Tiotropium Is Predicted to Be a Promising Drug for COVID-19 Through Transcriptome-Based Comprehensive Molecular Pathway Analysis

Abstract
:1. Introduction
2. Materials and Methods
2.1. RNA-seq Data Analysis
2.2. Network Analysis
2.3. Gene Set Enrichment Analysis
2.4. Transcription Factor Binding Motif Prediction
2.5. Gene Ontology Analysis
2.6. Visualization
3. Results
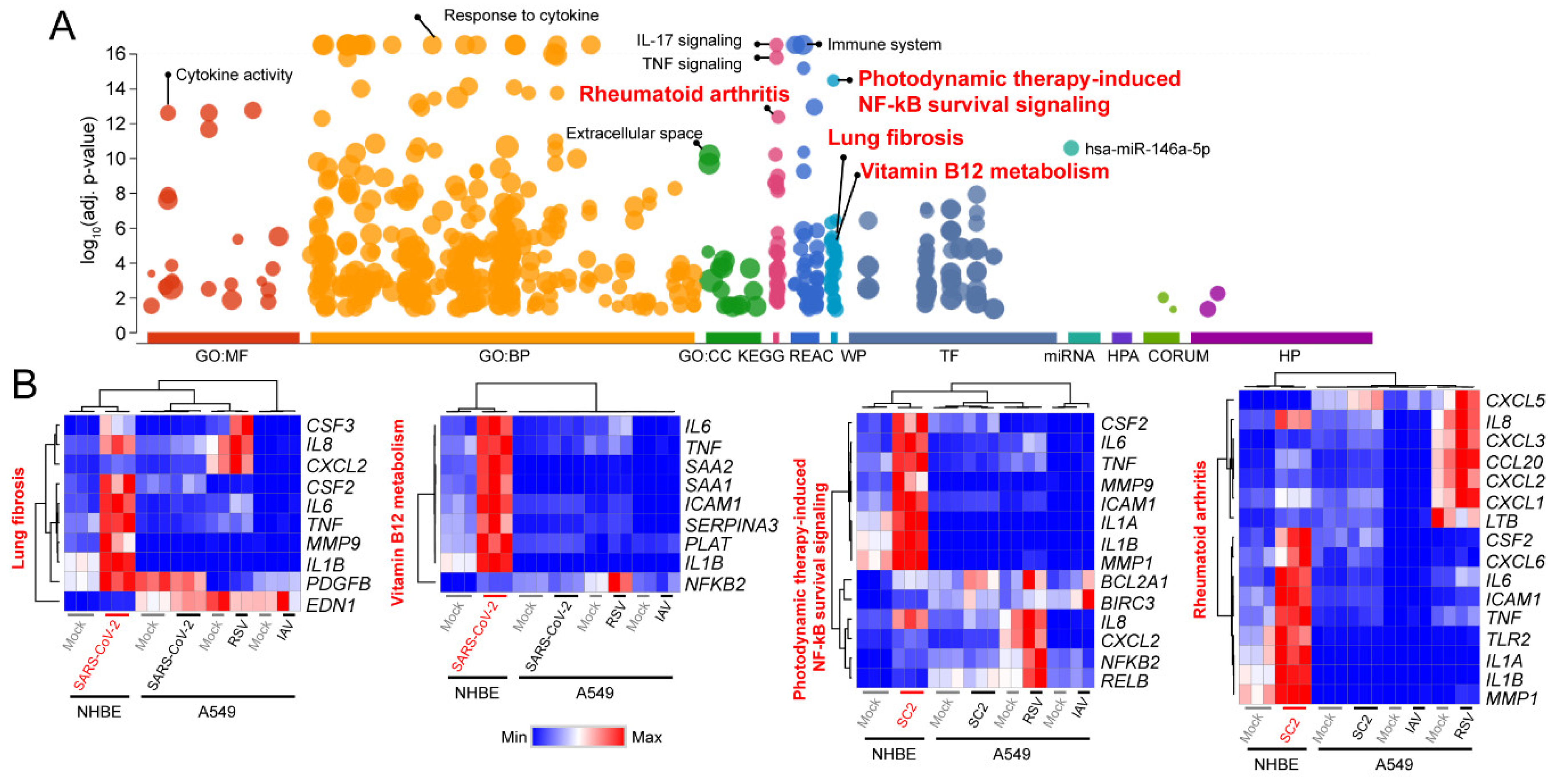
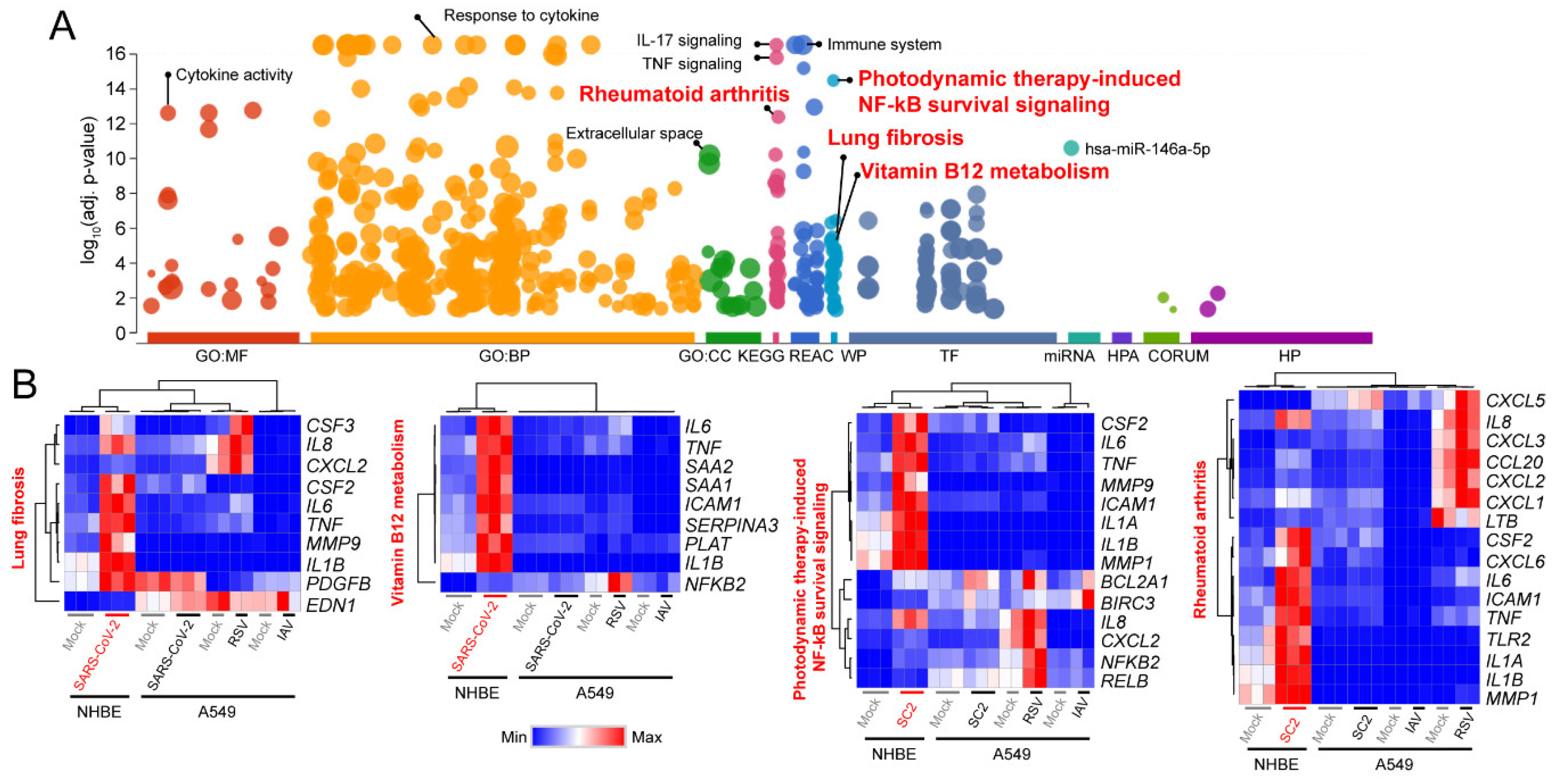
3.1. Signaling Pathways Upregulated by SARS-CoV-2 Infection in Normal Human Bronchial Epithelial Cells
3.2. Decoding Upregulated Signaling Pathways Caused by SARS-CoV-2
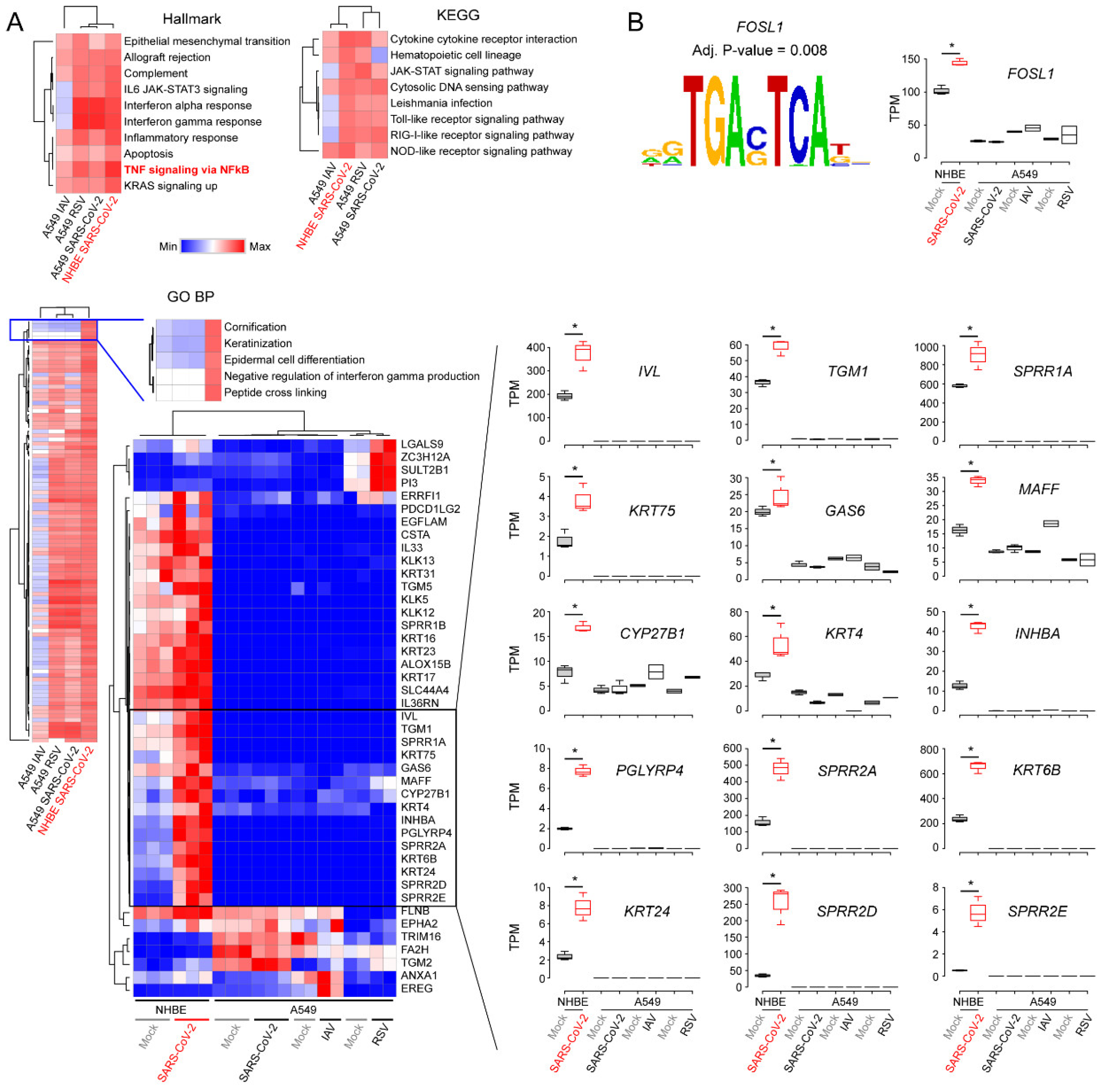
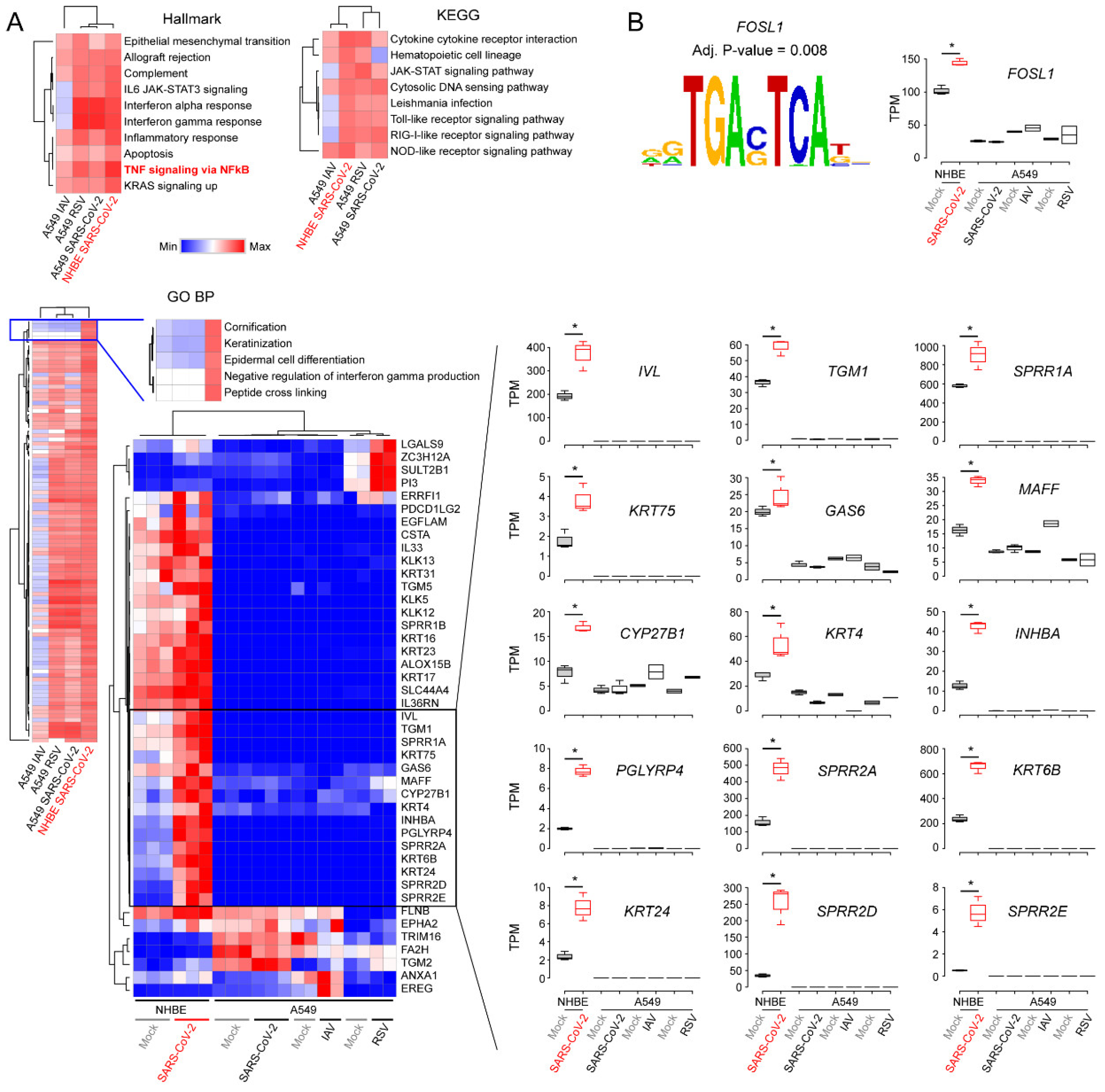
3.3. Comparison of Transcriptomic Changes Caused by SARS-CoV-2, Respiratory Syncytial Virus and Influenza A Virus
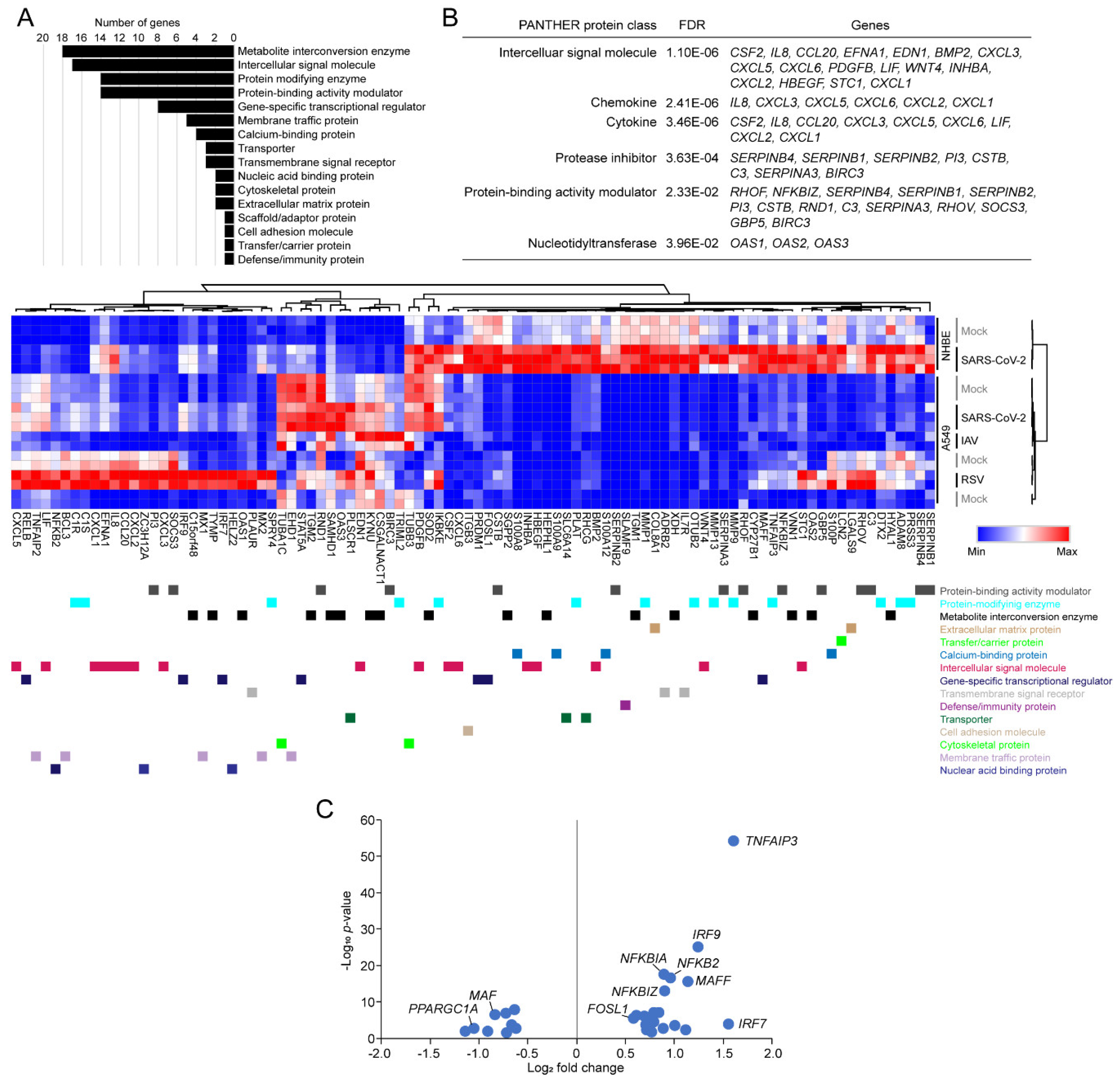
3.4. Functional Classification of SARS-CoV-2-Activating Genes
3.5. Tiotropium as a Promising Drug Candidate for COVID-19
