
Turnover of SARS-CoV-2 Lineages Shaped the Pandemic and Enabled the Emergence of New Variants in the State of Rio de Janeiro, Brazil

 , , , , ,
, , , , ,  , , , , , and add
Show full author list
, , , , , and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling, Genome Extraction, Sequencing, and Assembly
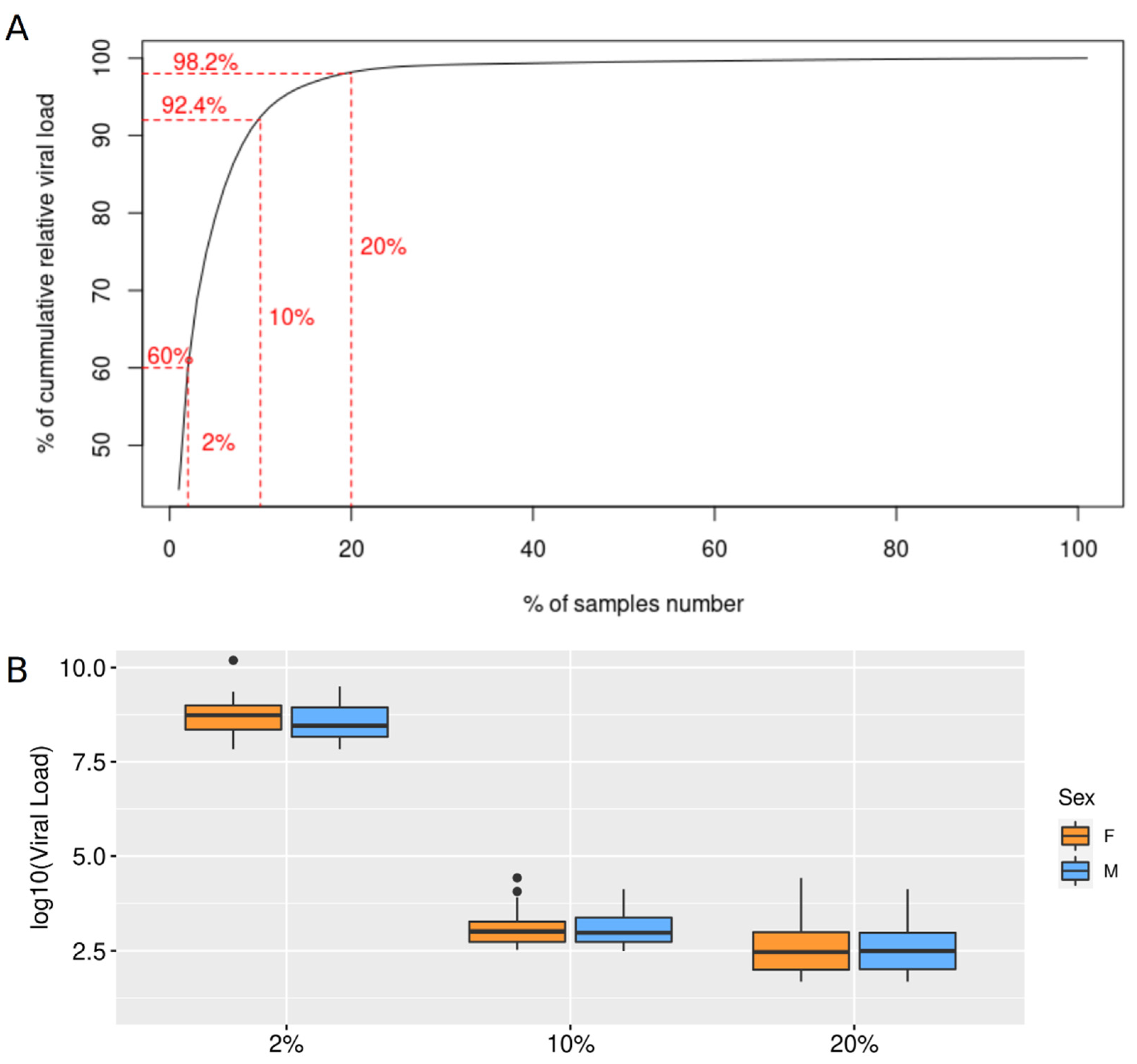
2.2. Epidemiology and Viral Load Analysis
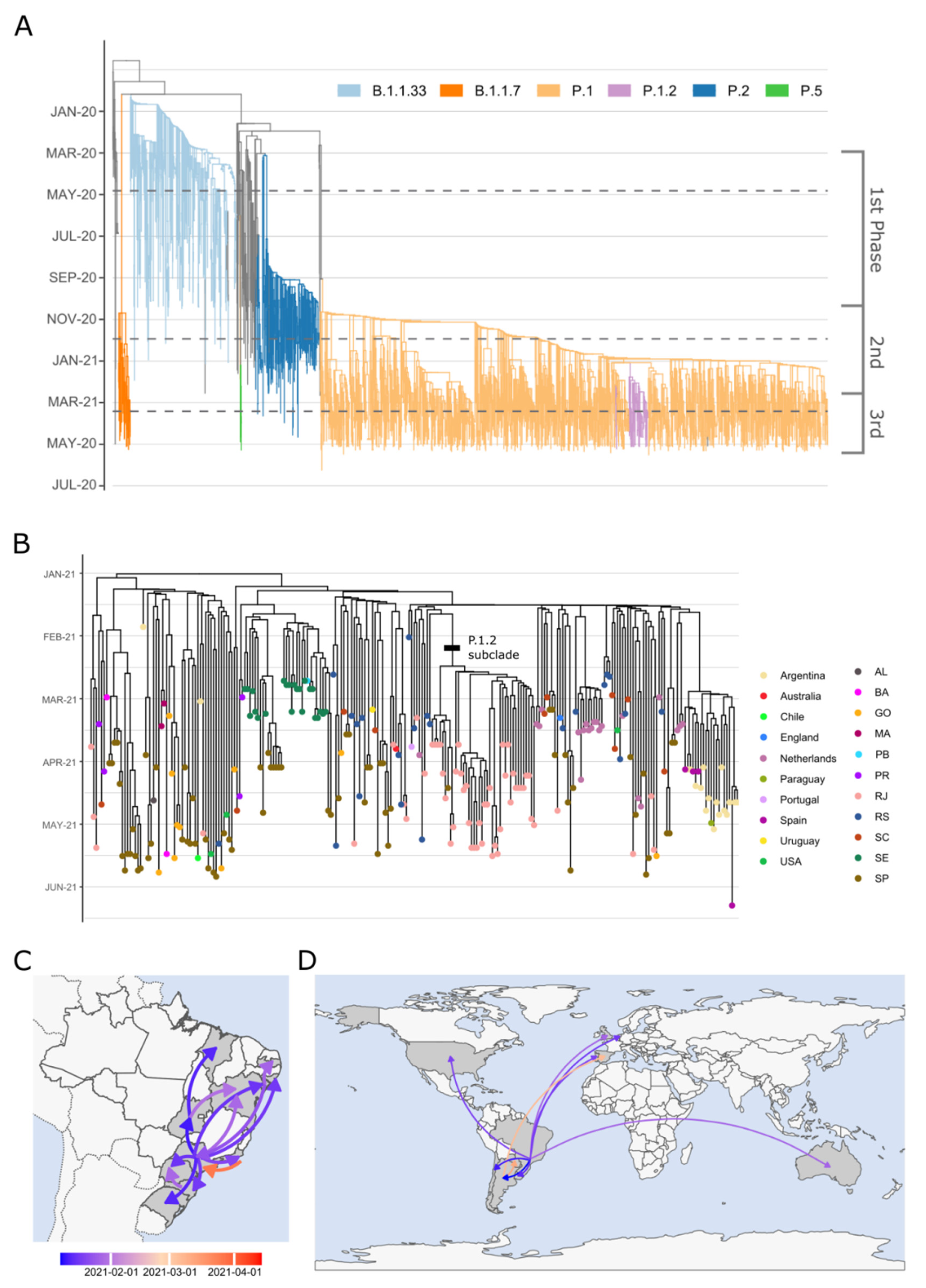
2.3. Evolutionary Analyses
2.4. Structural Analysis of Spike Protein of P.1 and P.1.2 Lineages
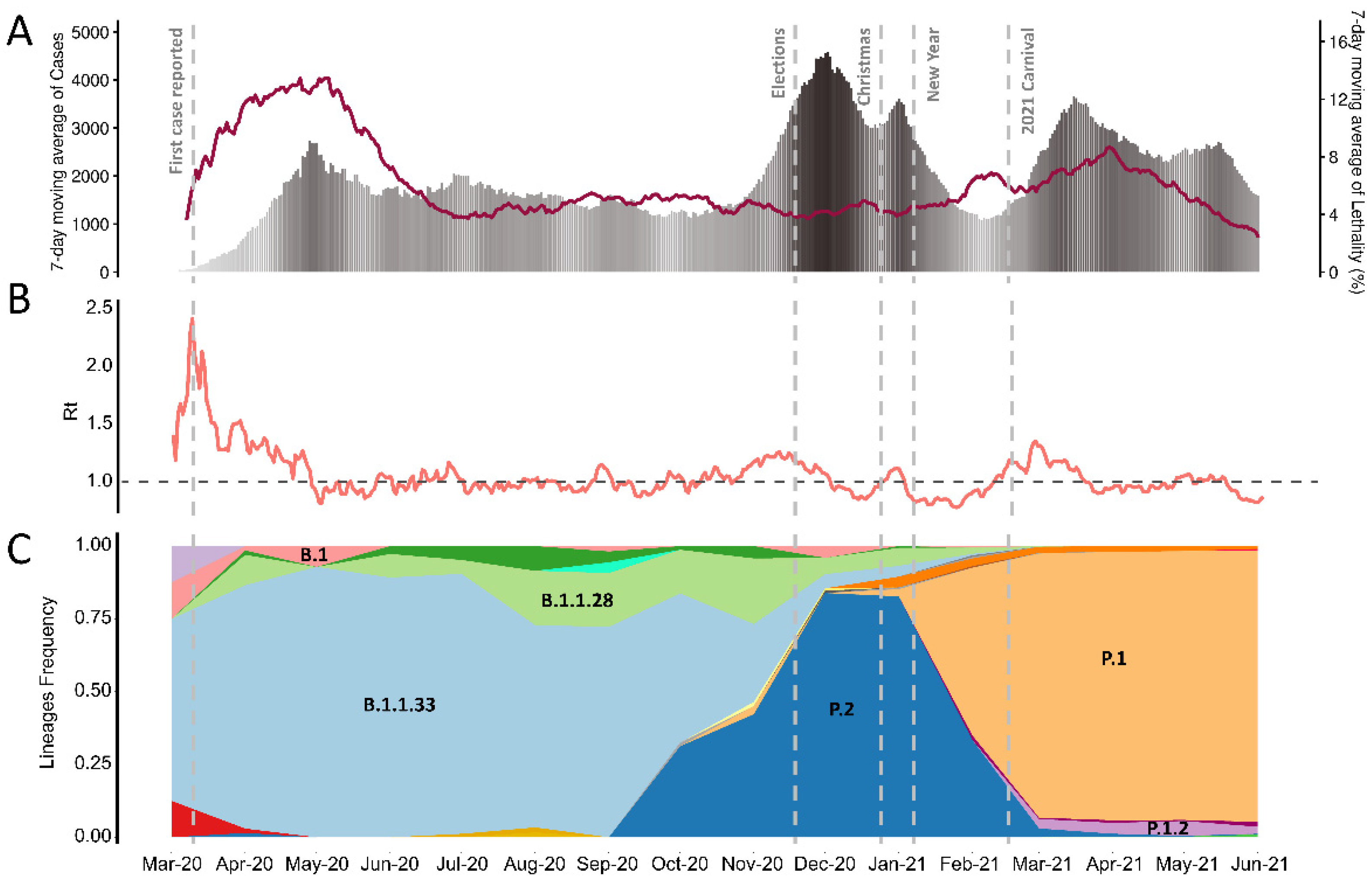
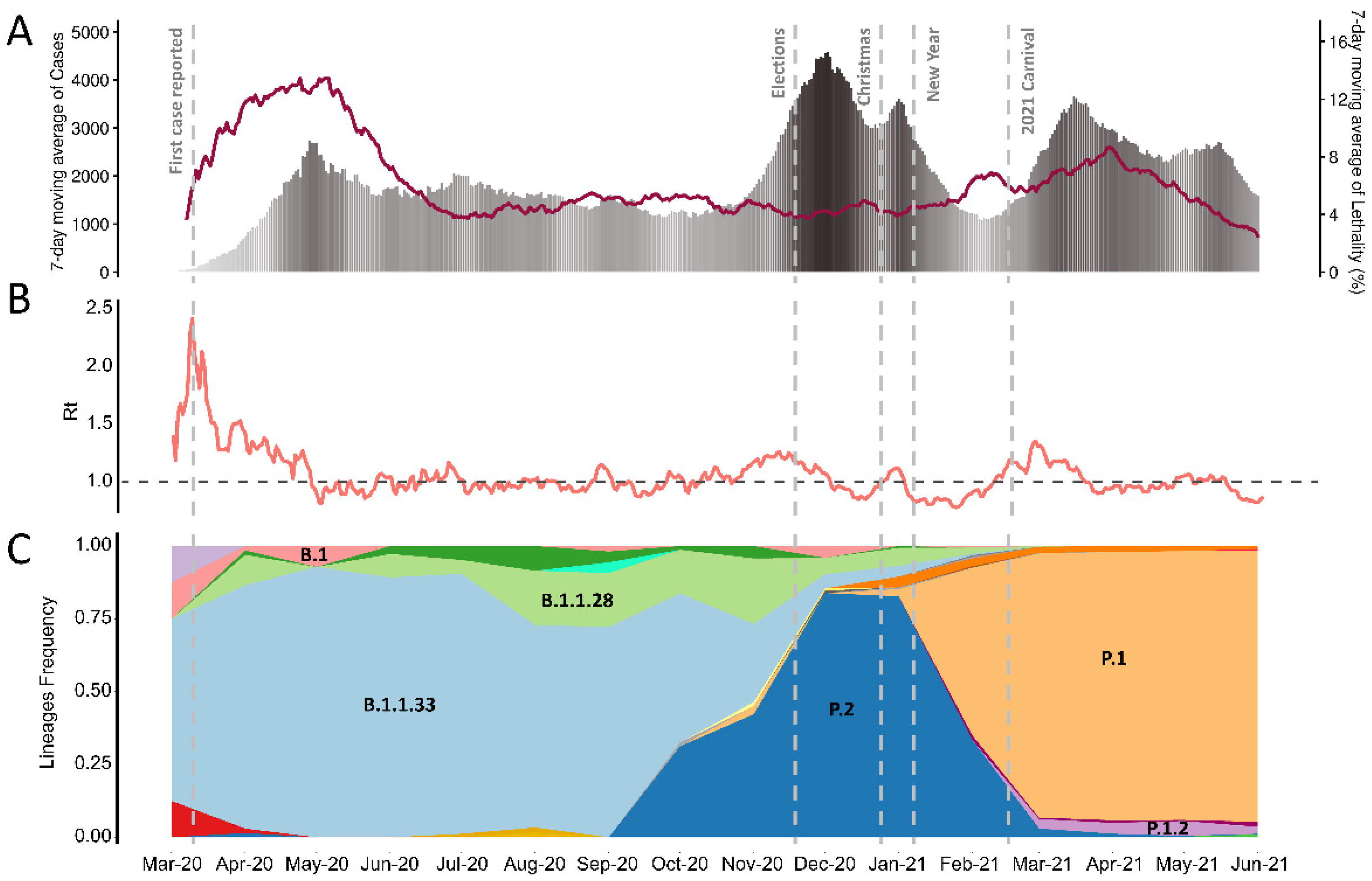
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. Addendum: A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2021, 6, 415. [Google Scholar] [CrossRef]
- Davis, C.; Logan, N.; Tyson, G.; Orton, R.; Harvey, W.; Haughney, J.; Perkins, J.; Peacock, T.P.; Barclay, W.S.; Cherepanov, P.; et al. Reduced Neutralisation of the Delta (B.1.617.2) SARS-CoV-2 Variant of Concern Following Vaccination. bioRxiv 2021. [Google Scholar] [CrossRef]
- Goes, L.R.; Siqueira, J.D.; Garrido, M.M.; Alves, B.M.; Pereira, A.C.P.M.; Cicala, C.; Arthos, J.; Viola, J.P.B.; Soares, M.A.; INCA COVID-19 Task Force. New Infections by SARS-CoV-2 Variants of Concern after Natural Infections and Post-Vaccination in Rio de Janeiro, Brazil. Infect. Genet. Evol. 2021, 94, 104998. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; Lam, E.C.; St Denis, K.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 Variants Escape Neutralization by Vaccine-Induced Humoral Immunity. Cell 2021, 184, 2372–2383.e9. [Google Scholar] [CrossRef]
