
Structure-Based Modeling of Complement C4 Mediated Neutralization of Adenovirus


Abstract
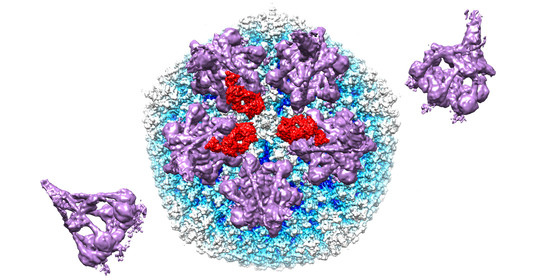
:
1. Introduction
2. Methods
2.1. Model Building
2.2. Molecular Dynamics Simulations
2.3. Calculation of Non-Bonded Interaction Energies
3. Results
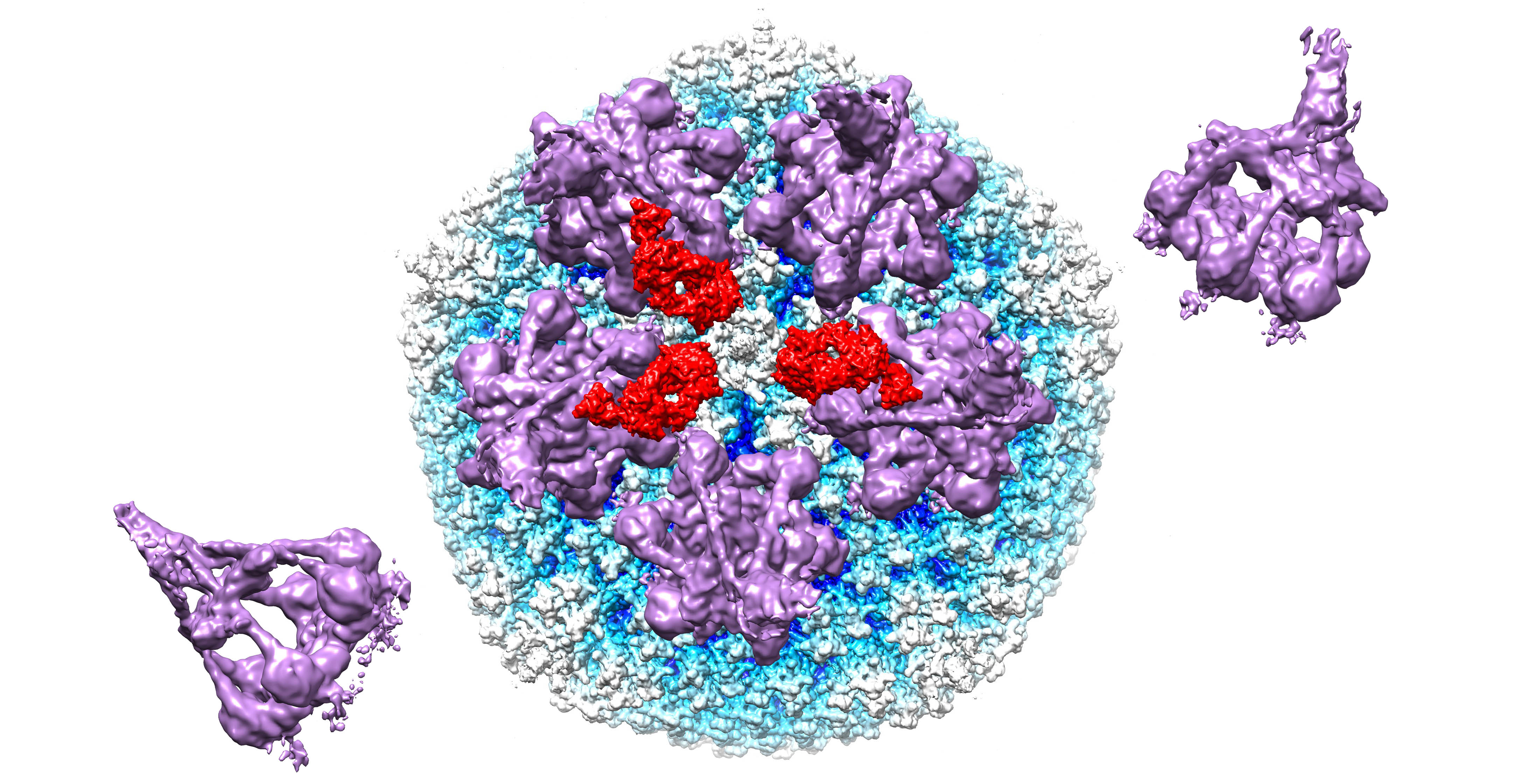
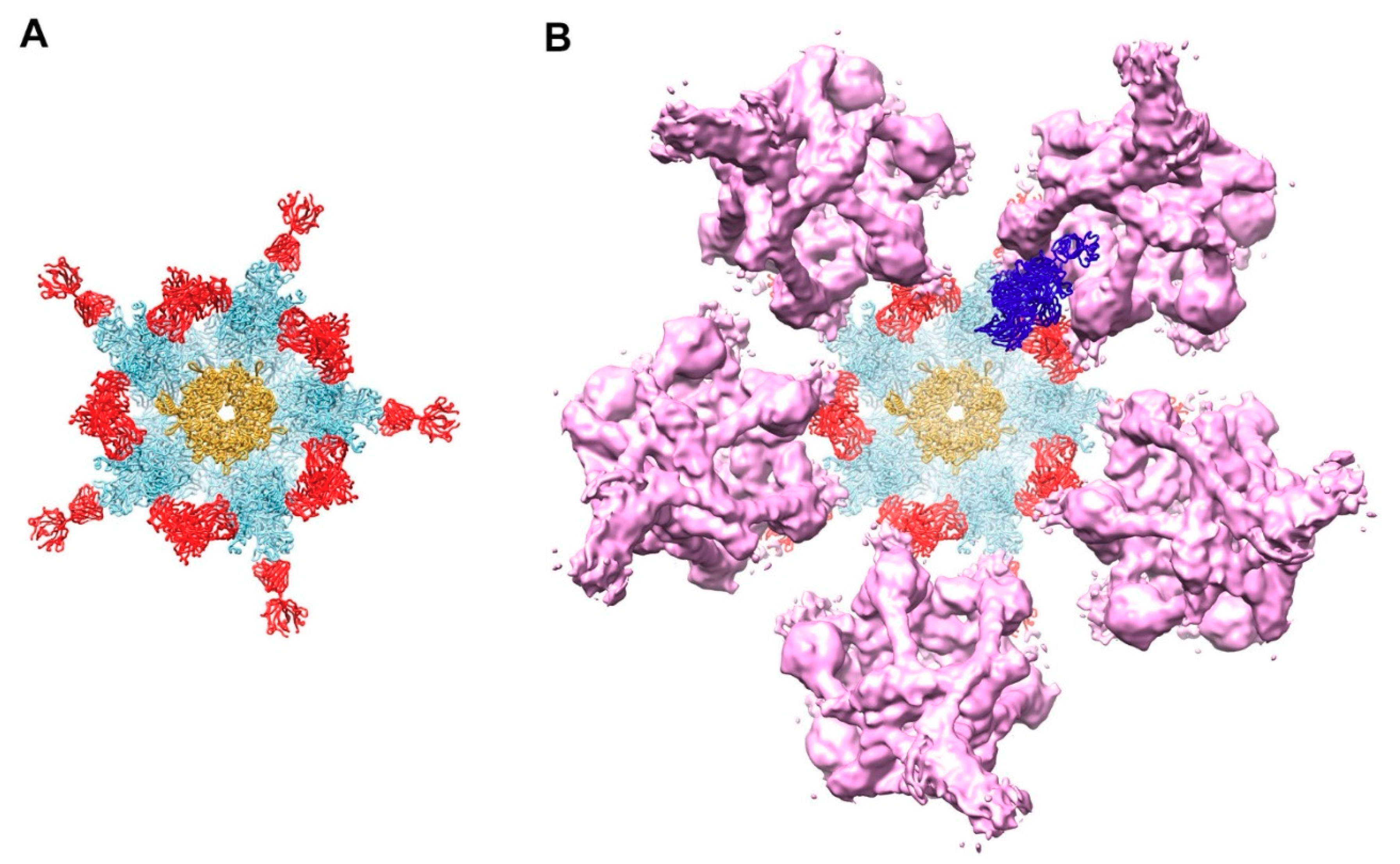
3.1. Modeling of HAdV-C5 with Antihexon Neutralizing Antibody
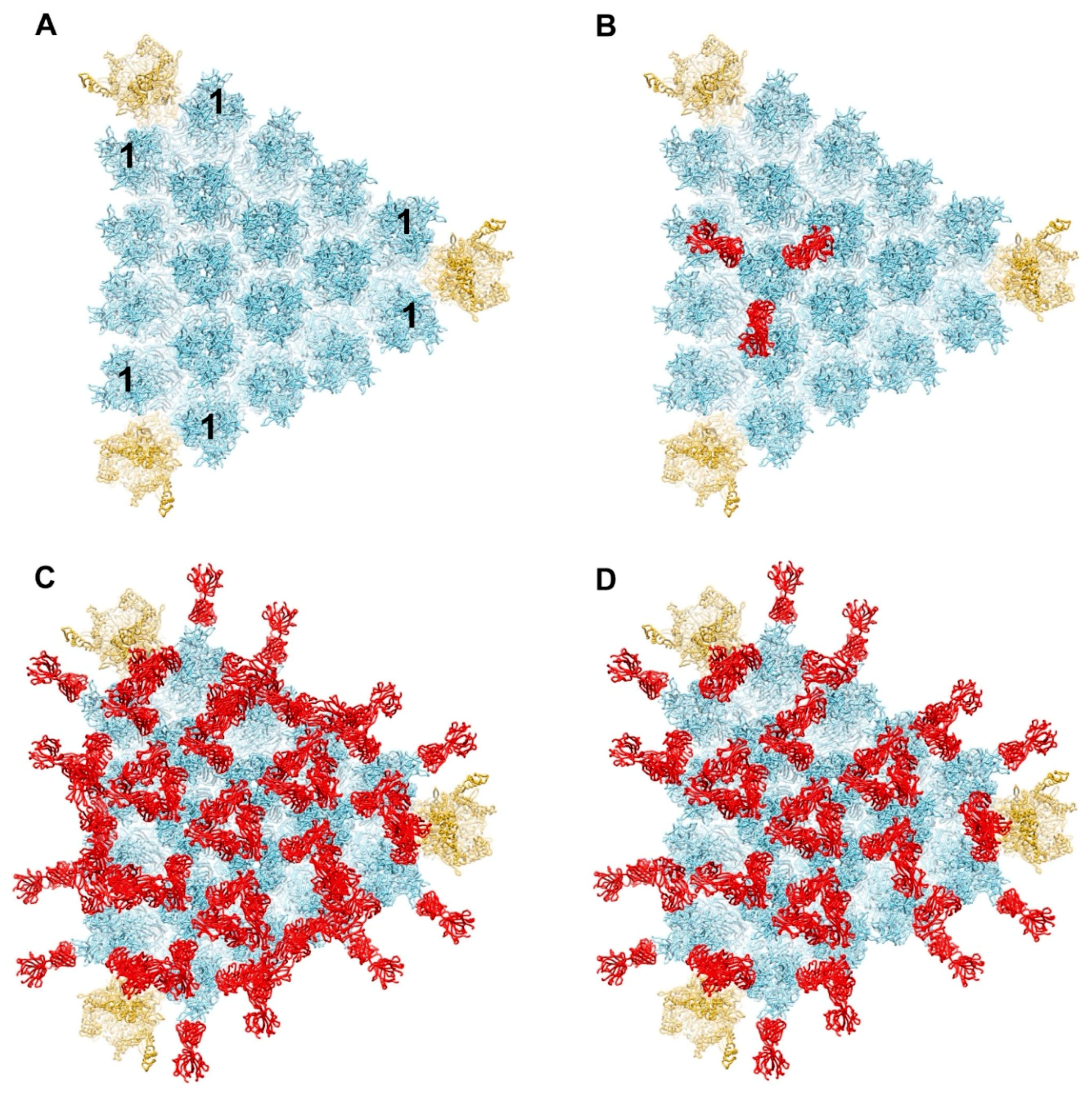
3.2. Modeling of HAdV-C5 with IgG and Complement Components C1 and C4b
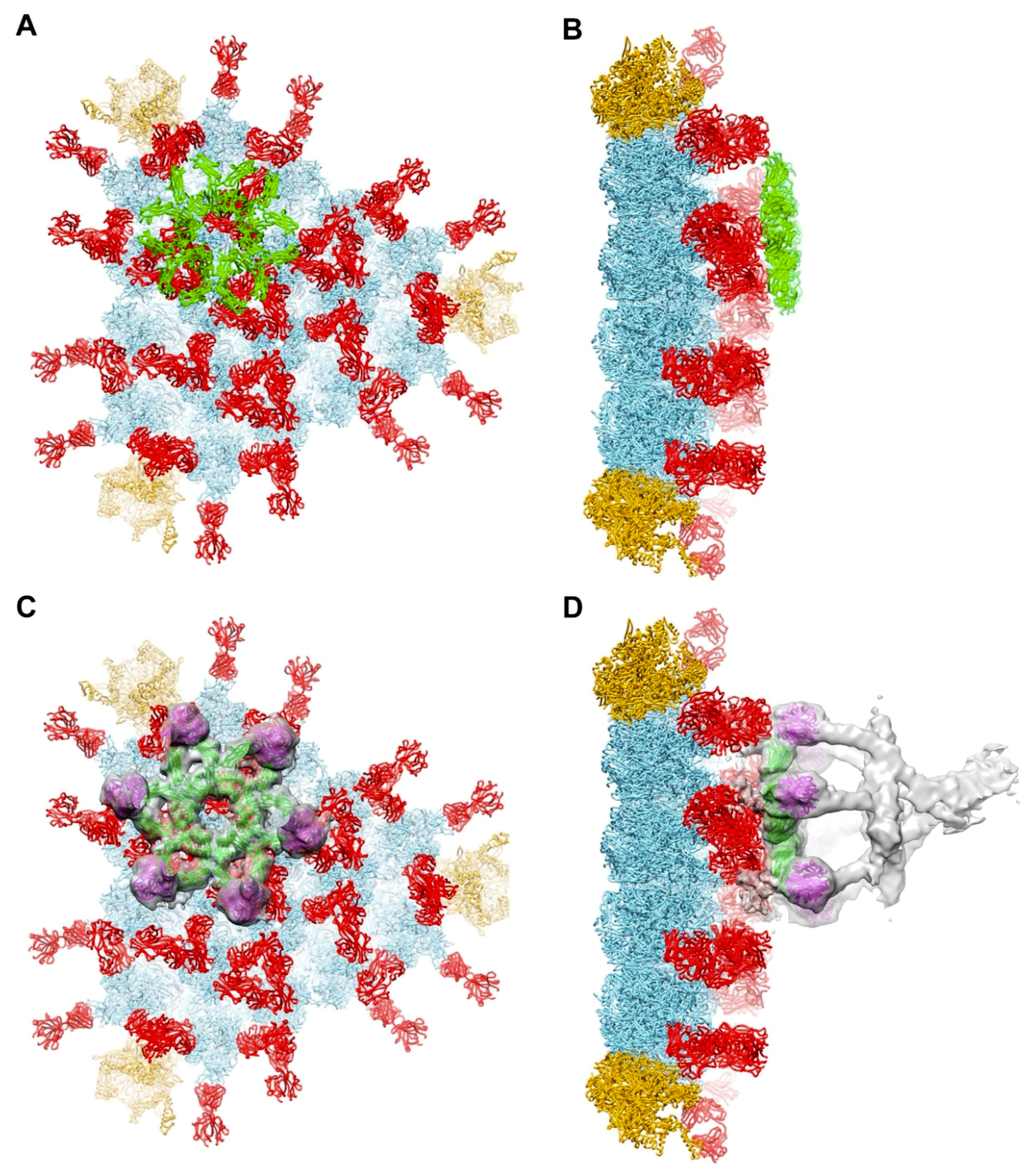
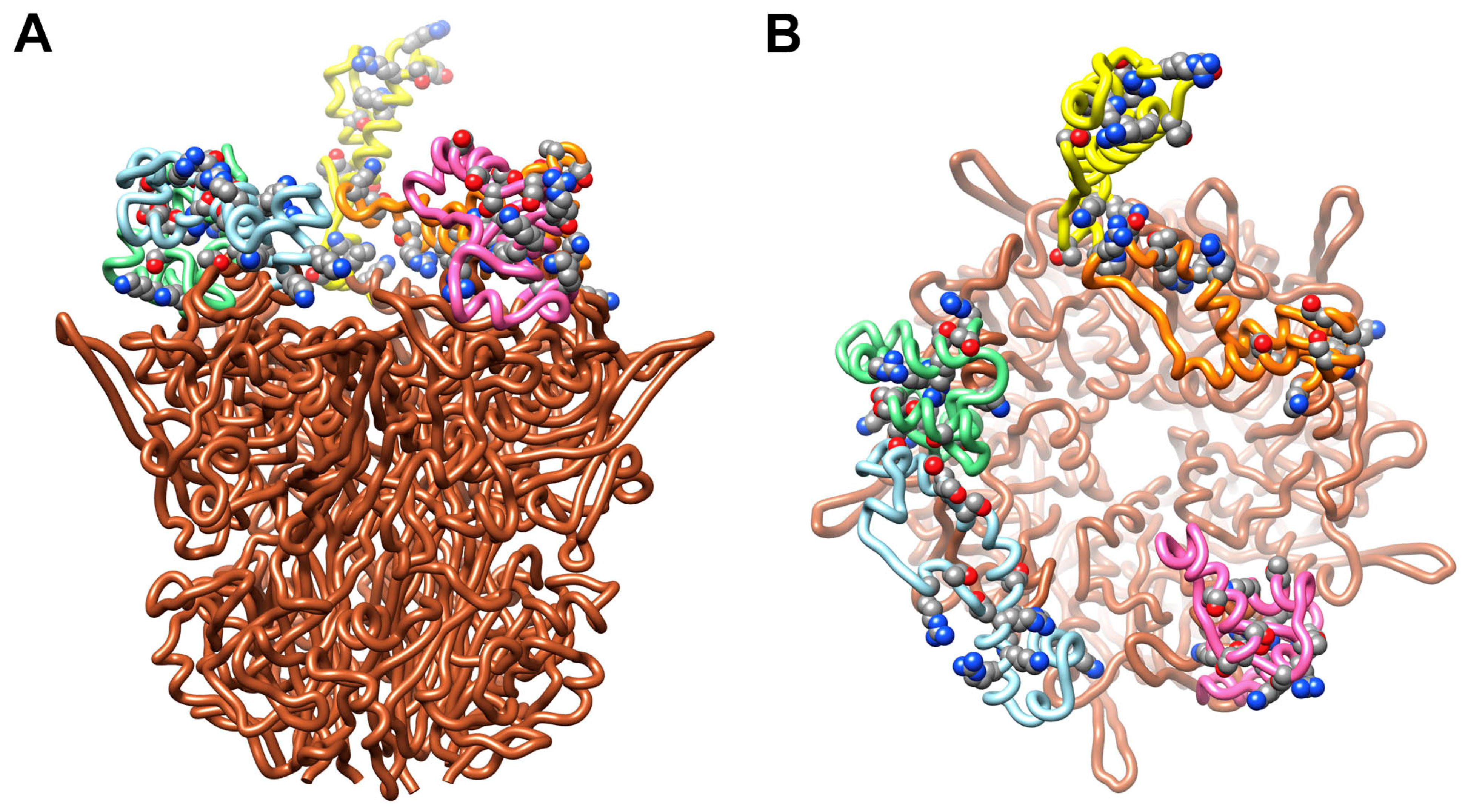
3.3. Possible Covalent Binding Sites for C4b on HAdV-C5 Penton Base
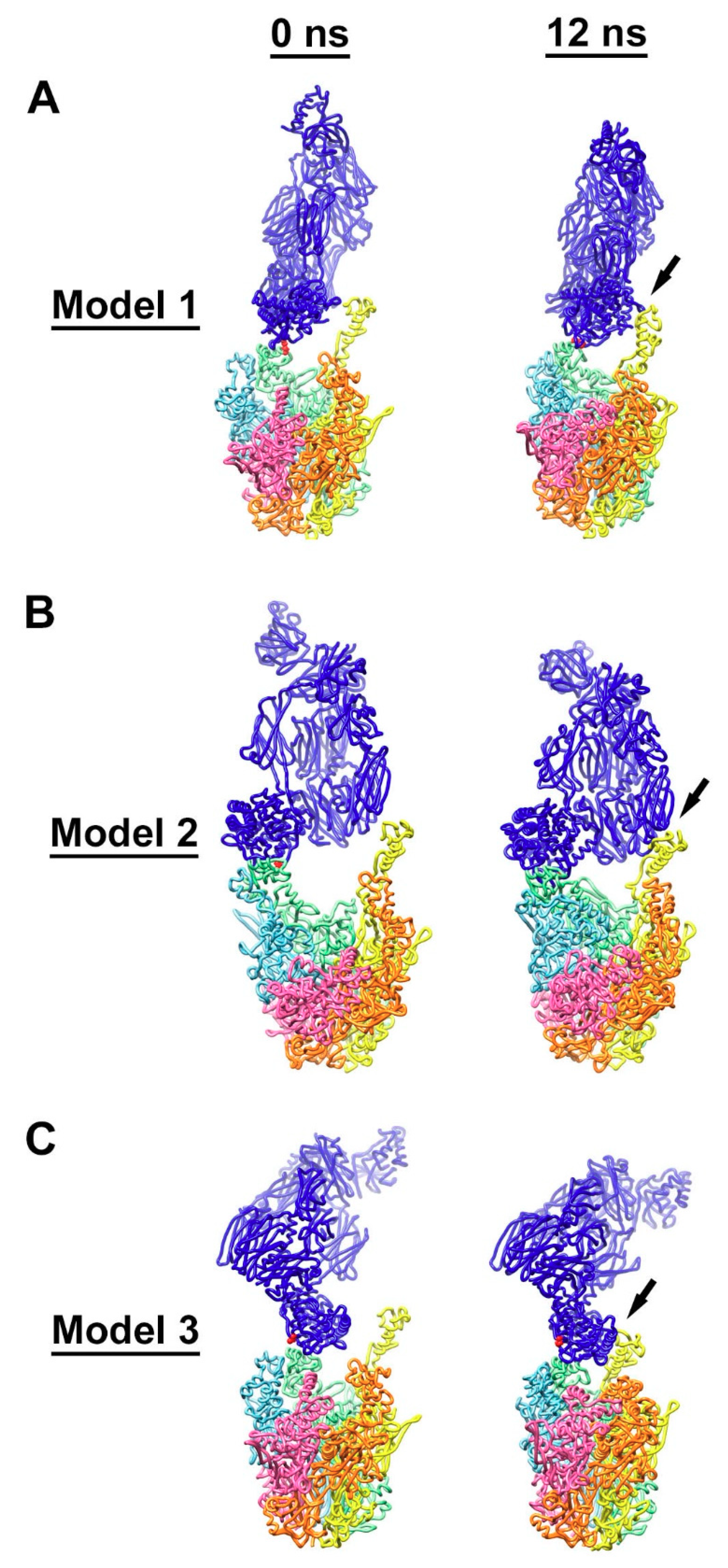
3.4. Molecular Dynamics Simulations with HAdV-C5 Penton Base and C4b
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Niemann, J.; Woller, N.; Brooks, J.; Fleischmann-Mundt, B.; Martin, N.T.; Kloos, A.; Knocke, S.; Ernst, A.M.; Manns, M.P.; Kubicka, S.; et al. Molecular retargeting of antibodies converts immune defense against oncolytic viruses into cancer immunotherapy. Nat. Commun. 2019, 10, 3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, R.J.; Byrnes, A.P. Interaction of adenovirus with antibodies, complement, and coagulation factors. FEBS Lett. 2019, 593, 3449–3460. [Google Scholar] [CrossRef] [PubMed]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.M.; Nanda, A.; Havenga, M.J.; Abbink, P.; Lynch, D.M.; Ewald, B.A.; Liu, J.; Thorner, A.R.; Swanson, P.E.; Gorgone, D.A.; et al. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature 2006, 441, 239–243. [Google Scholar] [CrossRef]