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Taubenberger, J.K.; Morens, D.M. Influenza: The mother of all pandemics. Emerg. Infect Dis. 2006, 12, 15–22. [Google Scholar] [CrossRef]
- Lin, L.; Lu, L.; Cao, W.; Li, T. Hypothesis for potential pathogenesis of SARS-CoV-2 infection–A review of immune changes in patients with viral pneumonia. Emerg. Microbes Infect. 2020, 9, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and Clinical Characteristics of 99 Cases of 2019-Novel Coronavirus (2019-nCoV) Pneumonia in Wuhan, China. SSRN Electron. J. 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the Eye of the Cytokine Storm. Microbiol. Mol. Boil. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; HLH Across Speciality Collaboration. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- De Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.; Chau, T.N.B.; Hoang, D.M.; Chau, N.V.V.; Khanh, T.H.; Dong, V.C.; et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaparianos, A.; Argyropoulou, E. Local renin-angiotensin II systems, angiotensin-converting enzyme and its homologue ACE2: Their potential role in the pathogenesis of chronic obstructive pulmonary diseases, pulmonary hypertension and acute respiratory distress syndrome. Curr. Med. Chem. 2011, 18, 3506–3515. [Google Scholar] [CrossRef] [PubMed]
- Halpin, D.M.G.; Faner, R.; Sibila, O.; Badia, J.R.; Agusti, A. Do chronic respiratory diseases or their treatment affect the risk of SARS-CoV-2 infection? Lancet Respir. Med. 2020, 8, 436–438. [Google Scholar] [CrossRef]
- Finney, L.J.; Glanville, N.; Farne, H.; Aniscenko, J.; Fenwick, P.; Kemp, S.V.; Trujillo-Torralbo, M.-B.; Calderazzo, M.A.; Wedzicha, J.A.; Mallia, P.; et al. Inhaled corticosteroids downregulate the SARS-CoV-2 receptor ACE2 in COPD through suppression of type I interferon. bioRxiv 2020. [Google Scholar] [CrossRef]
- Iqbal, A.; Barnes, N.; Brooks, J. Is Blood Eosinophil Count a Predictor of Response to Bronchodilators in Chronic Obstructive Pulmonary Disease? Results from Post Hoc Subgroup Analyses. Clin. Drug Investig. 