- Bolze, A.; Cirulli, E.T.; Luo, S.; White, S.; Wyman, D.; Dei Rossi, A.; Cassens, T.; Jacobs, S.; Nguyen, J.; Ramirez, J.M., III; et al. Rapid Displacement of SARS-CoV-2 Variant B.1.1.7 by B.1.617.2 and P.1 in the United States. bioRxiv 2021. [Google Scholar] [CrossRef]
- Chen, Z.; Chong, K.C.; Wong, M.C.S.; Boon, S.S.; Huang, J.; Wang, M.H.; Ng, R.W.Y.; Lai, C.K.C.; Chan, P.K.S. A Global Analysis of Replacement of Genetic Variants of SARS-CoV-2 in Association with Containment Capacity and Changes in Disease Severity. Clin. Microbiol. Infect. 2021, 27, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 Variant of Concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Resende, P.C.; Delatorre, E.; Gräf, T.; Mir, D.; Motta, F.C.; Appolinario, L.R.; da Paixão, A.C.D.; da Fonseca Mendonça, A.C.; Ogrzewalska, M.; Caetano, B.; et al. Evolutionary Dynamics and Dissemination Pattern of the SARS-CoV-2 Lineage B.1.1.33 During the Early Pandemic Phase in Brazil. Front. Microbiol. 2020, 11, 615280. [Google Scholar] [CrossRef]
- Candido, D.S.; Claro, I.M.; de Jesus, J.G.; Souza, W.M.; Moreira, F.R.R.; Dellicour, S.; Mellan, T.A.; du Plessis, L.; Pereira, R.H.M.; Sales, F.C.S.; et al. Evolution and Epidemic Spread of SARS-CoV-2 in Brazil. Science 2020, 369, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.P.; de Almeida, L.G.P.; da Silva Francisco, R.; Lima, L.F.A.; Scortecci, K.C.; Perez, V.P.; Brustolini, O.J.; Sousa, E.S.S.; Secco, D.A.; Santos, A.M.G.; et al. Genomic Surveillance of SARS-CoV-2 Tracks Early Interstate Transmission of P.1 Lineage and Diversification within P.2 Clade in Brazil. medRxiv 2021. [Google Scholar] [CrossRef]