- Zhu, F.C.; Li, Y.H.; Guan, X.H.; Hou, L.H.; Wang, W.J.; Li, J.X.; Wu, S.P.; Wang, B.S.; Wang, Z.; Wang, L.; et al. Safety, tolerability, and immunogenicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: A dose-escalation, open-label, non-randomised, first-in-human trial. Lancet 2020, 395, 1845–1854. [Google Scholar] [CrossRef]
- Ferguson, M.S.; Lemoine, N.R.; Wang, Y. Systemic delivery of oncolytic viruses: Hopes and hurdles. Adv. Virol. 2012, 2012, 805629. [Google Scholar] [CrossRef]
- Khare, R.; Hillestad, M.L.; Xu, Z.; Byrnes, A.P.; Barry, M.A. Circulating antibodies and macrophages as modulators of adenovirus pharmacology. J. Virol. 2013, 87, 3678–3686. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Tian, J.; Smith, J.S.; Byrnes, A.P. Clearance of adenovirus by Kupffer cells is mediated by scavenger receptors, natural antibodies, and complement. J. Virol. 2008, 82, 11705–11713. [Google Scholar] [CrossRef] [Green Version]
- Kalyuzhniy, O.; Di Paolo, N.C.; Silvestry, M.; Hofherr, S.E.; Barry, M.A.; Stewart, P.L.; Shayakhmetov, D.M. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 5483–5488. [Google Scholar] [CrossRef] [Green Version]
- Waddington, S.N.; McVey, J.H.; Bhella, D.; Parker, A.L.; Barker, K.; Atoda, H.; Pink, R.; Buckley, S.M.; Greig, J.A.; Denby, L.; et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 2008, 132, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Atasheva, S.; Emerson, C.C.; Yao, J.; Young, C.; Stewart, P.L.; Shayakhmetov, D.M. Systemic cancer therapy with engineered adenovirus that evades innate immunity. