in seed potato certification. J. Plant Dis. Prot. 2015, 122, 109–119. [Google Scholar] [CrossRef]
- Rigotti, S.; Balmelli, C.; Gugerli, P. Census Report of the Potato Virus Y (PVY) Population in Swiss Seed Potato Production in 2003 and 2008. Potato Res. 2011, 54, 105–117. [Google Scholar] [CrossRef]
- Allala-Messaoudi, L.; Glais, L.; Kerkoud, M.; Boukhris-Bouhachem, S.; Bouznad, Z. Preliminary characterization of potato virus Y (PVY) populations in Algerian potato fields. J. Plant Pathol. 2019, 101, 1–14. [Google Scholar] [CrossRef]
- Funke, C.N.; Nikolaeva, O.V.; Green, K.J.; Tran, L.T.; Chikh-Ali, M.; Quintero-Ferrer, A.; Cating, R.A.; Frost, K.E.; Hamm, P.B.; Olsen, N.; et al. Strain-specific resistance to potato virus Y (PVY) in potato and its effect on the relative abundance of pvy strains in commercial potato fields. Plant Dis. 2017, 101, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Blanchard, A.; Rolland, M.; Lacroix, C.; Kerlan, C.; Jacquot, E. Potato virus Y: A century of evolution. Curr. Top. Virol. 2008, 7, 21–32. [Google Scholar]
- Hu, X.; Karasev, A.V.; Brown, C.J.; Lorenzen, J.H. Sequence characteristics of potato virus Y recombinants. J. Gen. Virol. 2009, 90, 3033–3041. [Google Scholar] [CrossRef]
- Hutton, F.; Spink, J.H.; Griffin, D.; Kildea, S.; Bonner, D.; Doherty, G.; Hunter, A. Distribution and incidence of viruses in Irish seed potato crops. Irish J. Agric. Food Res. 2015, 54, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Hutton, F.; Kildea, S.; Griffin, D.; Spink, J.; Doherty, G.; Hunter, A. First report of potato tuber necrotic ringspot disease associated with PVYrecombinantstrains in Ireland. New Dis. Rep. 2013, 28, 12. [Google Scholar] [CrossRef] [Green Version]
- Glais, L.; Bellstedt, D.U.; Lacomme, C. Diversity, characterisation and classification of PVY. In Potato Virus Y: Biodiversity, Pathogenicity, Epidemiology and Management; Springer International Publishing: Basel, Switzerland, 2017; pp. 43–76. ISBN 9783319588605. [Google Scholar]
- Du, Z.Y.; Chen, J.S.; Hiruki, C. Optimization and application of a multiplex RT-PCR system for simultaneous detection of five potato viruses using 18S rRNA as an internal control. Plant Dis. 2006, 90, 185–189. [Google Scholar] [CrossRef] [Green Version]
- Chikh Ali, M.; Maoka, T.; Natsuaki, K.T.; Natsuaki, T. The simultaneous differentiation of Potato virus Y strains including the newly described strain PVYNTN-NW by multiplex PCR assay. J. Virol. Methods 2010, 165, 15–20. [Google Scholar] [CrossRef]
- Lorenzen, J.H.; Piche, L.M.; Gudmestad, N.C.; Meacham, T.; Shiel, P. A multiplex PCR assay to characterize Potato virus Y isolates and identify strain mixtures. Plant Dis. 2006, 90, 935–940. [Google Scholar] [CrossRef] [Green Version]
- Rigotti, S.; Gugerli, P. Rapid identification of potato virus Y strains by one-step triplex RT-PCR. J. Virol. Methods 2007, 140, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Singh, R.P. A new approach for the simultaneous differentiation of biological and geographical strains of Potato virus Y by uniplex and multiplex RT-PCR. J. Virol. Methods 2002, 104, 41–54. [Google Scholar] [CrossRef]
- Kogovšek, P.; Gow, L.; Pompe-Novak, M.; Gruden, K.; Foster, G.D.; Boonham, N.; Ravnikar, M. Single-step RT real-time PCR for sensitive detection and discrimination of Potato virus Y isolates. J. Virol. Methods 2008, 149, 1–11. [Google Scholar] [CrossRef]