- Moreira, F.R.R.; D’arc, M.; Mariani, D.; Herlinger, A.L.; Schiffler, F.B.; Rossi, Á.D.; de Carvalho Leitão, I.; dos Santos Miranda, T.; Cosentino, M.A.C.; de Paula Tôrres, M.C.; et al. Epidemiological Dynamics of SARS-CoV-2 VOC Gamma in Rio de Janeiro, Brazil. medRxiv 2021. [Google Scholar] [CrossRef]
- Voloch, C.M.; da Silva Francisco, R.; de Almeida, L.G.P.; Cardoso, C.C.; Brustolini, O.J.; Gerber, A.L.; Guimarães, A.P.D.C.; Mariani, D.; da Costa, R.M.; Ferreira, O.C.; et al. Genomic Characterization of a Novel SARS-CoV-2 Lineage from Rio de Janeiro, Brazil. J. Virol. 2021, 95, 10. [Google Scholar] [CrossRef]
- Cori, A.; Ferguson, N.M.; Fraser, C.; Cauchemez, S. A New Framework and Software to Estimate Time-Varying Reproduction Numbers During Epidemics. Am. J. Epidemiol. 2013, 178, 1505–1512. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. Available online: https://www.R-project.org/ (accessed on 20 July 2021).
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Frith, M.C. Adding Unaligned Sequences into an Existing Alignment Using MAFFT and LAST. Bioinformatics 2012, 28, 3144–3146. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-Likelihood Phylodynamic Analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian Evolutionary Analysis by Sampling Trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Karcher, M.D.; Palacios, J.A.; Lan, S.; Minin, V.N. Phylodyn: An R Package for Phylodynamic Simulation and Inference. Mol. Ecol. Resour. 2017, 17, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Dellicour, S.; Rose, R.; Faria, N.R.; Lemey, P.; Pybus, O.G. SERAPHIM: Studying Environmental Rasters and Phylogenetically Informed Movements. Bioinformatics 2016, 32, 3204–3206. [Google Scholar] [CrossRef]
