
Therapy Implications of Hepatitis C Virus Genetic Diversity



Abstract
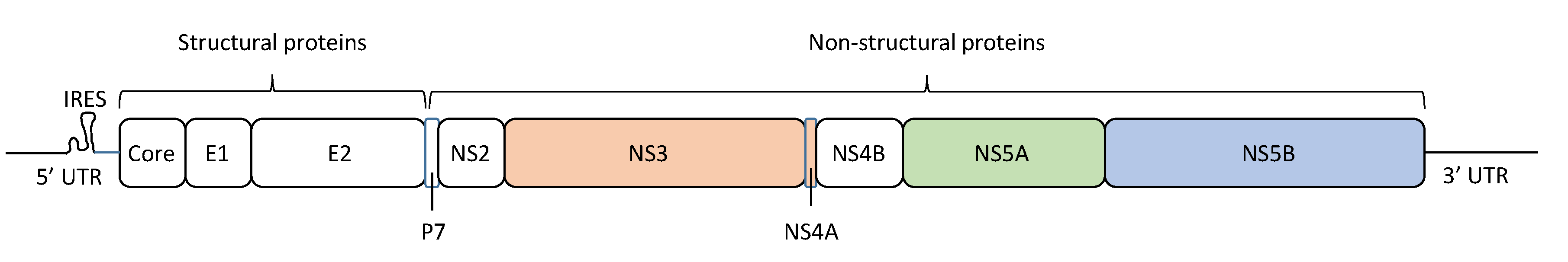
:1. Introduction
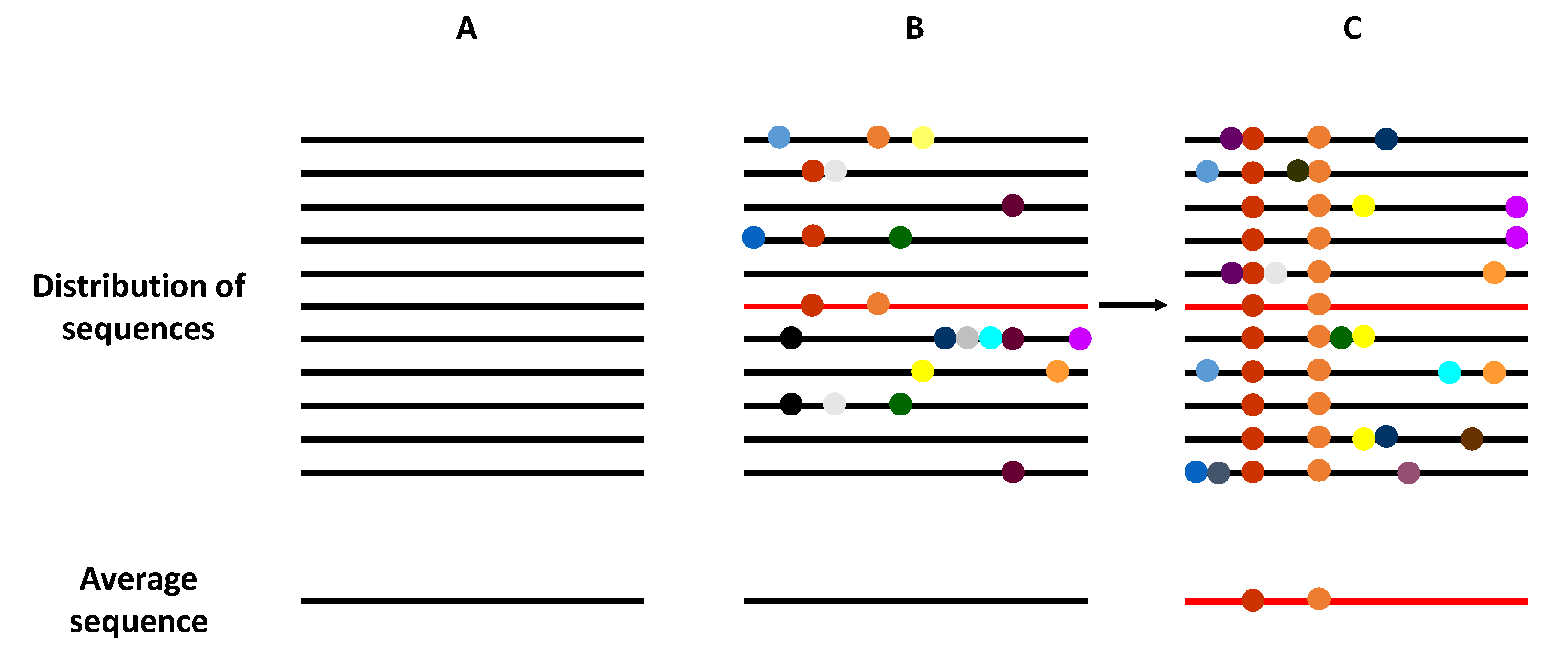
2. HCV Quasispecies and Diversity
3. Implications of HCV Diversity in Pathogenesis and Transmission
4. HCV Therapy
5. DAA Resistance
6. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Simmonds, P. Genetic diversity and evolution of hepatitis C virus—15 years on. J. Gen. Virol. 2004, 85, 3173–3188. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Simmonds, P.; Gerold, G.; Qaisar, N.; Jain, K.; Henriquez, J.A.; Firth, C.; Hirschberg, D.L.; Rice, C.M.; Shields, S.; et al. Characterization of a canine homolog of hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 11608–11613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burbelo, P.D.; Dubovi, E.J.; Simmonds, P.; Medina, J.L.; Henriquez, J.A.; Mishra, N.; Wagner, J.; Tokarz, R.; Cullen, J.M.; Iadarola, M.J.; et al. Serology-Enabled Discovery of Genetically Diverse Hepaciviruses in a New Host. J. Virol. 2012, 86, 6171–6178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinstone, S.M.; Kapikian, A.Z.; Purcell, R.H.; Alter, H.J.; Holland, P.V. Transfusion-Associated Hepatitis Not Due to Viral Hepatitis Type A or B. N. Engl. J. Med. 1975, 292, 767–770. [Google Scholar] [CrossRef]
- Kolykhalov, A.A.; Agapov, E.V.; Blight, K.J.; Mihalik, K.; Feinstone, S.M.; Rice, C.M. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 1997, 277, 570–574. [Google Scholar] [CrossRef]
- Kuo, G.; Choo, Q.L.; Alter, H.J.; Gitnick, G.L.; Redeker, A.G.; Purcell, R.H.; Miyamura, T.; Dienstag, J.L.; Alter, M.J.; Stevens, C.E.; et al. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science 1989, 244, 362–364. [Google Scholar] [CrossRef]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [Green Version]
- Grebely, J.; Dore, G.J.; Kim, A.Y.; Lloyd, A.; Shoukry, N.H.; Prins, M.; Page, K. Genetics of spontaneous clearance of hepatitis C virus infection: A complex topic with much to learn. Hepatology 2014, 60, 2127–2128. [Google Scholar] [CrossRef] [Green Version]
- Westbrook, R.H.; Dusheiko, G. Natural history of hepatitis C. J. Hepatol. 2014, 61, S58–S68. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Global Hepatitis Report 2017. Available online: http://apps.who.int/iris/bitstream/10665/255016/1/9789241565455-eng.pdf?ua=1 (accessed on 10 March 2017).
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef] [Green Version]
- Catanese, M.T.; Uryu, K.; Kopp, M.; Edwards, T.J.; Andrus, L.; Rice, W.J.; Silvestry, M.; Kuhn, R.J.; Rice, C.M. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. USA 2013, 110, 9505–9510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moradpour, D.; Penin, F. Hepatitis C Virus Proteins: From Structure to Function. In Current Topics in Microbiology and Immunology; Curr Top Microbiol Immunol, Springer: New York, NY, USA, 2013; Volume 369, pp. 113–142. [Google Scholar]
- Tabata, K.; Neufeldt, C.J.; Bartenschlager, R. Hepatitis C virus replication. Cold Spring Harb. Perspect. Med. 2020, 10, a037093. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V.; Körner, F.; Koch, J.O.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef] [Green Version]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Kräusslich, H.G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Más, A.; López-Galíndez, C.; Cacho, I.; Gómez, J.; Martínez, M.A. Unfinished stories on viral quasispecies and darwinian views of evolution. J. Mol. Biol. 2010, 397, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Perales, C. Viral quasispecies. PLoS Genet. 2019, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eigen, M. Selforganization of matter and the evolution of biological macromolecules. Naturwissenschaften 1971, 58, 465–523. [Google Scholar] [CrossRef]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef]
