Role of Viruses in the Pathogenesis of Multiple Sclerosis

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Herpesviruses
2.1. Alphaherpesviruses (VZV, HSV-1 and 2)
2.2. Beta-Herpesviruses (CMV)
2.3. Beta-Herpesviruses (HHV-6)
2.4. Gamma Herpesviruses (EBV)
3. Non-Herpes Viruses Associated with MS
3.1. JCV
3.2. HERVs
4. Antiviral Effects of MS Treatment
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Popescu, B.F.G.; Pirko, I.; Lucchinetti, C.F. Pathology of multiple sclerosis: Where do we stand? Contin. Lifelong Learn. Neurol. 2013, 19, 901–921. [Google Scholar] [CrossRef] [PubMed]
- Tarlinton, R.E.; Khaibullin, T.; Granatov, E.; Martynova, E.; Rizvanov, A.; Khaiboullina, S. The interaction between viral and environmental risk factors in the pathogenesis of multiple sclerosis. Int. J. Mol. Sci. 2019, 20, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horowitz, A.L.; Kaplan, R.D.; Grewe, G.; White, R.; Salberg, L.M. The ovoid lesion: A new MR observation in patients with multiple sclerosis. Am. J. Neuroradiol. 1989, 10, 303–305. [Google Scholar] [PubMed]
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Geurts, J.J.; Bö, L.; Pouwels, P.J.; Castelijns, J.A.; Polman, C.H.; Barkhof, F. Cortical lesions in multiple sclerosis: Combined postmortem MR imaging and histopathology. Am. J. Neuroradiol. 2005, 26, 572–577. [Google Scholar]
- Bø, L.; Vedeler, C.A.; Nyland, H.; Trapp, B.D.; Mørk, S.J. Intracortical multiple sclerosis lesions are not associated with increased lymphocyte infiltration. Mult. Scler. J. 2003, 9, 323–331. [Google Scholar] [CrossRef]
- Geurts, J.J.; Bö, L.; Roosendaal, S.D.; Hazes, T.; Daniëls, R.; Barkhof, F.; Witter, M.P.; Huitinga, I.; van der Valk, P. Extensive hippocampal demyelination in multiple sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Stone, S. The unfolded protein response in multiple sclerosis. Front. Neurosci. 2015, 9, 264. [Google Scholar]
- Calabrese, M.; De Stefano, N.; Atzori, M.; Bernardi, V.; Mattisi, I.; Barachino, L.; Morra, A.; Rinaldi, L.; Romualdi, C.; Perini, P. Detection of cortical inflammatory lesions by double inversion recovery magnetic resonance imaging in patients with multiple sclerosis. Arch. Neurol. 2007, 64, 1416–1422. [Google Scholar] [CrossRef] [Green Version]
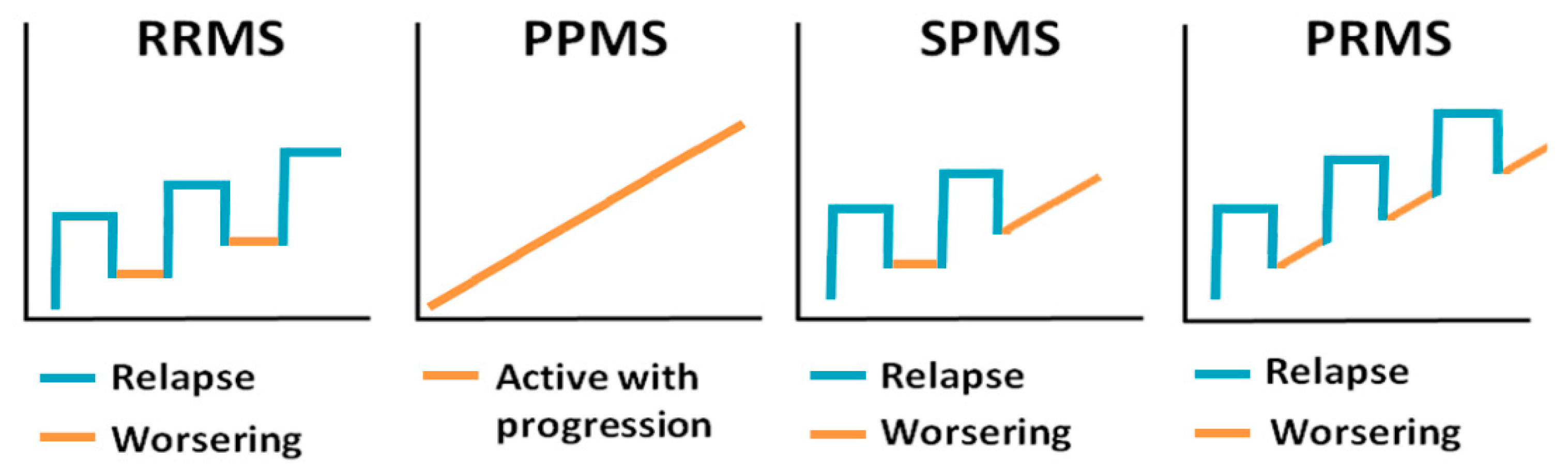
- Lublin, F.D.; Reingold, S.C. Defining the clinical course of multiple sclerosis: Results of an international survey. Neurology 1996, 46, 907–911. [Google Scholar] [CrossRef]
- Noseworthy, J.H. Progress in determining the causes and treatment of multiple sclerosis. Nature 1999, 399, A40. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Nave, K.-A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Kingwell, E.; Rieckmann, P.; Tremlett, H. The natural history of primary progressive multiple sclerosis. Neurology 2009, 73, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Khaleeli, Z.; Ciccarelli, O.; Manfredonia, F.; Barkhof, F.; Brochet, B.; Cercignani, M.; Dousset, V.; Filippi, M.; Montalban, X.; Polman, C. Predicting progression in primary progressive multiple sclerosis: A 10-year multicenter study. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2008, 63, 790–793. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [Green Version]
- Meyding-Lamadé, U.; Strank, C. Herpesvirus infections of the central nervous system in immunocompromised patients. Ther. Adv. Neurol. Disord. 2012, 5, 279–296. [Google Scholar] [CrossRef] [Green Version]
- Dubois-Dalcq, M.; Armstrong, R. The cellular and molecular events of central nervous system remyelination. Bioessays 1990, 12, 569–576. [Google Scholar] [CrossRef]
- Dubois-Dalcq, M.; Behar, T.; Hudson, L.; Lazzarini, R. Emergence of three myelin proteins in oligodendrocytes cultured without neurons. J. Cell Biol. 1986, 102, 384–392. [Google Scholar] [CrossRef] [Green Version]
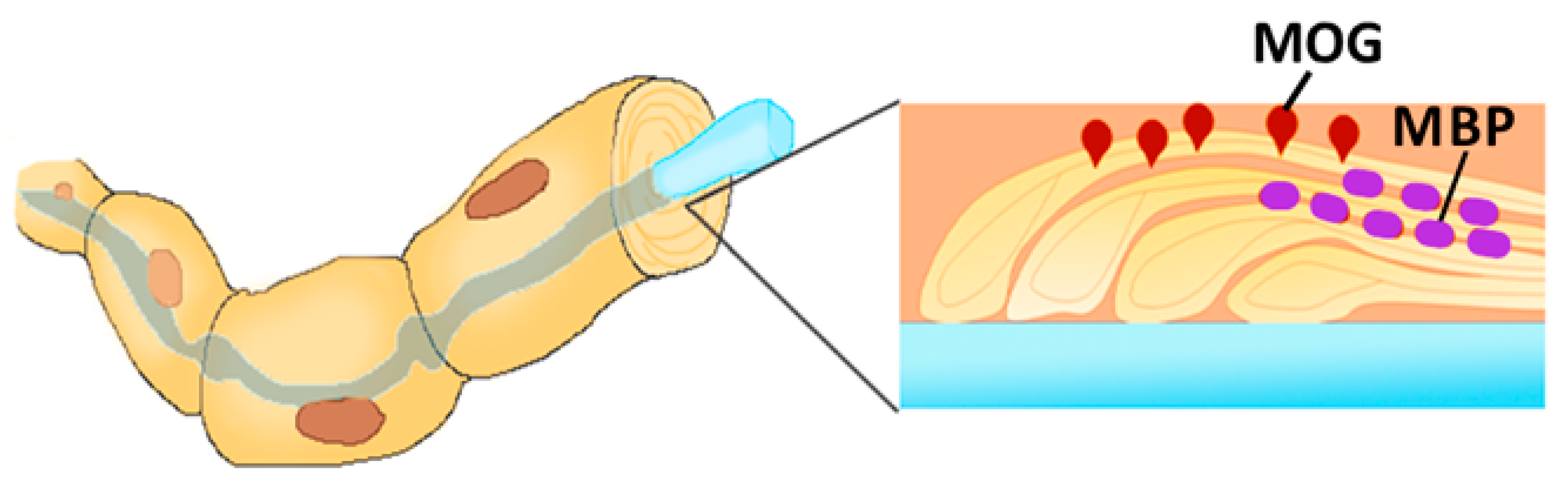
- Peschl, P.; Bradl, M.; Höftberger, R.; Berger, T.; Reindl, M. Myelin oligodendrocyte glycoprotein: Deciphering a target in inflammatory demyelinating diseases. Front. Immunol. 2017, 8, 529. [Google Scholar] [CrossRef]
- Madsen, C. The innovative development in interferon beta treatments of relapsing-remitting multiple sclerosis. Brain Behav. 2017, 7, e00696. [Google Scholar] [CrossRef] [Green Version]
- Morandi, E.; Tanasescu, R.; Tarlinton, R.E.; Constantinescu, C.S.; Zhang, W.; Tench, C.; Gran, B. The association between human endogenous retroviruses and multiple sclerosis: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0172415. [Google Scholar] [CrossRef] [PubMed]
- tHart, B.A.; Kap, Y.S.; Morandi, E.; Laman, J.D.; Gran, B. EBV Infection and Multiple Sclerosis: Lessons from a Marmoset Model. Trends Mol. Med. 2016, 22, 1012–1024. [Google Scholar] [CrossRef] [Green Version]
- Bartsch, T.; Rempe, T.; Leypoldt, F.; Riedel, C.; Jansen, O.; Berg, D.; Deuschl, G. The spectrum of progressive multifocal leukoencephalopathy: A practical approach. Eur. J. Neurol. 2019, 26, 566-e41. [Google Scholar] [CrossRef] [PubMed]
- Makhani, N.; Banwell, B.; Tellier, R.; Yea, C.; McGovern, S.; O’Mahony, J.; Ahorro, J.M.; Arnold, D.; Sadovnick, A.D.; Marrie, R.A.; et al. Viral exposures and MS outcome in a prospective cohort of children with acquired demyelination. Mult. Scler. 2016, 22, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S., Jr.; Taylor, B.; Dwyer, D.E.; Taylor, J.; Blizzard, L.; Ponsonby, A.L.; Pittas, F.; Dwyer, T.; van der Mei, I. Anti-HHV-6 IgG titer significantly predicts subsequent relapse risk in multiple sclerosis. Mult. Scler. 2012, 18, 799–806. [Google Scholar] [CrossRef]