- Buß, O.; Rudat, J.; Ochsenreither, K. FoldX as Protein Engineering Tool: Better Than Random Based Approaches? Comput. Struct. Biotechnol. J. 2018, 16, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Sarakatsannis, J.N.; Duan, Y. Statistical Characterization of Salt Bridges in Proteins. Proteins 2005, 60, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Tao, H.; He, J.; Huang, S.-Y. The HDOCK Server for Integrated Protein–protein Docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef] [PubMed]
- Raybould, M.I.J.; Kovaltsuk, A.; Marks, C.; Deane, C.M. CoV-AbDab: The Coronavirus Antibody Database. Bioinformatics 2021, 37, 734–735. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A Web Server for Predicting the Binding Affinity of Protein-Protein Complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef]
- Darnell, S.J.; LeGault, L.; Mitchell, J.C. KFC Server: Interactive Forecasting of Protein Interaction Hot Spots. Nucleic Acids Res. 2008, 36, W265–W269. [Google Scholar] [CrossRef] [Green Version]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An Automated Pipeline for the Setup of Poisson-Boltzmann Electrostatics Calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Ciceri, F.; Ruggeri, A.; Lembo, R.; Puglisi, R.; Landoni, G.; Zangrillo, A. COVID-BioB Study Group Decreased in-Hospital Mortality in Patients with COVID-19 Pneumonia. Pathog. Glob. Health 2020, 114, 281–282. [Google Scholar] [CrossRef]