- Ogata, N.; Alter, H.J.; Miller, R.H.; Purcell, R.H. Nucleotide sequence and mutation rate of the H strain of hepatitis C virus. Proc. Natl. Acad. Sci. USA 1991, 88, 3392–3396. [Google Scholar] [CrossRef] [Green Version]
- Franco, S.; Parera, M.; Aparicio, E.; Clotet, B.; Martinez, M.A. Genetic and catalytic efficiency structure of an HCV protease quasispecies. Hepatology 2007, 45, 899–910. [Google Scholar] [CrossRef]
- Martinez, M.A.; Nevot, M.; Jordan-Paiz, A.; Franco, S. Similarities between Human Immunodeficiency Virus Type 1 and Hepatitis C Virus Genetic and Phenotypic Protease Quasispecies Diversity. J. Virol. 2015, 89, 9758–9764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martell, M.; Esteban, J.I.; Quer, J.; Vargas, V.; Esteban, R.; Guardia, J.; Gómez, J. Dynamic behavior of hepatitis C virus quasispecies in patients undergoing orthotopic liver transplantation. J. Virol. 1994, 68, 3425–3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuevas, J.M.; González-Candelas, F.; Moya, A.; Sanjuán, R. Effect of Ribavirin on the Mutation Rate and Spectrum of Hepatitis C Virus In Vivo. J. Virol. 2009, 83, 5760–5764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, R.M.; Li, H.; Wang, S.; Stoddard, M.B.; Learn, G.H.; Korber, B.T.; Bhattacharya, T.; Guedj, J.; Parrish, E.H.; Hahn, B.H.; et al. Quantifying the Diversification of Hepatitis C Virus (HCV) during Primary Infection: Estimates of the In Vivo Mutation Rate. PLoS Pathog. 2012, 8, e1002881. [Google Scholar] [CrossRef] [PubMed]
- Borgia, S.M.; Hedskog, C.; Parhy, B.; Hyland, R.H.; Stamm, L.M.; Brainard, D.M.; Subramanian, M.G.; McHutchison, J.G.; Mo, H.; Svarovskaia, E.; et al. Identification of a novel hepatitis C virus genotype from Punjab, India: Expanding classification of hepatitis C virus into 8 genotypes. J. Infect. Dis. 2018, 218, 1722–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blach, S.; Zeuzem, S.; Manns, M.; Altraif, I.; Duberg, A.S.; Muljono, D.H.; Waked, I.; Alavian, S.M.; Lee, M.H.; Negro, F.; et al. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: A modelling study. Lancet Gastroenterol. Hepatol. 2017, 2, 161–176. [Google Scholar] [CrossRef] [Green Version]
- Galli, A.; Bukh, J. Comparative analysis of the molecular mechanisms of recombination in hepatitis C virus. Trends Microbiol. 2014, 22, 354–364. [Google Scholar] [CrossRef]
- Farci, P.; Shimoda, A.; Coiana, A.; Diaz, G.; Peddis, G.; Melpolder, J.C.; Strazzera, A.; Chien, D.Y.; Munoz, S.J.; Balestrieri, A.; et al. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science 2000, 288, 339–344. [Google Scholar] [CrossRef]
- Geller, R.; Estada, Ú.; Peris, J.B.; Andreu, I.; Bou, J.V.; Garijo, R.; Cuevas, J.M.; Sabariegos, R.; Mas, A.; Sanjuán, R. Highly heterogeneous mutation rates in the hepatitis C virus genome. Nat. Microbiol. 2016, 1, 16045. [Google Scholar] [CrossRef]
- Raghwani, J.; Rose, R.; Sheridan, I.; Lemey, P.; Suchard, M.A.; Santantonio, T.; Farci, P.; Klenerman, P.; Pybus, O.G. Exceptional Heterogeneity in Viral Evolutionary Dynamics Characterises Chronic Hepatitis C Virus Infection. PLoS Pathog. 2016, 12, e1005894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, R.R.; Tanaka, Y.; Takebe, Y.; Magiorkinis, G.; Buskell, Z.; Seeff, L.; Alter, H.J.; Pybus, O.G. Evolutionary analysis of hepatitis C virus gene sequences from 1953. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 2013.0168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farci, P.; Wollenberg, K.; Diaz, G.; Engle, R.E.; Lai, M.E.; Klenerman, P.; Purcell, R.H.; Pybus, O.G.; Alter, H.J. Profibrogenic chemokines and viral evolution predict rapid progression of hepatitis C to cirrhosis. Proc. Natl. Acad. Sci. USA 2012, 109, 14562–14567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemey, P.; Kosakovsky Pond, S.L.; Drummond, A.J.; Pybus, O.G.; Shapiro, B.; Barroso, H.; Taveira, N.; Rambaut, A. Synonymous substitution rates predict HIV disease progression as a result of underlying replication dynamics. PLoS Comput. Biol. 2007, 3, 0282–0292. [Google Scholar] [CrossRef]