- Niedobitek, G.; Meru, N.; Delecluse, H.J. Epstein-Barr virus infection and human malignancies. Int. J. Exp. Pathol. 2001, 82, 149–170. [Google Scholar] [CrossRef]
- Sehrawat, S.; Kumar, D.; Rouse, B.T. Herpesviruses: Harmonious pathogens but relevant cofactors in other diseases? Front. Cell. Infect. Microbiol. 2018, 8, 177. [Google Scholar] [CrossRef]
- Whitley, R. Herpesviruses. In Medical Microbiology, 4th ed.; University of Texas Medical Branch: Galveston, TX, USA, 1996. [Google Scholar]
- Kelly, G.L.; Milner, A.E.; Baldwin, G.S.; Bell, A.I.; Rickinson, A.B. Three restricted forms of Epstein–Barr virus latency counteracting apoptosis in c-myc-expressing Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 2006, 103, 14935–14940. [Google Scholar] [CrossRef] [Green Version]
- Baird, N.L.; Yu, X.; Cohrs, R.J.; Gilden, D. Varicella zoster virus (VZV)-human neuron interaction. Viruses 2013, 5, 2106–2115. [Google Scholar] [CrossRef]
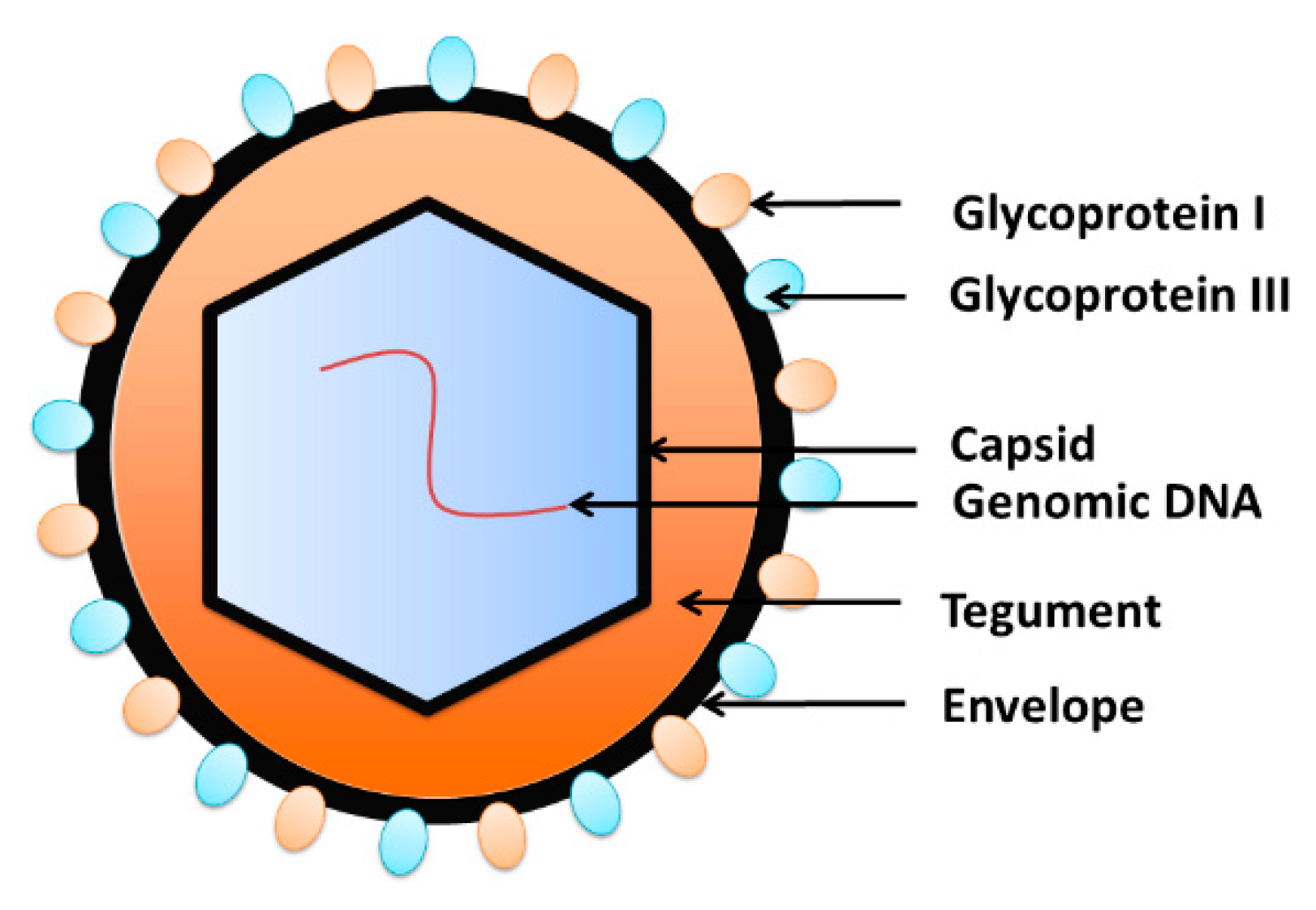
- Chen, D.H.; Jiang, H.; Lee, M.; Liu, F.; Zhou, Z.H. Three-dimensional visualization of tegument/capsid interactions in the intact human cytomegalovirus. Virology 1999, 260, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Mocarski, E.S.; Shenk, T.; Pass, R.F. Cytomegaloviruses . In Fields Virology, 5th ed.; Fields, B.M., Knipe, D.M., Howley, P., Eds.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 2, pp. 2701–2702. [Google Scholar]
- Wang, B.; Nishimura, M.; Tang, H.; Kawabata, A.; Mahmoud, N.F.; Khanlari, Z.; Hamada, D.; Tsuruta, H.; Mori, Y. Crystal structure of human herpesvirus 6B tegument protein U14. PLoS Pathog. 2016, 12, e1005594. [Google Scholar] [CrossRef] [PubMed]
- Smatti, M.K.; Al-Sadeq, D.W.; Ali, N.H.; Pintus, G.; Abou-Saleh, H.; Nasrallah, G.K. Epstein–Barr virus epidemiology, serology, and genetic variability of LMP-1 oncogene among healthy population: An update. Front. Oncol. 2018, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Crough, T.; Khanna, R. Immunobiology of human cytomegalovirus: From bench to bedside. Clin. Microbiol. Rev. 2009, 22, 76–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, W. Structure and formation of the cytomegalovirus virion. In Human Cytomegalovirus; Springer: Berlin/Heidelberg, Germany, 2008; pp. 187–204. [Google Scholar]
- Hammerschmidt, W.; Sugden, B. Replication of Epstein–Barr Viral DNA. Cold Spring Harb. Perspect. Biol. 2013, 5, a013029. [Google Scholar] [CrossRef] [Green Version]
- Arbuckle, J.H.; Medveczky, M.M.; Luka, J.; Hadley, S.H.; Luegmayr, A.; Ablashi, D.; Lund, T.C.; Tolar, J.; De Meirleir, K.; Montoya, J.G. The latent human herpesvirus-6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 5563–5568. [Google Scholar] [CrossRef] [Green Version]
- Kondo, K.; Kondo, T.; Okuno, T.; Takahashi, M.; Yamanishi, K. Latent human herpesvirus 6 infection of human monocytes/macrophages. J. Gen. Virol. 1991, 72, 1401–1408. [Google Scholar] [CrossRef]
- Manouchehrinia, A.; Tanasescu, R.; Kareem, H.; Jerca, O.P.; Jabeen, F.; Shafei, R.; Breuer, J.; Neal, K.; Irving, W.; Constantinescu, C.S. Prevalence of a history of prior varicella/herpes zoster infection in multiple sclerosis. J. Neurovirol. 2017, 23, 839–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarius, S.; Eichhorn, P.; Franciotta, D.; Petereit, H.F.; Akman-Demir, G.; Wick, M.; Wildemann, B. The MRZ reaction as a highly specific marker of multiple sclerosis: Re-evaluation and structured review of the literature. J. Neurol. 2017, 264, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Hernández-González, O.; Martínez-Palomo, A.; Sotelo, J.; Chávez-Munguía, B.; Ordoñez, G.; Talamás-Lara, D.; Pineda, B.; de Jesús Flores-Rivera, J.; Espinosa-Cantellano, M. Varicella-Zoster Virus in Cerebrospinal Fluid at Relapses of Multiple Sclerosis is Infective in Vitro. Arch. Med Res. 2018, 49, 350–355. [Google Scholar] [CrossRef]
- Sotelo, J.; Ordoñez, G.; Pineda, B.; Flores, J. The participation of varicella zoster virus in relapses of multiple sclerosis. Clin. Neurol. Neurosurg. 2014, 119, 44–48. [Google Scholar] [CrossRef]
- Pfender, N.; Jelcic, I.; Linnebank, M.; Schwarz, U.; Martin, R. Reactivation of herpesvirus under fingolimod: A case of severe herpes simplex encephalitis. Neurology 2015, 84, 2377–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perini, P.; Rinaldi, F.; Puthenparampil, M.; Marcon, M.; Perini, F.; Gallo, P. Herpes simplex virus encephalitis temporally associated with dimethyl fumarate-induced lymphopenia in a multiple sclerosis patient. Mult. Scler. Relat. Disord. 2018, 26, 68–70. [Google Scholar] [CrossRef] [PubMed]
- Dudek, M.I.R.; Thies, K.; Kammenhuber, S.; Bösel, J.; Rösche, J. HSV-2-encephalitis in a patient with multiple sclerosis treated with ocrelizumab. J. Neurol. 2019, 266, 2322–2323. [Google Scholar] [CrossRef] [PubMed]
- Nourbakhsh, B.; Rutatangwa, A.; Waltz, M.; Rensel, M.; Moodley, M.; Graves, J.; Casper, T.C.; Waldman, A.; Belman, A.; Greenberg, B.; et al. Heterogeneity in association of remote herpesvirus infections and pediatric MS. Ann. Clin. Transl. Neurol. 2018, 5, 1222–1228. [Google Scholar] [CrossRef] [Green Version]