2015, 35, 685–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decramer, M.; Celli, B.; Kesten, S.; Lystig, T.; Mehra, S.; Tashkin, D.P. Effect of tiotropium on outcomes in patients with moderate chronic obstructive pulmonary disease (UPLIFT): A prespecified subgroup analysis of a randomised controlled trial. Lancet 2009, 374, 1171–1178. [Google Scholar] [CrossRef]
- Tashkin, D.; Celli, B.; Senn, S.; Burkhart, D.; Kesten, S.; Menjoge, S.; Decramer, M. A 4-Year Trial of Tiotropium in Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2008, 359, 1543–1554. [Google Scholar] [CrossRef] [Green Version]
- Halpin, D.M.G. Tiotropium in asthma: What is the evidence and how does it fit in? World Allergy Organ. J. 2016, 9, 29. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhong, N.-S.; Li, X.; Chen, S.; Zheng, J.P.; Zhao, D.; Yao, W.; Zhi, R.; Wei, L.; He, B.; et al. Tioropium in Early-Stage Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2017, 377, 923–935. [Google Scholar] [CrossRef]
- Buels, K.S.; Fryer, A.D. Muscarinic receptor antagonists: Effects on pulmonary function. Handb. Exp. Pharmacol. 2012, 208, 317–341. [Google Scholar] [CrossRef] [Green Version]
- Kerstjens, H.A.; O’Byrne, P.M. Tiotropium for the treatment of asthma: A drug safety evaluation. Expert Opin. Drug Saf. 2016, 15, 1115–1124. [Google Scholar] [CrossRef] [Green Version]
- Radovanovic, D.; Santus, P.; Blasi, F.; Mantero, M. The evidence on tiotropium bromide in asthma: From the rationale to the bedside. Multidiscip. Respir. Med. 2017, 12, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, B.R.; Shin, B.; Choi, Y.; Park, S.; Kang, K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput. Struct. Biotechnol. J. 2020, 18, 784–790. [Google Scholar] [CrossRef]
- Shin, B.; Park, S.; Kang, K.; Ho, J.C. Self-Attention Based Molecule Representation for Predicting Drug-Target Interaction. Proc. Mach. Learn. Res. 2019, 106, 230–248. [Google Scholar]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 002832. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Soufan, O.; Ewald, J.D.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [Green Version]
- Korotkevich, G.; Sukhov, V.; Sergushichev, A. Fast gene set enrichment analysis. bioRxiv 2019.