- Alter, H.J.; Farci, P.; Bukh, J.; Purcell, R.H. Reflections on the History of HCV: A Posthumous Examination. Clin. Liver Dis. 2020, 15, S64–S71. [Google Scholar] [CrossRef]
- Fafi-Kremer, S.; Fofana, I.; Soulier, E.; Carolla, P.; Meuleman, P.; Leroux-Roels, G.; Patel, A.H.; Cosset, F.L.; Pessaux, P.; Doffoël, M.; et al. Viral entry and escape from antibody-mediated neutralization influence hepatitis C virus reinfection in liver transplantation. J. Exp. Med. 2010, 207, 2019–2031. [Google Scholar] [CrossRef] [Green Version]
- Harouaka, D.; Engle, R.E.; Wollenberg, K.; Diaz, G.; Tice, A.B.; Zamboni, F.; Govindarajan, S.; Alter, H.; Kleiner, D.E.; Farci, P. Diminished viral replication and compartmentalization of hepatitis C virus in hepatocellular carcinoma tissue. Proc. Natl. Acad. Sci. USA 2016, 113, 1375–1380. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, S.; Perez-del-Pulgar, S.; Carrion, J.A.; Costa, J.; Gonzalez, P.; Massaguer, A.; Fondevila, C.; Garcia-Valdecasas, J.C.; Navasa, M.; Forns, X. Hepatitis C Virus Compartmentalization and Infection Recurrence after Liver Transplantation. Am. J. Transplant. 2009, 9, 1591–1601. [Google Scholar] [CrossRef]
- Hedegaard, D.L.; Tully, D.C.; Rowe, I.A.; Reynolds, G.M.; Bean, D.J.; Hu, K.; Davis, C.; Wilhelm, A.; Ogilvie, C.B.; Power, K.A.; et al. High resolution sequencing of hepatitis C virus reveals limited intra-hepatic compartmentalization in end-stage liver disease. J. Hepatol. 2017, 66, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Stoddard, M.B.; Wang, S.; Blair, L.M.; Giorgi, E.E.; Parrish, E.H.; Learn, G.H.; Hraber, P.; Goepfert, P.A.; Saag, M.S.; et al. Elucidation of Hepatitis C Virus Transmission and Early Diversification by Single Genome Sequencing. PLoS Pathog. 2012, 8, e1002880. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Stoddard, M.B.; Wang, S.; Giorgi, E.E.; Blair, L.M.; Learn, G.H.; Hahn, B.H.; Alter, H.J.; Busch, M.P.; Fierer, D.S.; et al. Single-Genome Sequencing of Hepatitis C Virus in Donor-Recipient Pairs Distinguishes Modes and Models of Virus Transmission and Early Diversification. J. Virol. 2016, 90, 152–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abayasingam, A.; Leung, P.; Eltahla, A.; Bull, R.A.; Luciani, F.; Grebely, J.; Dore, G.J.; Applegate, T.; Page, K.; Bruneau, J.; et al. Genomic characterization of hepatitis C virus transmitted founder variants with deep sequencing. Infect. Genet. Evol. 2019, 71, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, C.; Leung, P.; Lloyd, A.R.; Bull, R.A.; Luciani, F.; Grebely, J.; Dore, G.J.; Applegate, T.; Page, K.; Bruneau, J.; et al. Genomic variability of within-host hepatitis C variants in acute infection. J. Viral Hepat. 2019, 26, 476–484. [Google Scholar] [CrossRef]
- Bukh, J. The history of hepatitis C virus (HCV): Basic research reveals unique features in phylogeny, evolution and the viral life cycle with new perspectives for epidemic control. J. Hepatol. 2016, 65, S2–S21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, J.D.; Salinas, E.; Grakoui, A. Immune system control of hepatitis C virus infection. Curr. Opin. Virol. 2021, 46, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Farci, P.; Alter, H.J.; Govindarajan, S.; Wong, D.C.; Engle, R.; Lesniewski, R.R.; Mushahwar, I.K.; Desai, S.M.; Miller, R.H.; Ogata, N.; et al. Lack of protective immunity against reinfection with hepatitis C virus. Science 1992, 258, 135–140. [Google Scholar] [CrossRef]