- Etemadifar, M.; Izadi, A.; Sabeti, F.; Noorshargh, P. Anti-HSV-2 antibody in patients with MS and NMO. Mult. Scler. Relat. Disord. 2019, 28, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Czarnowska, A.; Kapica-Topczewska, K.; Zajkowska, O.; Świerzbińska, R.; Chorąży, M.; Tarasiuk, J.; Zajkowska, J.; Kochanowicz, J.; Kułakowska, A. Herpesviridae Seropositivity in Patients with Multiple Sclerosis: First Polish Study. Eur. Neurol. 2018, 80, 229–235. [Google Scholar] [CrossRef]
- Boukhvalova, M.S.; Mortensen, E.; Mbaye, A.; Lopez, D.; Kastrukoff, L.; Blanco, J.C.G. Herpes Simplex Virus 1 Induces Brain Inflammation and Multifocal Demyelination in the Cotton Rat Sigmodon hispidus. J. Virol. 2019, 94. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Zandian, M.; Kuo, J.; Mott, K.R.; Chen, S.; Arditi, M.; Ghiasi, H. Suppression of IL-12p70 formation by IL-2 or following macrophage depletion causes T-cell autoreactivity leading to CNS demyelination in HSV-1-infected mice. PLoS Pathog. 2017, 13, e1006401. [Google Scholar] [CrossRef] [Green Version]
- Najafi, S.; Ghane, M.; Poortahmasebi, V.; Jazayeri, S.M.; Yousefzadeh-Chabok, S. Prevalence of cytomegalovirus in patients with multiple sclerosis: A case-control study in northern Iran. Jundishapur J. Microbiol. 2016, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Sanadgol, N.; Ramroodi, N.; Ahmadi, G.A.; Komijani, M.; Moghtaderi, A.; Bouzari, M.; Rezaei, M.; Kardi, M.T.; Dabiri, S.; Moradi, M. Prevalence of cytomegalovirus infection and its role in total immunoglobulin pattern in Iranian patients with different subtypes of multiple sclerosis. Microbiol.-Q. J. Microbiol. Sci. 2011, 34, 263. [Google Scholar]
- Clerico, M.; De Mercanti, S.; Artusi, C.A.; Durelli, L.; Naismith, R.T. Active CMV infection in two patients with multiple sclerosis treated with alemtuzumab. Mult. Scler. 2017, 23, 874–876. [Google Scholar] [CrossRef] [PubMed]
- Vanheusden, M.; Stinissen, P.; Hart, B.A.t.; Hellings, N. Cytomegalovirus: A culprit or protector in multiple sclerosis? Trends Mol. Med. 2015, 21, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Sundqvist, E.; Bergström, T.; Daialhosein, H.; Nyström, M.; Sundström, P.; Hillert, J.; Alfredsson, L.; Kockum, I.; Olsson, T. Cytomegalovirus seropositivity is negatively associated with multiple sclerosis. Mult. Scler. J. 2014, 20, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Zivadinov, R.; Nasuelli, D.; Tommasi, M.A.; Serafin, M.; Bratina, A.; Ukmar, M.; Pirko, I.; Johnson, A.J.; Furlan, C.; Pozzi-Mucelli, R.S. Positivity of cytomegalovirus antibodies predicts a better clinical and radiological outcome in multiple sclerosis patients. Neurol. Res. 2006, 28, 262–269. [Google Scholar] [CrossRef]
- Waubant, E.; Mowry, E.M.; Krupp, L.; Chitnis, T.; Yeh, E.; Kuntz, N.; Ness, J.; Chabas, D.; Strober, J.; McDonald, J. Common viruses associated with lower pediatric multiple sclerosis risk. Neurology 2011, 76, 1989–1995. [Google Scholar] [CrossRef] [Green Version]
- Langer-Gould, A.; Wu, J.; Lucas, R.; Smith, J.; Gonzales, E.; Amezcua, L.; Haraszti, S.; Chen, L.H.; Quach, H.; James, J.A.; et al. Epstein-Barr virus, cytomegalovirus, and multiple sclerosis susceptibility: A multiethnic study. Neurology 2017, 89, 1330–1337. [Google Scholar] [CrossRef] [Green Version]
- Alari-Pahissa, E.; Moreira, A.; Zabalza, A.; Alvarez-Lafuente, R.; Munteis, E.; Vera, A.; Arroyo, R.; Alvarez-Cermeno, J.C.; Villar, L.M.; Lopez-Botet, M.; et al. Low cytomegalovirus seroprevalence in early multiple sclerosis: A case for the ‘hygiene hypothesis’? Eur. J. Neurol. 2018, 25, 925–933. [Google Scholar] [CrossRef] [Green Version]
- Pakpoor, J.; Pakpoor, J.; Disanto, G.; Giovannoni, G.; Ramagopalan, S.V. Cytomegalovirus and multiple sclerosis risk. J. Neurol. 2013, 260, 1658–1660. [Google Scholar] [CrossRef]
- Pirko, I.; Cardin, R.; Chen, Y.; Lohrey, A.K.; Lindquist, D.M.; Dunn, R.S.; Zivadinov, R.; Johnson, A.J. CMV infection attenuates the disease course in a murine model of multiple sclerosis. PLoS ONE 2012, 7, e32767. [Google Scholar] [CrossRef] [Green Version]
- Vanheusden, M.; Broux, B.; Welten, S.P.; Peeters, L.M.; Panagioti, E.; Van Wijmeersch, B.; Somers, V.; Stinissen, P.; Arens, R.; Hellings, N. Cytomegalovirus infection exacerbates autoimmune mediated neuroinflammation. Sci. Rep. 2017, 7, 663. [Google Scholar] [CrossRef]
- Dumitriu, I.E. The life (and death) of CD 4+ CD 28null T cells in inflammatory diseases. Immunology 2015, 146, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraussen, J.D.; Stinissen, P. Marielle Thewissen, Veerle Somers, Niels Hellings, Judith. J. Immunol. 2007, 179, 6514–6523. [Google Scholar]
- Leibovitch, E.C.; Jacobson, S. Evidence linking HHV-6 with multiple sclerosis: An update. Curr. Opin. Virol. 2014, 9, 127–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merelli, E.; Bedin, R.; Sola, P.; Barozzi, P.; Mancardi, G.; Ficarra, G.; Franchini, G. Human herpes virus 6 and human herpes virus 8 DNA sequences in brains of multiple sclerosis patients, normal adults and children. J. Neurol. 1997, 244, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, W.; Malessa, R.; Faustmann, P.M.; Eis-Hübinger, A.-M. Human herpesvirus 6 polymerase chain reaction findings in human immunodeficiency virus associated neurological disease and multiple sclerosis. J. Neurovirol. 1995, 1, 253–258. [Google Scholar] [CrossRef]