- Horwitz, L.I.; Jones, S.A.; Cerfolio, R.J.; Francois, F.; Greco, J.; Rudy, B.; Petrilli, C.A. Trends in Covid-19 Risk-Adjusted Mortality Rates in a Single Health System. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sun, Q.; Qiu, H.; Huang, M.; Yang, Y. Lower Mortality of COVID-19 by Early Recognition and Intervention: Experience from Jiangsu Province. Ann. Intensive Care 2020, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Victora, C.; Castro, M.C.; Gurzenda, S.; de Medeiros, A.C.; França, G.; Barros, A.J.D. Estimating the Early Impact of Vaccination against COVID-19 on Deaths among Elderly People in Brazil: Analyses of Routinely-Collected Data on Vaccine Coverage and Mortality. bioRxiv 2021. [Google Scholar] [CrossRef]
- de Souza, F.S.H.; Hojo-Souza, N.S.; da Silva, C.M.; Guidoni, D.L. Second Wave of COVID-19 in Brazil: Younger at Higher Risk. Eur. J. Epidemiol. 2021, 36, 441–443. [Google Scholar] [CrossRef] [PubMed]
- Bono, L.M.; Gensel, C.L.; Pfennig, D.W.; Burch, C.L. Competition and the Origins of Novelty: Experimental Evolution of Niche-Width Expansion in a Virus. Biol. Lett. 2013, 9, 20120616. [Google Scholar] [CrossRef] [Green Version]
- Freitas, A.R.R.; Beckedorff, O.A.; Cavalcanti, L.P.D.G.; Siqueira, A.M.; Castro, D.B.; Costa, C.F.D.; Lemos, D.R.Q.; Barros, E.N.C. The Emergence of Novel SARS-CoV-2 Variant P.1 in Amazonas (Brazil) Was Temporally Associated with a Change in the Age and Gender Profile of COVID-19 Mortality. SSRN Electron. J. 2021. [Google Scholar] [CrossRef]