- Bassett, S.E.; Guerra, B.; Brasky, K.; Miskovsky, E.; Houghton, M.; Klimpel, G.R.; Lanford, R.E. Protective immune response to hepatitis C virus in chimpanzees rechallenged following clearance of primary infection. Hepatology 2001, 33, 1479–1487. [Google Scholar] [CrossRef]
- Franco, S.; Tural, C.; Nevot, M.; Moltó, J.; Rockstroh, J.K.; Clotet, B.; Martinez, M.A. Detection of a sexually transmitted hepatitis C virus protease inhibitor-resistance variant in a human immunodeficiency virus-infected homosexual man. Gastroenterology 2014, 147, 599–601. [Google Scholar] [CrossRef]
- Wrensch, F.; Ligat, G.; Heydmann, L.; Schuster, C.; Zeisel, M.B.; Pessaux, P.; Habersetzer, F.; King, B.J.; Tarr, A.W.; Ball, J.K.; et al. Interferon-Induced Transmembrane Proteins Mediate Viral Evasion in Acute and Chronic Hepatitis C Virus Infection. Hepatology 2019, 70, 1506–1520. [Google Scholar] [CrossRef]
- Grakoui, A.; Shoukry, N.H.; Woollard, D.J.; Han, J.H.; Hanson, H.L.; Ghrayeb, J.; Murthy, K.K.; Rice, C.M.; Walker, C.M. HCV Persistence and Immune Evasion in the Absence of Memory T Cell Help. Science 2003, 302, 659–662. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.L.; Mosbruger, T.; Lauer, G.M.; Pardoll, D.; Thomas, D.L.; Ray, S.C. Comprehensive analyses of CD8+ T cell responses during longitudinal study of acute human hepatitis C. Hepatology 2005, 42, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Neumann-Haefelin, C.; Timm, J.; Spangenberg, H.C.; Wischniowski, N.; Nazarova, N.; Kersting, N.; Roggendorf, M.; Allen, T.M.; Blum, H.E.; Thimme, R. Virological and immunological determinants of intrahepatic virus-specific CD8+ T-cell failure in chronic hepatitis C virus infection. Hepatology 2008, 47, 1824–1836. [Google Scholar] [CrossRef] [PubMed]
- Neumann-Haefelin, C.; McKiernan, S.; Ward, S.; Viazov, S.; Spangenberg, H.C.; Killinger, T.; Baumert, T.F.; Nazarova, N.; Sheridan, I.; Pybus, O.; et al. Dominant influence of an HLA-B27 restricted CD8+ T cell response in mediating HCV clearance and evolution. Hepatology 2006, 43, 563–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzmaurice, K.; Petrovic, D.; Ramamurthy, N.; Simmons, R.; Merani, S.; Gaudieri, S.; Sims, S.; Dempsey, E.; Freitas, E.; Lea, S.; et al. Molecular footprints reveal the impact of the protective HLA-A*03 allele in hepatitis C virus infection. Gut 2011, 60, 1563–1571. [Google Scholar] [CrossRef] [Green Version]
- Thimme, R. T cell immunity to hepatitis C virus: Lessons for a prophylactic vaccine. J. Hepatol. 2021, 74, 220–229. [Google Scholar] [CrossRef]
- Cornberg, M.; Tacke, F.; Karlsen, T.H. Clinical Practice Guidelines of the European Association for the study of the Liver – Advancing methodology but preserving practicability. J. Hepatol. 2019, 70, 5–7. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; De Clercq, E. Current therapy for chronic hepatitis C: The role of direct-acting antivirals. Antiviral Res. 2017, 142, 83–122. [Google Scholar] [CrossRef]
- Ansaldi, F.; Orsi, A.; Sticchi, L.; Bruzzone, B.; Icardi, G. Hepatitis C virus in the new era: Perspectives in epidemiology, prevention, diagnostics and predictors of response to therapy. World J. Gastroenterol. 2014, 20, 9633–9652. [Google Scholar] [CrossRef]
- Jacobson, I.M.; McHutchison, J.G.; Dusheiko, G.; Di Bisceglie, A.M.; Reddy, K.R.; Bzowej, N.H.; Marcellin, P.; Muir, A.J.; Ferenci, P.; Flisiak, R.; et al. Telaprevir for Previously Untreated Chronic Hepatitis C Virus Infection. N. Engl. J. Med. 2011, 364, 2405–2416. [Google Scholar] [CrossRef]