- Opsahl, M.L.; Kennedy, P.G. Early and late HHV-6 gene transcripts in multiple sclerosis lesions and normal appearing white matter. Brain 2005, 128, 516–527. [Google Scholar] [CrossRef] [Green Version]
- Challoner, P.B.; Smith, K.T.; Parker, J.D.; MacLeod, D.L.; Coulter, S.N.; Rose, T.M.; Schultz, E.R.; Bennett, J.L.; Garber, R.L.; Chang, M. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc. Natl. Acad. Sci. USA 1995, 92, 7440–7444. [Google Scholar] [CrossRef] [Green Version]
- Hogestyn, J.M.; Mock, D.J.; Mayer-Proschel, M. Contributions of neurotropic human herpesviruses herpes simplex virus 1 and human herpesvirus 6 to neurodegenerative disease pathology. Neural Regen. Res. 2018, 13, 211–221. [Google Scholar] [CrossRef]
- Pormohammad, A.; Azimi, T.; Falah, F.; Faghihloo, E. Relationship of human herpes virus 6 and multiple sclerosis: A systematic review and meta-analysis. J. Cell. Physiol. 2018, 233, 2850–2862. [Google Scholar] [CrossRef]
- Pakpoor, J.; Disanto, G.; Gerber, J.E.; Dobson, R.; Meier, U.C.; Giovannoni, G.; Ramagopalan, S.V. The risk of developing multiple sclerosis in individuals seronegative for Epstein-Barr virus: A meta-analysis. Mult. Scler. J. 2013, 19, 162–166. [Google Scholar] [CrossRef]
- Santiago, O.; Gutierrez, J.; Sorlozano, A.; de Dios Luna, J.; Villegas, E.; Fernandez, O. Relation between Epstein-Barr virus and multiple sclerosis: Analytic study of scientific production. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2010, 29, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Hassani, A.; Corboy, J.R.; Al-Salam, S.; Khan, G. Epstein-Barr virus is present in the brain of most cases of multiple sclerosis and may engage more than just B cells. PLoS ONE 2018, 13, e0192109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giess, R.M.; Pfuhl, C.; Behrens, J.R.; Rasche, L.; Freitag, E.; Khalighy, N.; Otto, C.; Wuerfel, J.; Brandt, A.U.; Hofmann, J.; et al. Epstein-Barr virus antibodies in serum and DNA load in saliva are not associated with radiological or clinical disease activity in patients with early multiple sclerosis. PLoS ONE 2017, 12, e0175279. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, K.; Wildemann, B.; Jarius, S. Low intrathecal antibody production despite high seroprevalence of Epstein-Barr virus in multiple sclerosis: A review of the literature. J. Neurol. 2018, 265, 239–252. [Google Scholar] [CrossRef]
- Lang, H.L.; Jacobsen, H.; Ikemizu, S.; Andersson, C.; Harlos, K.; Madsen, L.; Hjorth, P.; Sondergaard, L.; Svejgaard, A.; Wucherpfennig, K. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat. Immunol. 2002, 3, 940–943. [Google Scholar] [CrossRef]
- van Noort, J.M.; Bajramovic, J.J.; Plomp, A.C.; van Stipdonk, M.J. Mistaken self, a novel model that links microbial infections with myelin-directed autoimmunity in multiple sclerosis. J. Neuroimmunol. 2000, 105, 46–57. [Google Scholar] [CrossRef]
- Pouly, S.; Antel, J.P. Multiple sclerosis and central nervous system demyelination. J. Autoimmun. 1999, 13, 297–306. [Google Scholar] [CrossRef]
- Lindsey, J.W. Antibodies to the Epstein-Barr virus proteins BFRF3 and BRRF2 cross-react with human proteins. J. Neuroimmunol. 2017, 310, 131–134. [Google Scholar] [CrossRef]
- Simpson, S., Jr.; Taylor, B.; Burrows, J.; Burrows, S.; Dwyer, D.E.; Taylor, J.; Ponsonby, A.L.; Blizzard, L.; Dwyer, T.; Pittas, F.; et al. EBV & HHV6 reactivation is infrequent and not associated with MS clinical course. Acta Neurol. Scand. 2014, 130, 328–337. [Google Scholar] [CrossRef]
- Kaiser, C.; Laux, G.; Eick, D.; Jochner, N.; Bornkamm, G.W.; Kempkes, B. The proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2. J. Virol. 1999, 73, 4481–4484. [Google Scholar] [CrossRef] [Green Version]
- Nikitin, P.A.; Yan, C.M.; Forte, E.; Bocedi, A.; Tourigny, J.P.; White, R.E.; Allday, M.J.; Patel, A.; Dave, S.S.; Kim, W. An ATM/Chk2-mediated DNA damage-responsive signaling pathway suppresses Epstein-Barr virus transformation of primary human B cells. Cell Host Microbe 2010, 8, 510–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.I. A region of herpes simplex virus VP16 can substitute for a transforming domain of Epstein-Barr virus nuclear protein 2. Proc. Natl. Acad. Sci. USA 1992, 89, 8030–8034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Grossman, S.R.; Kieff, E. Epstein–Barr virus nuclear protein 2 interacts with p300, CBP, and PCAF histone acetyltransferases in activation of the LMP1 promoter. Proc. Natl. Acad. Sci. USA 2000, 97, 430–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricigliano, V.A.; Handel, A.E.; Sandve, G.K.; Annibali, V.; Ristori, G.; Mechelli, R.; Cader, M.Z.; Salvetti, M. EBNA2 binds to genomic intervals associated with multiple sclerosis and overlaps with vitamin D receptor occupancy. PLoS ONE 2015, 10, e0119605. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Zou, J.; Wang, H.; Johannsen, E.; Peng, C.-w.; Quackenbush, J.; Mar, J.C.; Morton, C.C.; Freedman, M.L.; Blacklow, S.C. Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proc. Natl. Acad. Sci. USA 2011, 108, 14902–14907. [Google Scholar] [CrossRef] [Green Version]