- De Oliveira, M.H.S.; Lippi, G.; Henry, B.M. Sudden Rise in COVID-19 Case Fatality among Young and Middle-Aged Adults in the South of Brazil after Identification of the Novel B.1.1.28.1 (P.1) SARS-CoV-2 Strain: Analysis of Data from the State of Parana. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hanley, K.A.; Nelson, J.T.; Schirtzinger, E.E.; Whitehead, S.S.; Hanson, C.T. Superior Infectivity for Mosquito Vectors Contributes to Competitive Displacement among Strains of Dengue Virus. BMC Ecol. 2008, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Yuen, J.E. Modelling Pathogen Competition and Displacement– Phytophthora Infestans in Scandinavia. Eur. J. Plant Pathol. 2012, 133, 25–32. [Google Scholar] [CrossRef]
- Wolf, Y.I.; Viboud, C.; Holmes, E.C.; Koonin, E.V.; Lipman, D.J. Long Intervals of Stasis Punctuated by Bursts of Positive Selection in the Seasonal Evolution of Influenza A Virus. Biol. Direct 2006, 1, 34. [Google Scholar] [CrossRef] [Green Version]
- Campbell, F.; Archer, B.; Laurenson-Schafer, H.; Jinnai, Y.; Konings, F.; Batra, N.; Pavlin, B.; Vandemaele, K.; Van Kerkhove, M.D.; Jombart, T.; et al. Increased Transmissibility and Global Spread of SARS-CoV-2 Variants of Concern as at June 2021. Eurosurveillance 2021, 26, 2100509. [Google Scholar] [CrossRef]
- Di Giallonardo, F.; Puglia, I.; Curini, V.; Cammà, C.; Mangone, I.; Calistri, P.; Cobbin, J.C.A.; Holmes, E.C.; Lorusso, A. Emergence and Spread of SARS-CoV-2 Lineages B.1.1.7 and P.1 in Italy. Viruses 2021, 13, 794. [Google Scholar] [CrossRef]