- Zambelli, F.; Pesole, G.; Pavesi, G. Pscan: Finding over-represented transcription factor binding site motifs in sequences from co-regulated or co-expressed genes. Nucleic Acids Res. 2009, 37, W247–W252. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A Library of Protein Families and Subfamilies Indexed by Function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.K.; Nandakumar, K.; Ross, A. Score normalization in multimodal biometric systems. Pattern Recognit. 2005, 38, 2270–2285. [Google Scholar] [CrossRef]
- Székely, G.J.; Rizzo, M.L. Hierarchical Clustering via Joint between-within Distances: Extending Ward’s Minimum Variance Method. J. Classif. 2005, 22, 151–183. [Google Scholar] [CrossRef]
- Datta, S.; Datta, S.J.B. Methods for evaluating clustering algorithms for gene expression data using a reference set of functional classes. BMC Bioinformatics 2006, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Litonjua, A.A.; Gong, L.; Duan, Q.L.; Shin, J.; Moore, M.J.; Weiss, S.T.; Johnson, J.A.; Klein, T.E.; Altman, R.B. Very important pharmacogene summary ADRB2. Pharmacogenetics Genom. 2010, 20, 64–69. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Correction to: Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensiv. Care Med. 2020, 46, 1294–1297. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Matthay, M.A.; Aldrich, J.M.; Gotts, J.E. Treatment for severe acute respiratory distress syndrome from COVID-19. Lancet Respir. Med. 2020, 8, 433–434. [Google Scholar] [CrossRef] [Green Version]
- Yamaya, M.; Nishimura, H.; Hatachi, Y.; Yasuda, H.; Deng, X.; Sasaki, T.; Kubo, H.; Nagatomi, R. Inhibitory effects of tiotropium on rhinovirus infection in human airway epithelial cells. Eur. Respir. J. 2012, 40, 122–132. [Google Scholar] [CrossRef]
- Iesato, K.; Tatsumi, K.; Saito, K.; Ogasawara, T.; Sakao, S.; Tada, Y.; Kasahara, Y.; Kurosu, K.; Tanabe, N.; Takiguchi, Y.; et al. Tiotropium Bromide Attenuates Respiratory Syncytial Virus Replication in Epithelial Cells. Respir. 2008, 76, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Bucher, H.; Duechs, M.J.; Tilp, C.; Jung, B.; Erb, K.J. Tiotropium Attenuates Virus-Induced Pulmonary Inflammation in Cigarette Smoke-Exposed Mice. J. Pharmacol. Exp. Ther. 2016, 357, 606–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fielhaber, J.A.; Carroll, S.F.; Dydensborg, A.B.; Shourian, M.; Triantafillopoulos, A.; Harel, S.; Hussain, S.N.; Bouchard, M.; Qureshi, S.T.; Kristof, A.S. Inhibition of Mammalian Target of Rapamycin Augments Lipopolysaccharide-Induced Lung Injury and Apoptosis. J. Immunol. 2012, 188, 4535–4542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houssaini, A.; Breau, M.; Kebe, K.; Abid, S.; Marcos, E.; Lipskaia, L.; Rideau, D.; Parpaleix, A.; Huang, J.; Amsellem, V.; et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerstjens, H.A.; Engel, M.; Dahl, R.; Paggiaro, P.; Beck, E.; Vandewalker, M.; Sigmund, R.; Seibold, W.; Moroni-Zentgraf, P.; Bateman, E.D. Tiotropium in Asthma Poorly Controlled with Standard Combination Therapy. N. Engl. J. Med. 2012, 367, 1198–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yum, H.-K.; Park, I.-N. Effect of Inhaled Tiotropium on Spirometric Parameters in Patients with Tuberculous Destroyed Lung. Tuberc. Respir. Dis. 2014, 77, 167–171. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Xiaoyu, Z.; Zhixin, S.; Di, Q.; Xinyu, D.; Jing, X.; Jing, H.; Wang, D.; Xi, Z.; Chunrong, Z.; et al. Rapamycin attenuates acute lung injury induced by LPS through inhibition of Th17 cell proliferation in mice. Sci. Rep. 2016, 6, 20156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kindrachuk, J.; Ork, B.; Hart, B.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H.; et al. Antiviral Potential of ERK/MAPK and PI3K/AKT/mTOR Signaling Modulation for Middle East Respiratory Syndrome Coronavirus Infection as Identified by Temporal Kinome Analysis. Antimicrob. Agents Chemother. 2014, 59, 1088–1099. [Google Scholar] [CrossRef] [Green Version]