- Poordad, F.; McCone, J.; Bacon, B.R.; Bruno, S.; Manns, M.P.; Sulkowski, M.S.; Jacobson, I.M.; Reddy, K.R.; Goodman, Z.D.; Boparai, N.; et al. Boceprevir for Untreated Chronic HCV Genotype 1 Infection. N. Engl. J. Med. 2011, 364, 1195–1206. [Google Scholar] [CrossRef] [Green Version]
- Manns, M.; Marcellin, P.; Poordad, F.; De Araujo, E.S.A.; Buti, M.; Horsmans, Y.; Janczewska, E.; Villamil, F.; Scott, J.; Peeters, M.; et al. Simeprevir with pegylated interferon alfa 2a or 2b plus ribavirin in treatment-naive patients with chronic hepatitis C virus genotype 1 infection (QUEST-2): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2014, 384, 414–426. [Google Scholar] [CrossRef]
- Lawitz, E.; Mangia, A.; Wyles, D.; Rodriguez-Torres, M.; Hassanein, T.; Gordon, S.C.; Schultz, M.; Davis, M.N.; Kayali, Z.; Reddy, K.R.; et al. Sofosbuvir for Previously Untreated Chronic Hepatitis C Infection. N. Engl. J. Med. 2013, 368, 1878–1887. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, I.M.; Gordon, S.C.; Kowdley, K.V.; Yoshida, E.M.; Rodriguez-Torres, M.; Sulkowski, M.S.; Shiffman, M.L.; Lawitz, E.; Everson, G.; Bennett, M.; et al. Sofosbuvir for Hepatitis C Genotype 2 or 3 in Patients without Treatment Options. N. Engl. J. Med. 2013, 368, 1867–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowdley, K.V.; Gordon, S.C.; Reddy, K.R.; Rossaro, L.; Bernstein, D.E.; Lawitz, E.; Shiffman, M.L.; Schiff, E.; Ghalib, R.; Ryan, M.; et al. Ledipasvir and Sofosbuvir for 8 or 12 Weeks for Chronic HCV without Cirrhosis. N. Engl. J. Med. 2014, 370, 1879–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afdhal, N.; Zeuzem, S.; Kwo, P.; Chojkier, M.; Gitlin, N.; Puoti, M.; Romero-Gomez, M.; Zarski, J.-P.; Agarwal, K.; Buggisch, P.; et al. Ledipasvir and Sofosbuvir for Untreated HCV Genotype 1 Infection. N. Engl. J. Med. 2014, 370, 1889–1898. [Google Scholar] [CrossRef] [Green Version]
- Afdhal, N.; Reddy, K.R.; Nelson, D.R.; Lawitz, E.; Gordon, S.C.; Schiff, E.; Nahass, R.; Ghalib, R.; Gitlin, N.; Herring, R.; et al. Ledipasvir and Sofosbuvir for Previously Treated HCV Genotype 1 Infection. N. Engl. J. Med. 2014, 370, 1483–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawitz, E.; Sulkowski, M.S.; Ghalib, R.; Rodriguez-Torres, M.; Younossi, Z.M.; Corregidor, A.; Dejesus, E.; Pearlman, B.; Rabinovitz, M.; Gitlin, N.; et al. Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non-responders to pegylated interferon and ribavirin and treatment-naive patients: The COSMOS randomised study. Lancet 2014, 384, 1756–1765. [Google Scholar] [CrossRef]
- Poordad, F.; Hezode, C.; Trinh, R.; Kowdley, K.V.; Zeuzem, S.; Agarwal, K.; Shiffman, M.L.; Wedemeyer, H.; Berg, T.; Yoshida, E.M.; et al. ABT-450/r–Ombitasvir and Dasabuvir with Ribavirin for Hepatitis C with Cirrhosis. N. Engl. J. Med. 2014, 370, 1973–1982. [Google Scholar] [CrossRef] [Green Version]
- Feld, J.J.; Kowdley, K.V.; Coakley, E.; Sigal, S.; Nelson, D.R.; Crawford, D.; Weiland, O.; Aguilar, H.; Xiong, J.; Pilot-Matias, T.; et al. Treatment of HCV with ABT-450/r–Ombitasvir and Dasabuvir with Ribavirin. N. Engl. J. Med. 2014, 370, 1594–1603. [Google Scholar] [CrossRef] [Green Version]
- Li, D.K.; Chung, R.T. Overview of direct-acting antiviral drugs and drug resistance of hepatitis C virus. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2019; Volume 1911, pp. 3–32. [Google Scholar]
- Lazarus, J.V.; Roel, E.; Elsharkawy, A.M. Hepatitis c virus epidemiology and the impact of interferon-free hepatitis c virus therapy. Cold Spring Harb. Perspect. Med. 2020, 10, a036913. [Google Scholar] [CrossRef]