- Sallam, M.; Mahafzah, A. Molecular Analysis of SARS-CoV-2 Genetic Lineages in Jordan: Tracking the Introduction and Spread of COVID-19 UK Variant of Concern at a Country Level. Pathogens 2021, 10, 302. [Google Scholar] [CrossRef]
- Challen, R.; Dyson, L.; Overton, C.E.; Guzman-Rincon, L.M.; Hill, E.M.; Stage, H.B.; Brooks-Pollock, E.; Pellis, L.; Scarabel, F.; Pascall, D.J.; et al. Early Epidemiological Signatures of Novel SARS-CoV-2 Variants: Establishment of B.1.617.2 in England. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wilkinson, E.; Giovanetti, M.; Tegally, H.; San, J.E.; Lessels, R.; Cuadros, D.; Martin, D.P.; Zekri, A.-R.N.; Sangare, A.K.; Ouedraogo, A.-S.; et al. A Year of Genomic Surveillance Reveals How the SARS-CoV-2 Pandemic Unfolded in Africa. medRxiv 2021. [Google Scholar] [CrossRef]
- Morato, M.M.; Bastos, S.B.; Cajueiro, D.O.; Normey-Rico, J.E. An Optimal Predictive Control Strategy for COVID-19 (SARS-CoV-2) Social Distancing Policies in Brazil. Annu. Rev. Control 2020, 50, 417–431. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Lau, E.H.Y.; Wu, P.; Deng, X.; Wang, J.; Hao, X.; Lau, Y.C.; Wong, J.Y.; Guan, Y.; Tan, X.; et al. Temporal Dynamics in Viral Shedding and Transmissibility of COVID-19. Nat. Med. 2020, 26, 672–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, M.; Millat-Martinez, P.; Ouchi, D.; Roberts, C.H.; Alemany, A.; Corbacho-Monné, M.; Ubals, M.; Tobias, A.; Tebé, C.; Ballana, E.; et al. Transmission of COVID-19 in 282 Clusters in Catalonia, Spain: A Cohort Study. Lancet Infect. Dis. 2021, 21, 629–636. [Google Scholar] [CrossRef]