- Sintzel, M.B.; Rametta, M.; Reder, A.T. Vitamin D and multiple sclerosis: A comprehensive review. Neurol. Ther. 2018, 7, 59–85. [Google Scholar] [CrossRef] [Green Version]
- Harley, J.B.; Chen, X.; Pujato, M.; Miller, D.; Maddox, A.; Forney, C.; Magnusen, A.F.; Lynch, A.; Chetal, K.; Yukawa, M.; et al. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat. Genet. 2018, 50, 699–707. [Google Scholar] [CrossRef]
- Fox, D.M.; Mundinano, I.C.; Bourne, J.A. Prehensile kinematics of the marmoset monkey: Implications for the evolution of visually-guided behaviors. J. Comp. Neurol. 2019, 527, 1495–1507. [Google Scholar] [CrossRef]
- Ehlers, B.; Spieß, K.; Leendertz, F.; Peeters, M.; Boesch, C.; Gatherer, D.; McGeoch, D.J. Lymphocryptovirus phylogeny and the origins of Epstein-Barr virus. J. Gen. Virol. 2009, 91, 630–642. [Google Scholar] [CrossRef]
- Jagessar, S.A.; Fagrouch, Z.; Heijmans, N.; Bauer, J.; Laman, J.D.; Oh, L.; Migone, T.; Verschoor, E.J.; Bert, A. The different clinical effects of anti-BLyS, anti-APRIL and anti-CD20 antibodies point at a critical pathogenic role of γ-herpesvirus infected B cells in the marmoset EAE model. J. Neuroimmune Pharmacol. 2013, 8, 727–738. [Google Scholar] [CrossRef]
- Pender, M.P.; Csurhes, P.A.; Smith, C.; Douglas, N.L.; Neller, M.A.; Matthews, K.K.; Beagley, L.; Rehan, S.; Crooks, P.; Hopkins, T.J.; et al. Epstein-Barr virus-specific T cell therapy for progressive multiple sclerosis. JCI Insight 2018, 3, e124714. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.S.; Koralnik, I.J. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: Clinical features and pathogenesis. Lancet Neurol. 2010, 9, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Del Valle, L.; White, M.K.; Khalili, K. Potential mechanisms of the human polyomavirus JC in neural oncogenesis. J. Neuropathol. Exp. Neurol. 2008, 67, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Meneguzzi, G.; Pignatti, P.F.; Barbanti-Brodano, G.; Milanesi, G. Minichromosome from BK virus as a template for transcription in vitro. Proc. Natl. Acad. Sci. USA 1978, 75, 1126–1130. [Google Scholar] [CrossRef] [Green Version]
- Khalili, K.; Gordon, J.; White, M.K. The polyomavirus, JCV, and its involvement in human disease. In Polyomaviruses and Human Diseases; Springer: Berlin/Heidelberg, Germany, 2006; pp. 274–287. [Google Scholar]
- Walker, D.L. The epidemiology of human polyomaviruses. Prog. Clin. Biol. Res. 1983, 105, 99–106. [Google Scholar]
- Chang, H.; Wang, M.; Tsai, R.-T.; Lin, H.-S.; Huan, J.-S.; Wang, W.-C.; Chang, D. High incidence of JC viruria in JC-seropositive older individuals. J. Neurovirol. 2002, 8, 447–451. [Google Scholar] [CrossRef]
- Beltrami, S.; Gordon, J. Immune surveillance and response to JC virus infection and PML. J. Neurovirol. 2014, 20, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Saribaş, A.S.; Özdemir, A.; Lam, C.; Safak, M. JC virus-induced progressive multifocal leukoencephalopathy. Future Virol. 2010, 5, 313–323. [Google Scholar] [CrossRef] [Green Version]
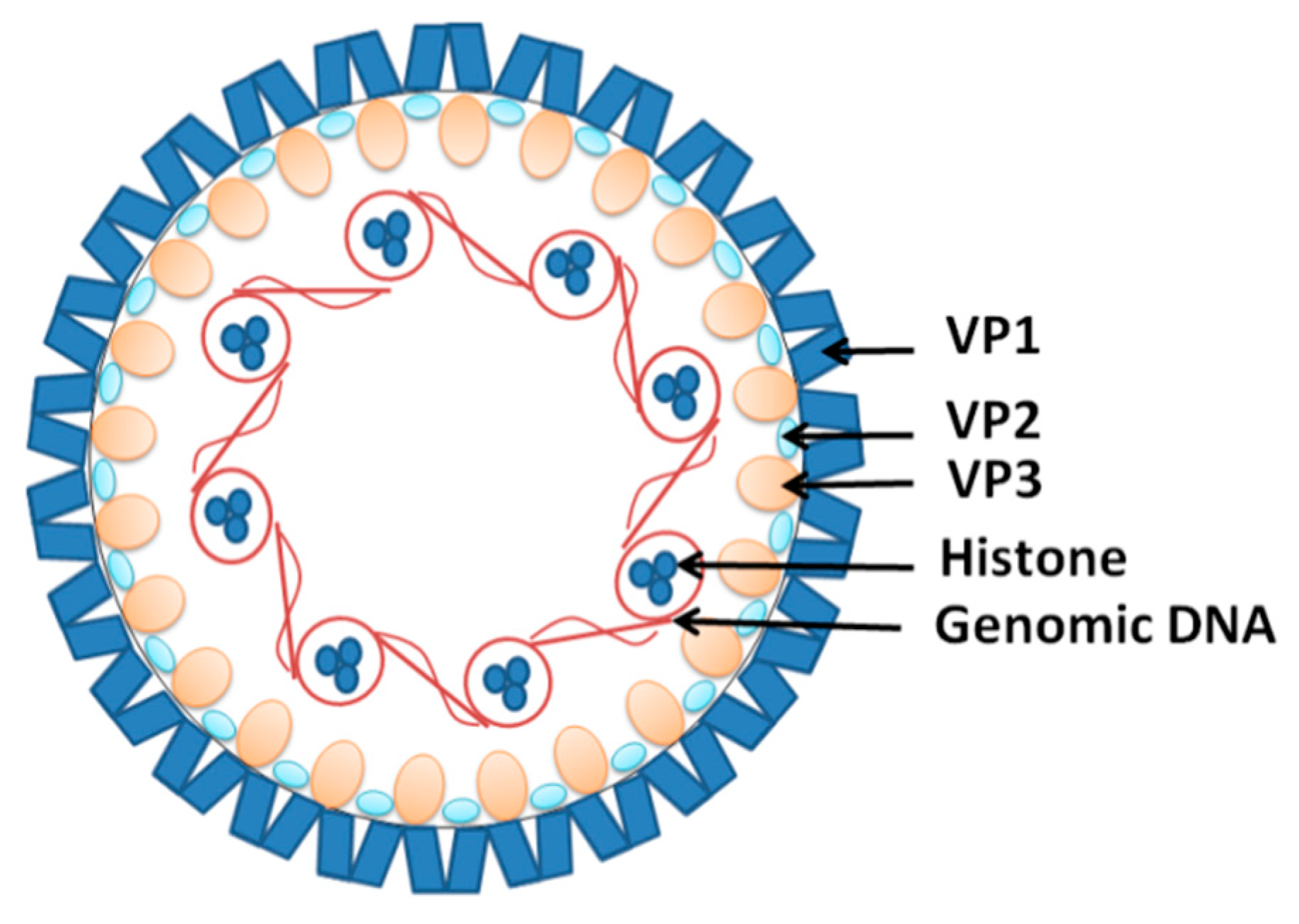
- Gorelik, L.; Reid, C.; Testa, M.; Brickelmaier, M.; Bossolasco, S.; Pazzi, A.; Bestetti, A.; Carmillo, P.; Wilson, E.; McAuliffe, M. Progressive multifocal leukoencephalopathy (PML) development is associated with mutations in JC virus capsid protein VP1 that change its receptor specificity. J. Infect. Dis. 2011, 204, 103–114. [Google Scholar] [CrossRef]
- Sabath, B.F.; Major, E.O. Traffic of JC virus from sites of initial infection to the brain: The path to progressive multifocal leukoencephalopathy. J. Infect. Dis. 2002, 186, S180–S186. [Google Scholar] [CrossRef] [Green Version]
- Kleinschmidt-DeMasters, B.; Tyler, K.L. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N. Engl. J. Med. 2005, 353, 369–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warnke, C.; Menge, T.; Hartung, H.-P.; Racke, M.K.; Cravens, P.D.; Bennett, J.L.; Frohman, E.M.; Greenberg, B.M.; Zamvil, S.S.; Gold, R. Natalizumab and progressive multifocal leukoencephalopathy: What are the causal factors and can it be avoided? Arch. Neurol. 2010, 67, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stüve, O.; Marra, C.M.; Jerome, K.R.; Cook, L.; Cravens, P.D.; Cepok, S.; Frohman, E.M.; Phillips, J.T.; Arendt, G.; Hemmer, B. Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2006, 59, 743–747. [Google Scholar]
- Assetta, B.; Atwood, W.J. The biology of JC polyomavirus. Biol. Chem. 2017, 398, 839–855. [Google Scholar] [CrossRef]
- Lander, E.; Linton, L.; Birren, B.; Nusbaum, C.; Zody, M.; Baldwin, J. Initial sequencing and analysis of the human genome. In Nature [Internet]; Nature Publishing Group: Berlin, Germany, 2001. [Google Scholar]
- Jern, P.; Coffin, J.M. Effects of retroviruses on host genome function. Annu. Rev. Genet. 2008, 42, 709–732. [Google Scholar] [CrossRef] [Green Version]
- Medstrand, P.; Landry, J.-R.; Mager, D.L. Long terminal repeats are used as alternative promoters for the endothelin B receptor and apolipoprotein CI genes in humans. J. Biol. Chem. 2001, 276, 1896–1903. [Google Scholar] [CrossRef] [Green Version]