- Wang, G.; Dyatkina, N.; Prhavc, M.; Williams, C.; Serebryany, V.; Hu, Y.; Huang, Y.; Wu, X.; Chen, T.; Huang, W.; et al. Synthesis and Anti-HCV Activity of Sugar-Modified Guanosine Analogues: Discovery of AL-611 as an HCV NS5B Polymerase Inhibitor for the Treatment of Chronic Hepatitis C. J. Med. Chem. 2020, 63, 10380–10395. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Dyatkina, N.; Prhavc, M.; Williams, C.; Serebryany, V.; Hu, Y.; Huang, Y.; Wan, J.; Wu, X.; Deval, J.; et al. Synthesis and Anti-HCV Activities of 4′-Fluoro-2′-Substituted Uridine Triphosphates and Nucleotide Prodrugs: Discovery of 4′-Fluoro-2′-C-methyluridine 5′-Phosphoramidate Prodrug (AL-335) for the Treatment of Hepatitis C Infection. J. Med. Chem. 2019, 62, 4555–4570. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.Y.; Shotwell, J.B.; Miller, J.; Price, D.J.; Maynard, A.; Voitenleitner, C.; Mathis, A.; Williams, S.; Pouliot, J.J.; Creech, K.; et al. Design of N-Benzoxaborole Benzofuran GSK8175—Optimization of Human Pharmacokinetics Inspired by Metabolites of a Failed Clinical HCV Inhibitor. J. Med. Chem. 2019, 62, 3254–3267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, P.; Kang, D.; Liu, X. Resurrecting the Condemned: Identification of N-Benzoxaborole Benzofuran GSK8175 as a Clinical Candidate with Reduced Metabolic Liability. J. Med. Chem. 2019, 62, 3251–3253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramdas, V.; Talwar, R.; Banerjee, M.; Joshi, A.A.; Das, A.K.; Walke, D.S.; Borhade, P.; Dhayagude, U.; Loriya, R.; Gote, G.; et al. Discovery and Characterization of Potent Pan-Genotypic HCV NS5A Inhibitors Containing Novel Tricyclic Central Core Leading to Clinical Candidate. J. Med. Chem. 2019, 62, 10563–10582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Gai, K.; Qin, H.; Wang, J.; Liu, X.; Cao, Y.; Lu, Q.; Lu, D.; Chen, D.; Shen, H.; et al. Discovery of a Silicon-Containing Pan-Genotype Hepatitis C Virus NS5A Inhibitor. J. Med. Chem. 2020, 63, 5312–5323. [Google Scholar] [CrossRef]
- Kazmierski, W.M.; Baskaran, S.; Walker, J.T.; Miriyala, N.; Meesala, R.; Beesu, M.; Adjabeng, G.; Grimes, R.M.; Hamatake, R.; Leivers, M.R.; et al. GSK2818713, a Novel Biphenylene Scaffold-Based Hepatitis C NS5A Replication Complex Inhibitor with Broad Genotype Coverage. J. Med. Chem. 2020, 63, 4155–4170. [Google Scholar] [CrossRef]
- Jiang, X.; Tan, J.; Wang, Y.; Chen, J.; Li, J.; Li, J.; Jiang, Z.; Quan, Y.; Jin, J.; Li, Y.; et al. 2-((4-Arylpiperazin-1-yl)methyl)benzonitrile Derivatives as Orally Available Inhibitors of Hepatitis C Virus with a Novel Mechanism of Action. J. Med. Chem. 2020, 63, 5972–5989. [Google Scholar] [CrossRef]
- Perales, C. Quasispecies dynamics and clinical significance of hepatitis C virus (HCV) antiviral resistance. Int. J. Antimicrob. Agents 2020, 56, 105562. [Google Scholar] [CrossRef]
- Dietz, J.; Susser, S.; Vermehren, J.; Peiffer, K.H.; Grammatikos, G.; Berger, A.; Ferenci, P.; Buti, M.; Müllhaupt, B.; Hunyady, B.; et al. Patterns of Resistance-Associated Substitutions in Patients With Chronic HCV Infection Following Treatment With Direct-Acting Antivirals. Gastroenterology 2018, 154, 976–988. [Google Scholar] [CrossRef] [Green Version]
- Pawlotsky, J.M. Hepatitis C Virus Resistance to Direct-Acting Antiviral Drugs in Interferon-Free Regimens. Gastroenterology 2016, 151, 70–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarrazin, C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J. Hepatol. 2016, 64, 486–504. [Google Scholar] [CrossRef] [PubMed]