- Rolland, M.; Glais, L.; Kerlan, C.; Jacquot, E. A multiple single nucleotide polymorphisms interrogation assay for reliable Potato virus Y group and variant characterization. J. Virol. Methods 2008, 147, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Ximba, S.P.F.; Ibaba, J.D.; Gubba, A. Potato virus Y strains infecting potatoes in the Msinga district in the province of KwaZulu-Natal, South Africa. Crop Prot. 2017, 96, 188–194. [Google Scholar] [CrossRef]
- Adams, I.P.; Glover, R.H.; Monger, W.A.; Mumford, R.; Jackeviciene, E.; Navalinskiene, M.; Samuitiene, M.; Boonham, N. Next-generation sequencing and metagenomic analysis: A universal diagnostic tool in plant virology. Mol. Plant Pathol. 2009, 10, 537–545. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Rowhani, A. Deep sequencing analysis of RNAs from a grapevine showing Syrah decline symptoms reveals a multiple virus infection that includes a novel virus. Virology 2009, 387, 395–401. [Google Scholar] [CrossRef] [Green Version]
- Kreuze, J.F.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I.; Simon, R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery and sequencing of viruses. Virology 2009, 388, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Fellers, J.P.; Webb, C.; Fellers, M.C.; Shoup Rupp, J.; De Wolf, E. Wheat Virus Identification Within Infected Tissue Using Nanopore Sequencing Technology. Plant Dis. 2019, 103, 2199–2203. [Google Scholar] [CrossRef]
- Filloux, D.; Fernandez, E.; Loire, E.; Claude, L.; Galzi, S.; Candresse, T.; Winter, S.; Jeeva, M.L.; Makeshkumar, T.; Martin, D.P.; et al. Nanopore-based detection and characterization of yam viruses. Sci. Rep. 2018, 8, 17879. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, L. Portable DNA sequencer helps farmers stymie devastating viruses. Proc. Natl. Acad. Sci. USA 2019, 116, 3351–3353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boykin, L.M.; Sseruwagi, P.; Alicai, T.; Ateka, E.; Mohammed, I.U.; Stanton, J.A.L.; Kayuki, C.; Mark, D.; Fute, T.; Erasto, J.; et al. Tree lab: Portable genomics for early detection of plant viruses and pests in sub-saharan africa. Genes 2019, 10, 632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badial, A.B.; Sherman, D.; Stone, A.; Gopakumar, A.; Wilson, V.; Schneider, W.; King, J. Nanopore sequencing as a surveillance tool for plant pathogens in plant and insect tissues. Plant Dis. 2018, 102, 1648–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rang, F.J.; Kloosterman, W.P.; de Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, M.F.; Adams, A.N. Characteristics of the microplate method of enzyme linked immunosorbent assay for the detection of plant viruses. J. Gen. Virol. 1977, 34, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Rott, M.E.; Jelkmann, W. Characterization and detection of several filamentous viruses of cherry: Adaptation of an alternative cloning method (DOP-PCR), and modification of an rna extraction protocol. Eur. J. Plant Pathol. 2001, 107, 411–420. [Google Scholar] [CrossRef]
- Tombácz, D.; Moldován, N.; Balázs, Z.; Gulyás, G.; Csabai, Z.; Boldogkői, M.; Snyder, M.; Boldogkői, Z. Multiple Long-Read Sequencing Survey of Herpes Simplex Virus Dynamic Transcriptome. Front. Genet. 2019, 10, 834. [Google Scholar] [CrossRef]
- Hardy, R.W.F.; Burns, R.C.; Holsten, R.D. Applications of the Acetylene-Ethylene Assay for Measurement of Nitrogen Fixation. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 December 2019).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- CLC Bio CLC Genomics Workbench 7.5 User Manual. Available online: http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/752/User_Manual.pdf (accessed on 11 February 2020).