- Ariza, M.-E.; Williams, M.V. A human endogenous retrovirus K dUTPase triggers a TH1, TH17 cytokine response: Does it have a role in psoriasis? J. Investig. Dermatol. 2011, 131, 2419–2427. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Miyagawa, K.; Chen, S.-Y.; Tamosiuniene, R.; Wang, L.; Sharpe, O.; Samayoa, E.; Harada, D.; Moonen, J.-R.A.; Cao, A. Upregulation of human endogenous retrovirus-K is linked to immunity and inflammation in pulmonary arterial hypertension. Circulation 2017, 136, 1920–1935. [Google Scholar] [CrossRef]
- Grandi, N.; Tramontano, E. Type W human endogenous retrovirus (HERV-W) integrations and their mobilization by L1 machinery: Contribution to the human transcriptome and impact on the host physiopathology. Viruses 2017, 9, 162. [Google Scholar] [CrossRef] [Green Version]
- Vargiu, L.; Rodriguez-Tomé, P.; Sperber, G.O.; Cadeddu, M.; Grandi, N.; Blikstad, V.; Tramontano, E.; Blomberg, J. Classification and characterization of human endogenous retroviruses; mosaic forms are common. Retrovirology 2016, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Christensen, T.; Sørensen, P.D.; Riemann, H.; Hansen, H.; Møller-Larsen, A. Expression of sequence variants of endogenous retrovirus RGH in particle form in multiple sclerosis. Lancet 1998, 352, 1033. [Google Scholar] [CrossRef]
- Perron, H.; Garson, J.; Bedin, F.; Beseme, F.; Paranhos-Baccala, G.; Komurian-Pradel, F.; Mallet, F.; Tuke, P.; Voisset, C.; Blond, J. Molecular identification of a novel retrovirus repeatedly isolated from patients with multiple sclerosis. Proc. Natl. Acad. Sci. USA 1997, 94, 7583–7588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, J.B.; Silva, C.; Holden, J.; Warren, K.G.; Clark, A.W.; Power, C. Monocyte activation and differentiation augment human endogenous retrovirus expression: Implications for inflammatory brain diseases. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2001, 50, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Perron, H.; Lazarini, F.; Ruprecht, K.; Péchoux-Longin, C.; Seilhean, D.; Sazdovitch, V.; Créange, A.; Battail-Poirot, N.; Sibaï, G.; Santoro, L. Human endogenous retrovirus (HERV)-W ENV and GAG proteins: Physiological expression in human brain and pathophysiological modulation in multiple sclerosis lesions. J. Neurovirol. 2005, 11, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Mameli, G.; Astone, V.; Arru, G.; Marconi, S.; Lovato, L.; Serra, C.; Sotgiu, S.; Bonetti, B.; Dolei, A. Brains and peripheral blood mononuclear cells of multiple sclerosis (MS) patients hyperexpress MS-associated retrovirus/HERV-W endogenous retrovirus, but not Human herpesvirus 6. J. Gen. Virol. 2007, 88, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Garson, J.; Tuke, P.; Giraud, P.; Paranhos-Baccala, G.; Perron, H. Detection of virion-associated MSRV-RNA in serum of patients with multiple sclerosis. Lancet 1998, 351, 33. [Google Scholar] [CrossRef]
- Serra, C.; Sotgiu, S.; Mameli, G.; Pugliatti, M.; Rosati, G.; Dolei, A. Multiple sclerosis and multiple sclerosis-associated retrovirus in Sardinia. Neurol. Sci. 2001, 22, 171–173. [Google Scholar] [CrossRef]
- Tarlinton, R.; Wang, B.; Morandi, E.; Gran, B.; Khaiboullin, T.; Martynova, E.; Rizvanov, A.; Khaiboullina, S. Differential Expression of HERV-W in Peripheral Blood in Multiple Sclerosis and Healthy Patients in Two Different Ethnic Groups. Front. Pharmacol. 2020, 10, 1645. [Google Scholar] [CrossRef] [Green Version]
- Sotgiu, S.; Arru, G.; Mameli, G.; Serra, C.; Pugliatti, M.; Rosati, G.; Dolei, A. Multiple sclerosis-associated retrovirus in early multiple sclerosis: A six-year follow-up of a Sardinian cohort. Mult. Scler. J. 2006, 12, 698–703. [Google Scholar] [CrossRef]
- Garcia-Montojo, M.; Dominguez-Mozo, M.; Arias-Leal, A.; Garcia-Martinez, Á.; De las Heras, V.; Casanova, I.; Faucard, R.; Gehin, N.; Madeira, A.; Arroyo, R. The DNA copy number of human endogenous retrovirus-W (MSRV-type) is increased in multiple sclerosis patients and is influenced by gender and disease severity. PLoS ONE 2013, 8, e53623. [Google Scholar] [CrossRef]
- García-Montojo, M.; de la Hera, B.; Varadé, J.; de la Encarnación, A.; Camacho, I.; Domínguez-Mozo, M.; Arias-Leal, A.; García-Martínez, Á.; Casanova, I.; Izquierdo, G. HERV-W polymorphism in chromosome X is associated with multiple sclerosis risk and with differential expression of MSRV. Retrovirology 2014, 11, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolei, A.; Serra, C.; Mameli, G.; Pugliatti, M.; Sechi, G.; Cirotto, M.; Rosati, G.; Sotgiu, S. Multiple sclerosis–associated retrovirus (MSRV) in Sardinian MS patients. Neurology 2002, 58, 471–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brudek, T.; Christensen, T.; Aagaard, L.; Petersen, T.; Hansen, H.J.; Møller-Larsen, A. B cells and monocytes from patients with active multiple sclerosis exhibit increased surface expression of both HERV-H Env and HERV-W Env, accompanied by increased seroreactivity. Retrovirology 2009, 6, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firouzi, R.; Rolland, A.; Michel, M.; Jouvin-Marche, E.; Hauw, J.; Malcus-Vocanson, C.; Lazarini, F.; Gebuhrer, L.; Seigneurin, J.; Touraine, J. Multiple sclerosis-associated retrovirus particles cause T lymphocyte-dependent death with brain hemorrhage in humanized SCID mice model. J. Neurovirol. 2003, 9, 79–93. [Google Scholar] [CrossRef] [PubMed]
- de Luca, V.; Higa, A.M.; Romano, C.M.; Mambrini, G.P.; Peroni, L.A.; Trivinho-Strixino, F.; Leite, F.L. Cross-reactivity between myelin oligodendrocyte glycoprotein and human endogenous retrovirus W protein: Nanotechnological evidence for the potential trigger of multiple sclerosis. Micron 2019, 120, 66–73. [Google Scholar] [CrossRef]