- Childs, K.; Davis, C.; Cannon, M.; Montague, S.; Filipe, A.; Tong, L.; Simmonds, P.; Smith, D.; Thomson, E.C.; Dusheiko, G.; et al. Suboptimal SVR rates in African patients with atypical genotype 1 subtypes: Implications for global elimination of hepatitis C. J. Hepatol. 2019, 71, 1099–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottwein, J.M.; Pham, L.V.; Mikkelsen, L.S.; Ghanem, L.; Ramirez, S.; Scheel, T.K.H.; Carlsen, T.H.R.; Bukh, J. Efficacy of NS5A Inhibitors Against Hepatitis C Virus Genotypes 1–7 and Escape Variants. Gastroenterology 2018, 154, 1435–1448. [Google Scholar] [CrossRef]
- Pawlotsky, J.M.; Negro, F.; Aghemo, A.; Berenguer, M.; Dalgard, O.; Dusheiko, G.; Marra, F.; Puoti, M.; Wedemeyer, H. EASL recommendations on treatment of hepatitis C: Final update of the series. J. Hepatol. 2020, 73, 1170–1218. [Google Scholar] [CrossRef]
- Aparicio-Puerta, E.; Lebrón, R.; Rueda, A.; Gómez-Martín, C.; Giannoukakos, S.; Jaspez, D.; Medina, J.M.; Zubkovic, A.; Jurak, I.; Fromm, B.; et al. SRNAbench and sRNAtoolbox 2019: Intuitive fast small RNA profiling and differential expression. Nucleic Acids Res. 2019, 47, W530–W535. [Google Scholar] [CrossRef] [Green Version]
- Chevaliez, S.; Rodriguez, C.; Pawlotsky, J.M. New virologic tools for management of chronic hepatitis B and C. Gastroenterology 2012, 142, 1303–1313. [Google Scholar] [CrossRef] [Green Version]
- Jensen, S.B.; Fahnøe, U.; Pham, L.V.; Serre, S.B.N.; Tang, Q.; Ghanem, L.; Pedersen, M.S.; Ramirez, S.; Humes, D.; Pihl, A.F.; et al. Evolutionary Pathways to Persistence of Highly Fit and Resistant Hepatitis C Virus Protease Inhibitor Escape Variants. Hepatology 2019, 70, 771–787. [Google Scholar] [CrossRef]
- Bourlière, M.; Gordon, S.C.; Flamm, S.L.; Cooper, C.L.; Ramji, A.; Tong, M.; Ravendhran, N.; Vierling, J.M.; Tran, T.T.; Pianko, S.; et al. Sofosbuvir, Velpatasvir, and Voxilaprevir for Previously Treated HCV Infection. N. Engl. J. Med. 2017, 376, 2134–2146. [Google Scholar] [CrossRef]
- Poordad, F.; Pol, S.; Asatryan, A.; Buti, M.; Shaw, D.; Hézode, C.; Felizarta, F.; Reindollar, R.W.; Gordon, S.C.; Pianko, S.; et al. Glecaprevir/Pibrentasvir in patients with hepatitis C virus genotype 1 or 4 and past direct-acting antiviral treatment failure. Hepatology 2018, 67, 1253–1260. [Google Scholar] [CrossRef]
- Nguyen, D.; Smith, D.; Vaughan-Jackson, A.; Magri, A.; Barnes, E.; Simmonds, P. Efficacy of NS5A inhibitors against unusual and potentially difficult-to-treat HCV subtypes commonly found in sub-Saharan Africa and South East Asia. J. Hepatol. 2020, 73, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, A.; Gimson, A.; Agarwal, K.; Aldersley, M.; Bathgate, A.; MacDonald, D.; McPherson, S.; Mutimer, D.; Gelson, W. Liver transplant listing for hepatitis C-associated cirrhosis and hepatocellular carcinoma has fallen in the United Kingdom since the introduction of direct-acting antiviral therapy. J. Viral Hepat. 2019, 26, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Carrat, F.; Fontaine, H.; Dorival, C.; Simony, M.; Diallo, A.; Hezode, C.; De Ledinghen, V.; Larrey, D.; Haour, G.; Bronowicki, J.P.; et al. Clinical outcomes in patients with chronic hepatitis C after direct-acting antiviral treatment: A prospective cohort study. Lancet 2019, 393, 1453–1464. [Google Scholar] [CrossRef]
- Pawlotsky, J.M. Retreatment of Hepatitis C Virus-Infected Patients with Direct-Acting Antiviral Failures. Semin. Liver Dis. 2019, 39, 354–368. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| DAA Virus Target | DAA Name |
|---|---|
| NS3/4A inhibitor | Telaprevir a |
| Boceprevir a | |
| Semiprevir | |
| Paritaprevir | |
| Grazoprevir | |
| Voxilaprevir | |
| Glecaprevir | |
| NS3/4A booster | Ritonavir |
| NS5A inhibitor | Ledivaspir |
| Ombitasvir | |
| Daclatasvir | |
| Elbasvir | |
| Velpatasvir | |