- Pecman, A.; Kutnjak, D.; Gutiérrez-Aguirre, I.; Adams, I.; Fox, A.; Boonham, N.; Ravnikar, M. Next generation sequencing for detection and discovery of plant viruses and viroids: Comparison of two approaches. Front. Microbiol. 2017, 8, 1998. [Google Scholar] [CrossRef] [Green Version]
- Kutnjak, D.; Rupar, M.; Gutierrez-Aguirre, I.; Curk, T.; Kreuze, J.F.; Ravnikar, M. Deep Sequencing of Virus-Derived Small Interfering RNAs and RNA from Viral Particles Shows Highly Similar Mutational Landscapes of a Plant Virus Population. J. Virol. 2015, 89, 4760–4769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [Green Version]
- GitHub—Rrwick/Porechop: Adapter Trimmer for Oxford Nanopore Reads. Available online: https://github.com/rrwick/Porechop (accessed on 12 December 2019).
- Xu, X.; Pan, S.; Cheng, S.; Zhang, B.; Mu, D.; Ni, P.; Zhang, G.; Yang, S.; Li, R.; Wang, J.; et al. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar] [PubMed]
- Potato Genome Sequencing Consortium PGSC Data Download. Available online: http://solanaceae.plantbiology.msu.edu/pgsc_download.shtml (accessed on 12 December 2019).
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canu FAQ—Canu 1.8 Documentation. Available online: https://canu.readthedocs.io/en/latest/faq.html#what-parameters-can-i-tweak (accessed on 13 December 2019).
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- GitHub—Alimanfoo/Pysamstats: A Fast Python and Command-Line Utility for Extracting Simple Statistics against Genome Positions Based on Sequence Alignments from a SAM or BAM File. Available online: https://github.com/alimanfoo/pysamstats (accessed on 14 December 2019).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: http://www.R-project.org (accessed on 16 December 2019).
- Zeileis, A.; Grothendieck, G. Zoo: S3 infrastructure for regular and irregular time series. J. Stat. Softw. 2005, 14, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Tyler, A.D.; Mataseje, L.; Urfano, C.J.; Schmidt, L.; Antonation, K.S.; Mulvey, M.R.; Corbett, C.R. Evaluation of Oxford Nanopore’s MinION Sequencing Device for Microbial Whole Genome Sequencing Applications. Sci. Rep. 2018, 8, 10931. [Google Scholar] [CrossRef]
- Batovska, J.; Lynch, S.E.; Rodoni, B.C.; Sawbridge, T.I.; Cogan, N.O. Metagenomic Arbovirus Detection Using MinION Nanopore Sequencing; Elsevier B.V.: Amsterdam, The Netherlands, 2017; pp. 79–84. [Google Scholar]
- McDonald, J.G.; Singh, R.P. Host range, symptomology, and serology of isolates of potato virus y (pvy) that share properties with both the PVYN and PVYO strain groups. Am. Potato J. 1996, 73, 309–315. [Google Scholar] [CrossRef]
- Nie, B.; Singh, M.; Murphy, A.; Sullivan, A.; Xie, C.; Nie, X. Response of potato cultivars to five isolates belonging to four strains of Potato virus Y. Plant Dis. 2012, 96, 1422–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzen, J.H.; Meacham, T.; Berger, P.H.; Shiel, P.J.; Crosslin, J.M.; Hamm, P.B.; Kopp, H. Whole genome characterization of Potato virus Y isolates collected in the western USA and their comparison to isolates from Europe and Canada. Arch. Virol. 2006, 151, 1055–1074. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.; McGeachy, K.D.; Toplak, N.; Gruden, K.; Žel, J.; Browning, I. Comparison of genome sequence of PVY isolates with biological properties. Am. J. Potato Res. 2009, 86, 227–238. [Google Scholar] [CrossRef]