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Xu, Z.; Qiu, Q.; Tian, J.; Smith, J.S.; Conenello, G.M.; Morita, T.; Byrnes, A.P. Coagulation factor X shields adenovirus type 5 from attack by natural antibodies and complement. Nat. Med. 2013, 19, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Doronin, K.; Flatt, J.W.; Di Paolo, N.C.; Khare, R.; Kalyuzhniy, O.; Acchione, M.; Sumida, J.P.; Ohto, U.; Shimizu, T.; Akashi-Takamura, S.; et al. Coagulation factor X activates innate immunity to human species C adenovirus. Science 2012, 338, 795–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, A.H.; Cardani, A.; Braciale, T.J. The host immune response in respiratory virus infection: Balancing virus clearance and immunopathology. Semin. Immunopathol. 2016, 38, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Barnum, S.R. Complement: A primer for the coming therapeutic revolution. Pharm. Ther. 2017, 172, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, D.R. Is IgM-like dislocation a common feature of antibody function? Immunol. Today 1986, 7, 165–167. [Google Scholar] [CrossRef]
- Feinstein, A.; Munn, E.A. Conformation of the free and antigen-bound IgM antibody molecules. Nature 1969, 224, 1307–1309. [Google Scholar] [CrossRef]
- Sharp, T.H.; Boyle, A.L.; Diebolder, C.A.; Kros, A.; Koster, A.J.; Gros, P. Insights into IgM-mediated complement activation based on in situ structures of IgM-C1-C4b. Proc. Natl. Acad. Sci. USA 2019, 116, 11900–11905. [Google Scholar] [CrossRef] [Green Version]
- de Jong, R.N.; Beurskens, F.J.; Verploegen, S.; Strumane, K.; van Kampen, M.D.; Voorhorst, M.; Horstman, W.; Engelberts, P.J.; Oostindie, S.C.; Wang, G.; et al. A Novel Platform for the Potentiation of Therapeutic Antibodies Based on Antigen-Dependent Formation of IgG Hexamers at the Cell Surface. PLoS Biol. 2016, 14, e1002344. [Google Scholar] [CrossRef] [Green Version]