| Pibrentasvir | |
| NS5B inhibitor | Sofosbuvir |
| Dasabuvir |
| Genome Region Drug Class a | Amino Acid Position | Genotype | ||||||
|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 2 | 3 | 4 | 5 | 6 | ||
| NS3 Protease Inhibitors: Semiprevir, Paritaprevir, Grazoprevir, Voxilaprevir and Glecaprevir b | ||||||||
| 36 | V36A/C/F/G/L/M | V36A/C/G/L/M | V36I | |||||
| 41 | Q41R | Q41R | Q41K | Q41R | Q41K/R | |||
| 43 | F43I/L/S/V | F43I/S/V | F43V | |||||
| 54 | T54A/S | T54A/C/G/S | ||||||
| 55 | V55I | V55A | V55A/I | |||||
| 56 | Y56H | Y56H/L/F | Y56H/F | Y56H | Y56H | Y56H | ||
| 80 | Q80K/L/R | Q80H/K/L/R | Q80K/R | Q80R | L80K/Q | |||
| 122 | S122G/N/R | S122A/D/G/I/N/R/T | S122T | |||||
| 155 | R155G/I/K/M/Q/S/T/V/W | R155C/G/I/K/L/Q/M/S/T/W | R155K | R155C/K | R155K | |||
| 156 | A156G/P/S/T/V | A156G/P/S/T/V | A156L/M/T/V | A156G/P/T/V | A156G/H/K/L/S/T/V | A156T/V | A156T/V | |
| 158 | V158I | V158I | ||||||
| 166 | A166S/T/Y | |||||||
| 168 | D168A/C/E/F/G/H/I/K/L/N/Q/R/T/V/Y | D168A/C/E/F/G/H/I/K/L/N/Q/T/V/Y | D168A/E/F/G/H/N/S/T/V/Y | Q168H/K/L/R | D168A/E/G/H/T/V | D168A/E/H/K/R/V/Y | D168A/E/G/H/V/Y | |
| 170 | I/V170T/V | I/V170A/L/T | I170V | |||||
| 175 | M175L | |||||||
| NS5A Inhibitors: Ledivaspir, Ombitasvir, Daclatasvir, Elbasvir, Velpatasvir and Pibrentasvir | ||||||||
| 24 | K24E/QR/T | Q24K | T24A/S | S24F | ||||
| 26 | K26E | |||||||
| 28 | M28A/G/S/T/V | L28A/M/T | L/F28C/S | M28T/K | L28M/S/T/V | L28I | F/L28A/I/L/M/T/V | |
| 29 | 29 P29R | P29S, del29 | P29S | |||||
| 30 | Q30C/D/E/G/H/K/L/N/R/T/Y, del30 | R30G/H/P/Q/S | L30H/S | A30D/E/K/S | L30F/G/H/R/S | Q30H | R30E/H/N/S | |
| 31 | L31I/F/M/P/V | L31F/I/M/V/W | L31I/M/V | L31F/I/M/P/V | M/L31I/V | L31F/I/V | L31I/M/V | |
| 32 | P32L/S, del32 | P32F/L/S, del32 | P32L | P32A/L/Q/R/S | ||||
| 38 | S38F | |||||||
| 58 | H58C/D/L/P/R | P58A/D/L/S/R/T | T58A/P/S | T58A/G/H/N/S | ||||
| 62 | Q/E62D | S62L | ||||||
| 92 | A92K/T | A92E/K/T/V | C92R/S/T/W | E92K | E92T | |||
| 93 | Y93C/F/H/L/N/R/S/T/W | Y93C/H/N/R/S/T | Y93F/N/H | Y93H/N/S | Y93C/H/N/S/R/W | T93A/H/N/S | ||
| NS5B Non-nucleoside Inhibitors: Dasabuvir | ||||||||
| 314 | L314H | |||||||
| 316 | C316Y | C316H/N/Y/W | ||||||
| 368 | S368T | |||||||
| 395 | A395G | |||||||
| 411 | ||||||||
| 414 | M414I/T/V | M414I/T/V | ||||||
| 445 | C445F/Y | |||||||
| 446 | E446K/Q | |||||||
| 448 | Y448C/H | Y448C/H | ||||||
| 553 | A553T/V | A553V | ||||||
| 554 | G554S | G554S | ||||||
| 555 | Y555H | |||||||
| 556 | S556G/R | S556G/R | ||||||
| 557 | G557R | |||||||
| 558 | G558R | G558R | ||||||
| 559 | D559G/N | D559G/N | ||||||
| 561 | Y561H/N | |||||||
| 565 | S565F | |||||||
| NS5B Nucleotide analogue Inhibitors: Sofosbuvir | ||||||||
| 150 | A150V | |||||||
| 159 | L159F | L159F | L159F | L159F | ||||
| 206 | K206E | |||||||
| 282 | S282G/R/T | S282G/R/T | S282G/R/T | S282G/R/T | S282C/G/R/T | S282G/R/T | S282G/R/T | |
| 316 | C316H/R | C316F/H/N | ||||||
| 320 | L320I/F/V | |||||||
| 321 | V321A | V321I | V321A | V321A | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez, M.A.; Franco, S. Therapy Implications of Hepatitis C Virus Genetic Diversity. Viruses 2021, 13, 41. https://doi.org/10.3390/v13010041
Martinez MA, Franco S. Therapy Implications of Hepatitis C Virus Genetic Diversity. Viruses. 2021; 13(1):41. https://doi.org/10.3390/v13010041
Chicago/Turabian StyleMartinez, Miguel Angel, and Sandra Franco. 2021. "Therapy Implications of Hepatitis C Virus Genetic Diversity" Viruses 13, no. 1: 41. https://doi.org/10.3390/v13010041
APA StyleMartinez, M. A., & Franco, S. (2021). Therapy Implications of Hepatitis C Virus Genetic Diversity. Viruses, 13(1), 41. https://doi.org/10.3390/v13010041