- Toumpanakis, D.; Loverdos, K.; Tzouda, V.; Vassilakopoulou, V.; Litsiou, E.; Magkou, C.; Karavana, V.; Pieper, M.; Vassilakopoulos, T. Tiotropium bromide exerts anti-inflammatory effects during resistive breathing, an experimental model of severe airway obstruction. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 2207–2220. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, G.; Gagliardo, R.; Bucchieri, F.; Albano, G.D.; Siena, L.; Montalbano, A.M.; Bonanno, A.; Riccobono, L.; Pieper, M.P.; Gjomarkaj, M.; et al. IL-17A induces chromatin remodeling promoting IL-8 release in bronchial epithelial cells: Effect of Tiotropium. Life Sci. 2016, 152, 107–116. [Google Scholar] [CrossRef]
- Asano, K.; Shikama, Y.; Shoji, N.; Hirano, K.; Suzaki, H.; Nakajima, H. Tiotropium bromide inhibits TGF-beta-induced MMP production from lung fibroblasts by interfering with Smad and MAPK pathways in vitro. Int. J. Chron. Obstruct. Pulmon. Dis. 2010, 5, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asano, K.; Suzaki, I.; Shikama, Y.; Hamasaki, T.; Kanei, A.; Suzaki, H. Suppression of IL-8 production from airway cells by tiotropium bromide in vitro. Int. J. Chronic Obstr. Pulm. Dis. 2011, 6, 439–448. [Google Scholar] [CrossRef] [Green Version]
- Vacca, G.; Randerath, W.; Gillissen, D.M.A. Inhibition of granulocyte migration by tiotropium bromide. Respir. Res. 2011, 12, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eraldemir, F.C.; Şengül, A.; Özkan, M.; Köktürk, S.; Özsoy, D.; Yildiz, F.A. The anti-inflammatory and anti-remodeling effect of tiotropium bromide in the subacute cigarette exposure mouse model. Int. J. Clin. Exp. Med. 2016, 9, 22824–22834. [Google Scholar]
- Kolahian, S.; Shahbazfar, A.A.; Tayefi-Nasrabadi, H.; Keyhanmanesh, R.; Ansarin, K.; Ghasemi, H.; Rashidi, A.H.; Gosens, R.; Hanifeh, M. Tiotropium effects on airway inflammatory events in the cat as an animal model for acute cigarette smoke-induced lung inflammation. Exp. Lung Res. 2014, 40, 272–287. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Lv, J.; Wu, Z.L.; Gan, Z.; Gui, P.; Yao, S. CXCL14 Overexpression Attenuates Sepsis-Associated Acute Kidney Injury by Inhibiting Proinflammatory Cytokine Production. Mediat. Inflamm. 2020, 2020, 2431705. [Google Scholar] [CrossRef]
- Babon, J.J.; Varghese, L.N.; Nicola, N.A. Inhibition of IL-6 family cytokines by SOCS3. Semin. Immunol. 2014, 26, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Ortega, M.; Marc, D.; Dupont, J.; Trapp, S.; Berri, M.; Meurens, F. SOCS proteins in infectious diseases of mammals. Veter- Immunol. Immunopathol. 2013, 151, 1–19. [Google Scholar] [CrossRef]
- Liu, S.; Yan, R.; Chen, B.; Pan, Q.; Chen, Y.; Hong, J.; Zhang, L.; Liu, W.; Wang, S.; Chen, J.-L. Influenza Virus-Induced Robust Expression of SOCS3 Contributes to Excessive Production of IL-6. Front. Immunol. 2019, 10, 1843. [Google Scholar] [CrossRef] [Green Version]
- Candi, E.; Schmidt, R.; Melino, G. The cornified envelope: A model of cell death in the skin. Nat. Rev. Mol. Cell Boil. 2005, 6, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Hackett, N.R.; Shaykhiev, R.; Walters, M.S.; Wang, R.; Zwick, R.K.; Ferris, B.; Witover, B.; Salit, J.; Crystal, R.G. The Human Airway Epithelial Basal Cell Transcriptome. PLoS ONE 2011, 6, e18378. [Google Scholar] [CrossRef] [PubMed]
- Eckhart, L.; Lippens, S.; Tschachler, E.; Declercq, W. Cell death by cornification. Biochim. Biophys. Acta (BBA)—Bioenerg. 2013, 1833, 3471–3480. [Google Scholar] [CrossRef]
- Lorand, L.; Graham, R.M. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Boil. 2003, 4, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Olsen, K.C.; Sapinoro, R.E.; Kottmann, R.M.; Kulkarni, A.A.; Iismaa, S.E.; Johnson, G.V.W.; Thatcher, T.H.; Phipps, R.P.; Sime, P.J. Transglutaminase 2 and Its Role in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 699–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Li, W.; Tong, Q.; Yi, J.; Zhao, L.; et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 Infected patients. EBioMedicine 2020, 55, 102763. [Google Scholar] [CrossRef]