- Diebolder, C.A.; Beurskens, F.J.; de Jong, R.N.; Koning, R.I.; Strumane, K.; Lindorfer, M.A.; Voorhorst, M.; Ugurlar, D.; Rosati, S.; Heck, A.J.; et al. Complement is activated by IgG hexamers assembled at the cell surface. Science 2014, 343, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugurlar, D.; Howes, S.C.; de Kreuk, B.J.; Koning, R.I.; de Jong, R.N.; Beurskens, F.J.; Schuurman, J.; Koster, A.J.; Sharp, T.H.; Parren, P.; et al. Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. Science 2018, 359, 794–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, K.B.; Porter, R.R. Subunit composition and structure of subcomponent C1q of the first component of human complement. Biochem. J. 1976, 155, 19–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottermann, M.; Foss, S.; Caddy, S.L.; Clift, D.; van Tienen, L.M.; Vaysburd, M.; Cruickshank, J.; O’Connell, K.; Clark, J.; Mayes, K.; et al. Complement C4 Prevents Viral Infection through Capsid Inactivation. Cell Host Microbe 2019, 25, 617–629.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, O.; Marvin, S.A.; Wodrich, H.; Campbell, E.M.; Wiethoff, C.M. Spatiotemporal dynamics of adenovirus membrane rupture and endosomal escape. J. Virol. 2012, 86, 10821–10828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiethoff, C.M.; Wodrich, H.; Gerace, L.; Nemerow, G.R. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 2005, 79, 1992–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef]