- Ramasamy, R.; Mohammed, F.; Meier, U.C. HLA DR2b-binding peptides from human endogenous retrovirus envelope, Epstein-Barr virus and brain proteins in the context of molecular mimicry in multiple sclerosis. Immunol. Lett. 2020, 217, 15–24. [Google Scholar] [CrossRef]
- Olival, G.S.d.; Faria, T.S.; Nali, L.H.; de Oliveira, A.C.; Casseb, J.; Vidal, J.E.; Cavenaghi, V.B.; Tilbery, C.P.; Moraes, L.; Fink, M.C.; et al. Genomic analysis of ERVWE2 locus in patients with multiple sclerosis: Absence of genetic association but potential role of human endogenous retrovirus type W elements in molecular mimicry with myelin antigen. Front. Microbiol. 2013, 4, 172. [Google Scholar] [CrossRef] [Green Version]
- Ramasamy, R.; Joseph, B.; Whittall, T. Potential molecular mimicry between the human endogenous retrovirus W family envelope proteins and myelin proteins in multiple sclerosis. Immunol. Lett. 2017, 183, 79–85. [Google Scholar] [CrossRef]
- Tu, X.; Li, S.; Zhao, L.; Xiao, R.; Wang, X.; Zhu, F. Human leukemia antigen-A* 0201-restricted epitopes of human endogenous retrovirus W family envelope (HERV-W env) induce strong cytotoxic T lymphocyte responses. Virol. Sin. 2017, 32, 280–289. [Google Scholar] [CrossRef]
- Rolland, A.; Jouvin-Marche, E.; Viret, C.; Faure, M.; Perron, H.; Marche, P.N. The envelope protein of a human endogenous retrovirus-W family activates innate immunity through CD14/TLR4 and promotes Th1-like responses. J. Immunol. 2006, 176, 7636–7644. [Google Scholar] [CrossRef]
- Duperray, A.; Barbe, D.; Raguenez, G.; Weksler, B.B.; Romero, I.A.; Couraud, P.O.; Perron, H.; Marche, P.N. Inflammatory response of endothelial cells to a human endogenous retrovirus associated with multiple sclerosis is mediated by TLR4. Int. Immunol. 2015, 27, 545–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perron, H.; Dougier-Reynaud, H.L.; Lomparski, C.; Popa, I.; Firouzi, R.; Bertrand, J.B.; Marusic, S.; Portoukalian, J.; Jouvin-Marche, E.; Villiers, C.L.; et al. Human endogenous retrovirus protein activates innate immunity and promotes experimental allergic encephalomyelitis in mice. PLoS ONE 2013, 8, e80128. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Montojo, M.; Rodriguez-Martin, E.; Ramos-Mozo, P.; Ortega-Madueño, I.; Dominguez-Mozo, M.I.; Arias-Leal, A.; García-Martínez, M.; Casanova, I.; Galan, V.; Arroyo, R.; et al. Syncytin-1/HERV-W envelope is an early activation marker of leukocytes and is upregulated in multiple sclerosis patients. Eur. J. Immunol. 2020, 50, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Gjelstrup, M.C.; Stilund, M.; Petersen, T.; Moller, H.J.; Petersen, E.L.; Christensen, T. Subsets of activated monocytes and markers of inflammation in incipient and progressed multiple sclerosis. Immunol. Cell Biol. 2018, 96, 160–174. [Google Scholar] [CrossRef] [Green Version]
- Carstensen, M.; Christensen, T.; Stilund, M.; Møller, H.J.; Petersen, E.L.; Petersen, T. Activated monocytes and markers of inflammation in newly diagnosed multiple sclerosis. Immunol. Cell Biol. 2020. [Google Scholar] [CrossRef]
- Azébi, S.; Batsché, E.; Michel, F.; Kornobis, E.; Muchardt, C. Expression of endogenous retroviruses reflects increased usage of atypical enhancers in T cells. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Kouwenhoven, M.; Teleshova, N.; Ozenci, V.; Press, R.; Link, H. Monocytes in multiple sclerosis: Phenotype and cytokine profile. J. Neuroimmunol. 2001, 112, 197–205. [Google Scholar] [CrossRef]
- Ruprecht, K.; Mayer, J. On the origin of a pathogenic HERV-W envelope protein present in multiple sclerosis lesions. Proc. Natl. Acad. Sci. USA 2019, 116, 19791–19792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremer, D.; Forster, M.; Schichel, T.; Gottle, P.; Hartung, H.P.; Perron, H.; Kury, P. The neutralizing antibody GNbAC1 abrogates HERV-W envelope protein-mediated oligodendroglial maturation blockade. Mult. Scler. 2014, 21, 1200–1203. [Google Scholar] [CrossRef]
- Diebold, M.; Derfuss, T. The monoclonal antibody GNbAC1: Targeting human endogenous retroviruses in multiple sclerosis. Adv. Neurol. Disord. 2019, 12, 1756286419833574. [Google Scholar] [CrossRef] [Green Version]
- Kremer, D.; Gruchot, J.; Weyers, V.; Oldemeier, L.; Göttle, P.; Healy, L.; Ho Jang, J.; Kang, T.X.Y.; Volsko, C.; Dutta, R.; et al. pHERV-W envelope protein fuels microglial cell-dependent damage of myelinated axons in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 15216–15225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irizar, H.; Muñoz-Culla, M.; Sepúlveda, L.; Sáenz-Cuesta, M.; Prada, Á.; Castillo-Triviño, T.; Zamora-López, G.; de Munain, A.L.; Olascoaga, J.; Otaegui, D. Transcriptomic profile reveals gender-specific molecular mechanisms driving multiple sclerosis progression. PLoS ONE 2014, 9, e90482. [Google Scholar] [CrossRef] [PubMed]
- Morandi, E.; Tanasescu, R.; Tarlinton, R.E.; Constantin-Teodosiu, D.; Gran, B. Do Antiretroviral Drugs Protect From Multiple Sclerosis by Inhibiting Expression of MS-Associated Retrovirus? Front. Immunol. 2018, 9, 3092. [Google Scholar] [CrossRef] [PubMed]