- Dupuis, B.; Bragard, C.; Schumpp, O. Resistance of Potato Cultivars as a Determinant Factor of Potato virus Y (PVY) Epidemiology. Potato Res. 2019, 62, 123–138. [Google Scholar] [CrossRef] [Green Version]
- Pooggin, M.M. Small RNA-omics for plant virus identification, virome reconstruction, and antiviral defense characterization. Front. Microbiol. 2018, 9, 2779. [Google Scholar] [CrossRef] [PubMed]
- Massart, S.; Olmos, A.; Jijakli, H.; Candresse, T. Current impact and future directions of high throughput sequencing in plant virus diagnostics. Virus Res. 2014, 188, 90–96. [Google Scholar] [CrossRef]
- Olmos, A.; Boonham, N.; Candresse, T.; Gentit, P.; Giovani, B.; Kutnjak, D.; Liefting, L.; Maree, H.J.; Minafra, A.; Moreira, A.; et al. High-throughput sequencing technologies for plant pest diagnosis: Challenges and opportunities. EPPO Bull. 2018, 48, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Maree, H.J.; Fox, A.; Al Rwahnih, M.; Boonham, N.; Candresse, T. Application of hts for routine plant virus diagnostics: State of the art and challenges. Front. Plant Sci. 2018, 9, 1082. [Google Scholar] [CrossRef] [Green Version]
- Adams, I.P.; Fox, A.; Boonham, N.; Massart, S.; De Jonghe, K. The impact of high throughput sequencing on plant health diagnostics. Eur. J. Plant Pathol. 2018, 152, 909–919. [Google Scholar] [CrossRef]
- Massart, S.; Candresse, T.; Gil, J.; Lacomme, C.; Predajna, L.; Ravnikar, M.; Reynard, J.S.; Rumbou, A.; Saldarelli, P.; Škoric, D.; et al. A framework for the evaluation of biosecurity, commercial, regulatory, and scientific impacts of plant viruses and viroids identified by NGS technologies. Front. Microbiol. 2017, 8, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roenhorst, J.W.; de Krom, C.; Fox, A.; Mehle, N.; Ravnikar, M.; Werkman, A.W. Ensuring validation in diagnostic testing is fit for purpose: A view from the plant virology laboratory. EPPO Bull. 2018, 48, 105–115. [Google Scholar] [CrossRef]
- Maliogka, V.I.; Minafra, A.; Saldarelli, P.; Ruiz-García, A.B.; Glasa, M.; Katis, N.; Olmos, A. Recent advances on detection and characterization of fruit tree viruses using high-throughput sequencing technologies. Viruses 2018, 10, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesanakurti, P.; Belton, M.; Saeed, H.; Rast, H.; Boyes, I.; Rott, M. Screening for plant viruses by next generation sequencing using a modified double strand RNA extraction protocol with an internal amplification control. J. Virol. Methods 2016, 236, 35–40. [Google Scholar] [CrossRef]