- Smith, J.G.; Nemerow, G.R. Mechanism of adenovirus neutralization by Human alpha-defensins. Cell Host Microbe 2008, 3, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Flatt, J.W.; Kim, R.; Smith, J.G.; Nemerow, G.R.; Stewart, P.L. An intrinsically disordered region of the adenovirus capsid is implicated in neutralization by human alpha defensin 5. PLoS ONE 2013, 8, e61571. [Google Scholar] [CrossRef] [Green Version]
- Gaboriaud, C.; Juanhuix, J.; Gruez, A.; Lacroix, M.; Darnault, C.; Pignol, D.; Verger, D.; Fontecilla-Camps, J.C.; Arlaud, G.J. The crystal structure of the globular head of complement protein C1q provides a basis for its versatile recognition properties. J. Biol. Chem. 2003, 278, 46974–46982. [Google Scholar] [CrossRef] [Green Version]
- Saphire, E.O.; Parren, P.W.; Pantophlet, R.; Zwick, M.B.; Morris, G.M.; Rudd, P.M.; Dwek, R.A.; Stanfield, R.L.; Burton, D.R.; Wilson, I.A. Crystal structure of a neutralizing human IGG against HIV-1: A template for vaccine design. Science 2001, 293, 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Bottermann, M.; Lode, H.E.; Watkinson, R.E.; Foss, S.; Sandlie, I.; Andersen, J.T.; James, L.C. Antibody-antigen kinetics constrain intracellular humoral immunity. Sci. Rep. 2016, 6, 37457. [Google Scholar] [CrossRef] [Green Version]
- Varghese, R.; Mikyas, Y.; Stewart, P.L.; Ralston, R. Postentry neutralization of adenovirus type 5 by an antihexon antibody. J. Virol. 2004, 78, 12320–12332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindert, S.; Silvestry, M.; Mullen, T.M.; Nemerow, G.R.; Stewart, P.L. Cryo-electron microscopy structure of an adenovirus-integrin complex indicates conformational changes in both penton base and integrin. J. Virol. 2009, 83, 11491–11501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, T.J.; Mathias, P.; Cheresh, D.A.; Nemerow, G.R. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 1993, 73, 309–319. [Google Scholar] [CrossRef]