- Araya, J.; Cambier, S.; Markovics, J.A.; Wolters, P.; Jablons, D.; Hill, A.; Finkbeiner, W.; Jones, K.; Broaddus, V.C.; Sheppard, D.; et al. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J. Clin. Investig. 2007, 117, 3551–3562. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Yu, H.; Gou, J.; Li, X.; Sun, Y.; Li, J.; Liu, L. Clinical Pathology of Critical Patient with Novel Coronavirus Pneumonia (COVID-19). Preprints 2020, 2020020407. [Google Scholar]
- Zhang, J.; Wu, L.; Qu, J.M.; Bai, C.X.; Merrilees, M.J.; Black, P.N. Pro-inflammatory phenotype of COPD fibroblasts not compatible with repair in COPD lung. J. Cell. Mol. Med. 2012, 16, 1522–1532. [Google Scholar] [CrossRef]
- Ahmed, S.S.; Paramasivam, P.; Raj, K. Regulatory cross talk between SARS-CoV-2 receptor binding and replication machinery in the human host. Front Physiol 2020, 11, 802. [Google Scholar] [CrossRef]
- Onder, G.; Rezza, G.; Brusaferro, S. Case-Fatality Rate and Characteristics of Patients Dying in Relation to COVID-19 in Italy. JAMA 2020, 323, 1775–1776. [Google Scholar] [CrossRef] [PubMed]
- Wollin, L.; Pieper, M. Tiotropium bromide exerts anti-inflammatory activity in a cigarette smoke mouse model of COPD. Pulm. Pharmacol. Ther. 2010, 23, 345–354. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Name | MT–DTI Affinity Score | Known Target or Phenotype (Effect) | Tissue or Cells (Species) | Reference |
|---|---|---|---|---|---|
| 1 | Rapamycin (Sirolimus) | 8.835 | IL-6 (decreased), IL-8 (decreased) | Pulmonary vascular endothelial cells and pulmonary-artery smooth muscle cells (human) | [44] |
| SOCS3 (increased) | Th17 cells (mouse) | [47] | |||
| NF-kB (decreased) | Lung tissue (mouse) | [43] | |||
| Neutrophilic inflammation (decreased) | |||||
| Lung injury (induced) | |||||
| MERS-CoV (inhibited) | Hepatocyte-derived epithelial-like Huh7 cell (human) | [48] | |||
| 2 | Tiotropium Bromide | 8.236 | NFKB1 (decreased), RELA (decreased), ICAM1 (decreased) | Rhinovirus-infected airway epithelial cells (human) | [40] |
| IL-6 (decreased), IL-8 (decreased), ICAM1 (decreased) | RSV-infected human epithelial type 2 cells (human) | [41] | |||
| IL-6 (decreased), IL-1B (decreased), IRB-induced lung inflammation (decreased) | Inspiratory resistive breathing (IRB)-induced lung tissue (rat) | [49] | |||
| IL-8 (decreased), proinflammation (decreased) | SV40 large T antigen-transformed 16HBE cells (human) | [50] | |||
| MMP1 (decreased) | Lung fibroblasts, which were obtained from patients’ healthy tissue area, induced by transforming growth factor beta (human) | [51] | |||
| IL-6 (decreased), TNF (decreased) | Lung tissue exposed to cigarette smoke and infected with RSV (mouse) | [42] | |||
| IL-8 (decreased) | LPS-stimulated BEAS-2B cells and lung fibroblasts from patient’s healthy tissue area (human) | [52] | |||
| TNF alpha-mediated chemotactic properties of stimulated alveolar macrophage (inhibited) | LPS-induced alveolar macrophage collected from COPD patients (human) | [53] | |||
| IL-1B (decreased), TNF (decreased), interstitial fibrosis and inflammation (decreased) | Cigarette smoked-exposed lung tissue (mouse) | [54] | |||
| IL-6 (decreased), IL-8 (decreased), TNF (decreased) | Cigarette smoked-exposed lung tissue (cat) | [55] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, K.; Kim, H.H.; Choi, Y. Tiotropium Is Predicted to Be a Promising Drug for COVID-19 Through Transcriptome-Based Comprehensive Molecular Pathway Analysis. Viruses 2020, 12, 776. https://doi.org/10.3390/v12070776
Kang K, Kim HH, Choi Y. Tiotropium Is Predicted to Be a Promising Drug for COVID-19 Through Transcriptome-Based Comprehensive Molecular Pathway Analysis. Viruses. 2020; 12(7):776. https://doi.org/10.3390/v12070776
Chicago/Turabian StyleKang, Keunsoo, Hoo Hyun Kim, and Yoonjung Choi. 2020. "Tiotropium Is Predicted to Be a Promising Drug for COVID-19 Through Transcriptome-Based Comprehensive Molecular Pathway Analysis" Viruses 12, no. 7: 776. https://doi.org/10.3390/v12070776
APA StyleKang, K., Kim, H. H., & Choi, Y. (2020). Tiotropium Is Predicted to Be a Promising Drug for COVID-19 Through Transcriptome-Based Comprehensive Molecular Pathway Analysis. Viruses, 12(7), 776. https://doi.org/10.3390/v12070776