- Jiang, L.; Schlesinger, F.; Davis, C.A.; Zhang, Y.; Li, R.; Salit, M.; Gingeras, T.R.; Oliver, B. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 2011, 21, 1543–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltimore, D. Expression of animal virus genomes. Bacteriol. Rev. 1971, 35, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Palatnick, A.; Zhou, B.; Ghedin, E.; Schatz, M.C. iGenomics: Comprehensive DNA Sequence Analysis on your Smartphone. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Berto, M.; Vecchi, E.; Baiamonte, L.; Condò, C.; Sensi, M.; Di Lauro, M.; Sola, M.; De Stradis, A.; Biscarini, F.; Minafra, A.; et al. Label free detection of plant viruses with organic transistor biosensors. Sens. Actuators B Chem. 2019, 281, 150–156. [Google Scholar] [CrossRef]
- Chiriacò, M.S.; Luvisi, A.; Primiceri, E.; Sabella, E.; De Bellis, L.; Maruccio, G. Development of a lab-on-a-chip method for rapid assay of Xylella fastidiosa subsp. pauca strain CoDiRO. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef]
- Jarocka, U.; Radecka, H.; Malinowski, T.; Michalczuk, L.; Radecki, J. Detection of Prunus Necrotic Ringspot Virus in Plant Extracts with Impedimetric Immunosensor based on Glassy Carbon Electrode. Electroanalysis 2013, 25, 433–438. [Google Scholar] [CrossRef]
- Jarocka, U.; Wasowicz, M.; Radecka, H.; Malinowski, T.; Michalczuk, L.; Radecki, J. Impedimetric immunosensor for detection of plum pox virus in plant extracts. Electroanalysis 2011, 23, 2197–2204. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PVY (Positive/Tested) | PVS (Positive/Tested) | PVX (Positive/Tested) | PLRV (Positive/Tested) | PVA (Positive/Tested) | |
|---|---|---|---|---|---|
| Germplasm collection | 74/90 | 0/90 | 0/90 | 1/90 | 10/90 |
| Seed-tuber Potato crops | 88/110 | 18/110 | 9/110 | 0/110 | 1/110 |
| Total | 162/200 | 18/200 | 9/200 | 1/200 | 11/200 |
| PVY Genotype | NTNa | NWi | Recombinant Mixed Infection 1 | O | NTNa+O | Na-N | O+NWi | O+Na-N | Eu-N |
|---|---|---|---|---|---|---|---|---|---|
| RT-PCR pattern (bp) | 441+633 +1307 | 441+853 | 441+633 +853+1307 | 532+853 | 441+532 +633+853 +1307 | 1307 | 441+532 +853 | 532+853 +1307 | 398+633 +1307 |
| Serotype | N | O/C | N + O/C | O/C | N + O/C | N | O/C | N + O/C | N |
| Germplasm collection | 28/88 | 32/88 | 13/88 | 6/88 | 4/88 | 0/88 | 3/88 | 2/88 | 0/88 |
| Seed-tuber Potato crops | 66/74 | 0/74 | 0/7 | 0/74 | 2/74 | 5/74 | 0/74 | 0/74 | 1/74 |
| Total | 94/162 | 32/162 | 13/162 | 6/162 | 6/162 | 5/162 | 3/162 | 2/162 | 1/162 |
| Sample ID | RT-PCR Virus | RT-PCR PVY Genotype | Total Reads | Virus Contigs/HSP Length (nt) 1 | NCBI BLASTn Accession 2 (Length nt) | Consensus Length 3 | Average Sequencing Depth | Assigned PVY Genotype 4 |
|---|---|---|---|---|---|---|---|---|
| P026 | PVY | O | 60.38 M | 1/9694 | PVY JX424837 (9699) | 9699 | 175,369 × | O |
| P097 | PVY | NTNa | 54.12 M | 1/9672 | PVY JF927763 (9701) | 9694 | 31,972 × | NTNa |
| P099 | PVY | NWi | 52.89 M | 1/9683 | PVY JF927753 (9697) | 9697 | 51,748 × | NWi |
| P156 | PVY | Eu-N | 53.56 M | 1/9687 | PVY X97895 (9701) | 9701 | 569,071 × | Eu-N |
| P141 | PVY | Na-N | 48.48 M | 1/9690 | PVY AB331517 (9701) | 9701 | 593,890 × | Na-N |
| P221 | PVY | recombinant mixed infection | 52.61 M | a. 2/2217–3490 b. 2/2217–3453 | PVY JF927763 (9701) PVY JF927754 (9697) | 9701 9697 | 124,208 × 95,512 × | NTNa NWi |
| P166 | PLRV | 54.28 M | 1/5868 | PLRV AY138970 (5884) | 5884 | 599,408 × | ||