- Dai, X.; Wu, L.; Sun, R.; Zhou, Z.H. Atomic Structures of Minor Proteins VI and VII in Human Adenovirus. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, S.; Kidmose, R.T.; Petersen, S.V.; Szilagyi, A.; Prohaszka, Z.; Andersen, G.R. Structural Basis for the Function of Complement Component C4 within the Classical and Lectin Pathways of Complement. J. Immunol. 2015, 194, 5488–5496. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Henin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi (1) and chi (2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhang, L.; Tong, H.; Peng, B.; Rames, M.J.; Zhang, S.; Ren, G. 3D Structural Fluctuation of IgG1 Antibody Revealed by Individual Particle Electron Tomography. Sci. Rep. 2015, 5, 9803. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; de Jong, R.N.; van den Bremer, E.T.; Beurskens, F.J.; Labrijn, A.F.; Ugurlar, D.; Gros, P.; Schuurman, J.; Parren, P.W.; Heck, A.J. Molecular Basis of Assembly and Activation of Complement Component C1 in Complex with Immunoglobulin G1 and Antigen. Mol. Cell 2016, 63, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Law, S.K. The covalent binding reaction of C3 and C4. Ann. N. Y. Acad. Sci 1983, 421, 246–258. [Google Scholar] [CrossRef]
- Zubieta, C.; Schoehn, G.; Chroboczek, J.; Cusack, S. The structure of the human adenovirus 2 penton. Mol. Cell 2005, 17, 121–135. [Google Scholar] [CrossRef]