- Mameli, G.; Madeddu, G.; Mei, A.; Uleri, E.; Poddighe, L.; Delogu, L.G.; Maida, I.; Babudieri, S.; Serra, C.; Manetti, R.; et al. Activation of MSRV-type endogenous retroviruses during infectious mononucleosis and Epstein-Barr virus latency: The missing link with multiple sclerosis? PLoS ONE 2013, 8, e78474. [Google Scholar] [CrossRef] [PubMed]
- Charvet, B.; Reynaud, J.M.; Gourru-Lesimple, G.; Perron, H.; Marche, P.N.; Horvat, B. Induction of Proinflammatory Multiple Sclerosis-Associated Retrovirus Envelope Protein by Human Herpesvirus-6A and CD46 Receptor Engagement. Front. Immunol. 2018, 9, 2803. [Google Scholar] [CrossRef]
- Ruprecht, K.; Obojes, K.; Wengel, V.; Gronen, F.; Kim, K.S.; Perron, H.; Schneider-Schaulies, J.; Rieckmann, P. Regulation of human endogenous retrovirus W protein expression by herpes simplex virus type 1: Implications for multiple sclerosis. J. Neurovirol. 2006, 12, 65–71. [Google Scholar] [CrossRef]
- Nellaker, C.; Yao, Y.; Jones-Brando, L.; Mallet, F.; Yolken, R.H.; Karlsson, H. Transactivation of elements in the human endogenous retrovirus W family by viral infection. Retrovirology 2006, 3, 44. [Google Scholar] [CrossRef]
- Lee, W.J.; Kwun, H.J.; Kim, H.S.; Jang, K.L. Activation of the human endogenous retrovirus W long terminal repeat by herpes simplex virus type 1 immediate early protein 1. Mol. Cells 2003, 15, 75–80. [Google Scholar]
- Brudek, T.; Luhdorf, P.; Christensen, T.; Hansen, H.J.; Moller-Larsen, A. Activation of endogenous retrovirus reverse transcriptase in multiple sclerosis patient lymphocytes by inactivated HSV-1, HHV-6 and VZV. J. Neuroimmunol. 2007, 187, 147–155. [Google Scholar] [CrossRef]
- Fox, E.J.; Buckle, G.J.; Singer, B.; Singh, V.; Boster, A. Lymphopenia and DMTs for relapsing forms of MS: Considerations for the treating neurologist. Neurol. Clin. Pract. 2019, 9, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wootla, B.; Watzlawik, J.O.; Stavropoulos, N.; Wittenberg, N.J.; Dasari, H.; Abdelrahim, M.A.; Henley, J.R.; Oh, S.-H.; Warrington, A.E.; Rodriguez, M. Recent advances in monoclonal antibody therapies for multiple sclerosis. Expert Opin. Biol. Ther. 2016, 16, 827–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossowski, A.; Urban, M.; Stasiak-Barmuta, A.; Turowski, D. Expression of very late antigen-4 and lymphocyte function-associated antigen-1 on peripheral blood lymphocytes from patients with graves disease. Pediatric Res. 2002, 52, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Yaldizli, Ö.; Putzki, N. Natalizumab in the treatment of multiple sclerosis. Ther. Adv. Neurol. Disord. 2009, 2, 115–128. [Google Scholar] [CrossRef] [Green Version]
- Yednock, T.A.; Cannon, C.; Fritz, L.C.; Sanchez-Madrid, F.; Steinman, L.; Karin, N. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4βl integrin. Nature 1992, 356, 63–66. [Google Scholar] [CrossRef]
- Burkly, L.C.; Jakubowski, A.; Newman, B.M.; Rosa, M.D.; Chi-Rosso, G.; Lobb, R.R. Signaling by vascular cell adhesion molecule-1 (VCAM-1) through VLA-4 promotes CD3-dependent T cell proliferation. Eur. J. Immunol. 1991, 21, 2871–2875. [Google Scholar] [CrossRef]
- Damle, N.K.; Aruffo, A. Vascular cell adhesion molecule 1 induces T-cell antigen receptor-dependent activation of CD4+ T lymphocytes. Proc. Natl. Acad. Sci. USA 1991, 88, 6403–6407. [Google Scholar] [CrossRef] [Green Version]
- Brandstadter, R.; Sand, I.K. The use of natalizumab for multiple sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1691–1702. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.P.; Sancho, J.; Campos-Rivera, J.; Boutin, P.M.; Severy, P.B.; Weeden, T.; Shankara, S.; Roberts, B.L.; Kaplan, J.M. Human peripheral blood mononuclear cells exhibit heterogeneous CD52 expression levels and show differential sensitivity to alemtuzumab mediated cytolysis. PLoS ONE 2012, 7, e39416. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Turner, M.J.; Shields, J.; Gale, M.S.; Hutto, E.; Roberts, B.L.; Siders, W.M.; Kaplan, J.M. Investigation of the mechanism of action of alemtuzumab in a human CD52 transgenic mouse model. Immunology 2009, 128, 260–270. [Google Scholar] [CrossRef]
- Malmeström, C.; Andersson, B.A.; Lycke, J. First reported case of diabetes mellitus type 1 as a possible secondary autoimmune disease following alemtuzumab treatment in MS. J. Neurol. 2014, 261, 2016–2018. [Google Scholar] [CrossRef] [PubMed]
- OCREVUS. (Ocrelizumab). Prescribing Information. Available online: https://www.ocrevus.com/ (accessed on 12 June 2020).




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarlinton, R.E.; Martynova, E.; Rizvanov, A.A.; Khaiboullina, S.; Verma, S. Role of Viruses in the Pathogenesis of Multiple Sclerosis. Viruses 2020, 12, 643. https://doi.org/10.3390/v12060643
Tarlinton RE, Martynova E, Rizvanov AA, Khaiboullina S, Verma S. Role of Viruses in the Pathogenesis of Multiple Sclerosis. Viruses. 2020; 12(6):643. https://doi.org/10.3390/v12060643
Chicago/Turabian StyleTarlinton, Rachael E., Ekaterina Martynova, Albert A. Rizvanov, Svetlana Khaiboullina, and Subhash Verma. 2020. "Role of Viruses in the Pathogenesis of Multiple Sclerosis" Viruses 12, no. 6: 643. https://doi.org/10.3390/v12060643
APA StyleTarlinton, R. E., Martynova, E., Rizvanov, A. A., Khaiboullina, S., & Verma, S. (2020). Role of Viruses in the Pathogenesis of Multiple Sclerosis. Viruses, 12(6), 643. https://doi.org/10.3390/v12060643