| P059 | PVY PVX PVS | O | 46.26 M | 1/9674 1/6427 1/8463 | PVY MH795851 (9700) PVX KR605396 (6435) PVS MF418030 (8499) | 9687 6435 8499 | 6384 × 556,895 × 79,253 × | O |
| Sample ID | Library | No. of Virus Contigs/ HSP Length (nt) 1 | Map to Reference (NCBI Accession) 2 | Consensus Length 3 | Average Sequencing Depth |
|---|---|---|---|---|---|
| P026 | PolyT | 1/9562 | PVY (JX424837) | 9684 nt | 1027.28 × |
| P097 | PolyT | 1/9685 | PVY (JF927763) | 9697 nt | 478.40 × |
| P099 | PolyT | 1/9680 | PVY (JF927753) | 9692 nt | 946.96 × |
| P156 | PolyT | 1/9693 | PVY (X97895) | 9697 nt | 1855.30 × |
| P141 | PolyT | 1/9678 | PVY (AB331517) | 9697 nt | 1426.61 × |
| P221 | PolyT | a. 1/9671 b. 4/1314–7239 | PVY (JF927763) PVY (JF927754) | 9698 nt 9688 nt | 501.54 × 734.40 × |
| P166 | PolyT | 2/2427–3796 | PLRV (AY138970) | 5882 nt | 81.49 × |
| P059 | PolyT | 3/1019–8478 11/1170–6434 1/9440 | PVS (MF418030) PVX (KR605396) PVY (MH795851) | 8498 nt 6434 nt 9682 nt | 832.57 × 6741.76 × 81.10 × |
| P026 | RH | 1/9676 | PVY (JX424837) | 9682 nt | 537.14 × |
| P097 | RH | 1/9675 | PVY (JF927763) | 9691 nt | 271.69 × |
| P099 | RH | 1/9668 | PVY (JF927753) | 9688 nt | 623.69 × |
| P156 | RH | 1/9690 | PVY (X97895) | 9694 nt | 962.31 × |
| P141 | RH | 1/9685 | PVY (AB331517) | 9689 nt | 1196.59 × |
| P221 | RH | a. 1/9684 b. 1/9625 | PVY (JF927763) PVY (JF927754) | 9699 nt 9688 nt | 399.30 × 553.74 × |
| P166 | RH | 1/5338 | PLRV (AY138970) | 5881 nt | 155.26 × |
| P059 | RH | 1/8476 10/1244–6435 1/9622 | PVS (MF418030) PVX (KR605396) PVY (MH795851) | 8497 nt 6433 nt 9689 nt | 1144.05 × 6614.26 × 71.62 × |
| PVY Sequence | Breakpoints Positions in Alignment | Established Breakpoints Previously Reported in Literature [16] | Non-Recombinant Parents | Genotype |
|---|---|---|---|---|
| P026 | - | - | O | |
| P097 | 2419, 5818, 9190 | 2390, 5850, 9200 | N, O | NTNa |
| P099 | 502, 2396 | 500, 2390 | O, N | NWi |
| P156 | - | - | Eu-N | |
| P141 | - | - | Na-N | |
| P221a | 2419, 5818, 9190 | 2390, 5850, 9200 | N, O | NTNa |
| P221b | 502, 2396 | 500, 2390 | O, N | NWi |
| P059 | - | - | O |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Della Bartola, M.; Byrne, S.; Mullins, E. Characterization of Potato Virus Y Isolates and Assessment of Nanopore Sequencing to Detect and Genotype Potato Viruses. Viruses 2020, 12, 478. https://doi.org/10.3390/v12040478
Della Bartola M, Byrne S, Mullins E. Characterization of Potato Virus Y Isolates and Assessment of Nanopore Sequencing to Detect and Genotype Potato Viruses. Viruses. 2020; 12(4):478. https://doi.org/10.3390/v12040478
Chicago/Turabian StyleDella Bartola, Michele, Stephen Byrne, and Ewen Mullins. 2020. "Characterization of Potato Virus Y Isolates and Assessment of Nanopore Sequencing to Detect and Genotype Potato Viruses" Viruses 12, no. 4: 478. https://doi.org/10.3390/v12040478
APA StyleDella Bartola, M., Byrne, S., & Mullins, E. (2020). Characterization of Potato Virus Y Isolates and Assessment of Nanopore Sequencing to Detect and Genotype Potato Viruses. Viruses, 12(4), 478. https://doi.org/10.3390/v12040478