- Agrawal, P.; Nawadkar, R.; Ojha, H.; Kumar, J.; Sahu, A. Complement Evasion Strategies of Viruses: An Overview. Front. Microbiol. 2017, 8, 1117. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Thangamani, S.; Ho, B.; Ding, J.L. The ancient origin of the complement system. EMBO J. 2005, 24, 382–394. [Google Scholar] [CrossRef] [Green Version]
- Mallery, D.L.; McEwan, W.A.; Bidgood, S.R.; Towers, G.J.; Johnson, C.M.; James, L.C. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc. Natl. Acad. Sci. USA 2010, 107, 19985–19990. [Google Scholar] [CrossRef] [Green Version]
- Foss, S.; Watkinson, R.; Sandlie, I.; James, L.C.; Andersen, J.T. TRIM21: A cytosolic Fc receptor with broad antibody isotype specificity. Immunol. Rev. 2015, 268, 328–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greber, U.F.; Flatt, J.W. Adenovirus Entry: From Infection to Immunity. Annu. Rev. Virol. 2019, 6, 177–197. [Google Scholar] [CrossRef] [PubMed]
- Mellors, J.; Tipton, T.; Longet, S.; Carroll, M. Viral Evasion of the Complement System and Its Importance for Vaccines and Therapeutics. Front. Immunol. 2020, 11, 1450. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, L.; Frank, L.; Wagner, J.; Andreozzi, P.; Hammer, B.; D’Alicarnasso, M.; Pelliccia, M.; Liu, W.; Chakrabortty, S.; et al. Patchy Amphiphilic Dendrimers Bind Adenovirus and Control Its Host Interactions and in Vivo Distribution. ACS Nano 2019, 13, 8749–8759. [Google Scholar] [CrossRef] [Green Version]
- Rojas, L.A.; Condezo, G.N.; Moreno, R.; Fajardo, C.A.; Arias-Badia, M.; San Martin, C.; Alemany, R. Albumin-binding adenoviruses circumvent pre-existing neutralizing antibodies upon systemic delivery. J. Control. Release 2016, 237, 78–88. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 0 ns (kcal/mol) | 12 ns (kcal/mol) | |
|---|---|---|
| Penton base Chain A (pink) | 0 | 0 |
| Penton base Chain B (blue) | 0 | 0 |
| Penton base Chain C (green) | −51 | −226 |
| Penton base Chain D (yellow) | 6 | −91 |
| Penton base Chain E (orange) | 0 | 0 |
| Penton base Chains A–E | −45 | −318 |
| 0 ns (kcal/mol) | 12 ns (kcal/mol) | |
|---|---|---|
| Penton base Chain A (pink) | 0 | 0 |
| Penton base Chain B (blue) | 4 | −10 |
| Penton base Chain C (green) | −121 | −517 |
| Penton base Chain D (yellow) | 1 | −67 |
| Penton base Chain E (orange) | 0 | 0 |
| Penton base Chains A–E | −116 | −594 |
| 0 ns (kcal/mol) | 12 ns (kcal/mol) | |
|---|---|---|
| Penton base Chain A (pink) | 0 | 0 |
| Penton base Chain B (blue) | 0 | 29 |
| Penton base Chain C (green) | −61 | −158 |
| Penton base Chain D (yellow) | 0 | −186 |
| Penton base Chain E (orange) | 0 | 0 |
| Penton base Chains A–E | −61 | −315 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emerson, C.C.; Stewart, P.L. Structure-Based Modeling of Complement C4 Mediated Neutralization of Adenovirus. Viruses 2021, 13, 111. https://doi.org/10.3390/v13010111
Emerson CC, Stewart PL. Structure-Based Modeling of Complement C4 Mediated Neutralization of Adenovirus. Viruses. 2021; 13(1):111. https://doi.org/10.3390/v13010111
Chicago/Turabian StyleEmerson, Corey C., and Phoebe L. Stewart. 2021. "Structure-Based Modeling of Complement C4 Mediated Neutralization of Adenovirus" Viruses 13, no. 1: 111. https://doi.org/10.3390/v13010111
APA StyleEmerson, C. C., & Stewart, P. L. (2021). Structure-Based Modeling of Complement C4 Mediated Neutralization of Adenovirus. Viruses, 13(1), 111. https://doi.org/10.3390/v13010111