- Bjorkman, K.K.; Saldi, T.K.; Lasda, E.; Bauer, L.C.; Kovarik, J.; Gonzales, P.K.; Fink, M.R.; Tat, K.L.; Hager, C.R.; Davis, J.C.; et al. Higher Viral Load Drives Infrequent SARS-CoV-2 Transmission between Asymptomatic Residence Hall Roommates. medRxiv 2021. [Google Scholar] [CrossRef]
- Yang, Q.; Saldi, T.K.; Gonzales, P.K.; Lasda, E.; Decker, C.J.; Tat, K.L.; Fink, M.R.; Hager, C.R.; Davis, J.C.; Ozeroff, C.D.; et al. Just 2% of SARS-CoV-2−positive Individuals Carry 90% of the Virus Circulating in Communities. Proc. Natl. Acad. Sci. USA 2021, 118, e2104547118. [Google Scholar] [CrossRef]
- Kissler, S.M.; Fauver, J.R.; Mack, C.; Olesen, S.W.; Tai, C.; Shiue, K.Y.; Kalinich, C.C.; Jednak, S.; Ott, I.M.; Vogels, C.B.F.; et al. Viral Dynamics of Acute SARS-CoV-2 Infection. medRxiv 2021. [Google Scholar] [CrossRef]
- Zou, L.; Ruan, F.; Huang, M.; Liang, L.; Huang, H.; Hong, Z.; Yu, J.; Kang, M.; Song, Y.; Xia, J.; et al. SARS-CoV-2 Viral Load in Upper Respiratory Specimens of Infected Patients. N. Engl. J. Med. 2020, 382, 1177–1179. [Google Scholar] [CrossRef]
- Frankham, R. Relationship of Genetic Variation to Population Size in Wildlife. Conserv. Biol. 1996, 10, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Stevens, M.H.H.; Sanchez, M.; Lee, J.; Finkel, S.E. Diversification Rates Increase with Population Size and Resource Concentration in an Unstructured Habitat. Genetics 2007, 177, 2243–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and Epidemiology of the P.1 SARS-CoV-2 Lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Naveca, F.G.; Nascimento, V.; de Souza, V.C.; Corado, A.L.; Nascimento, F.; Silva, G.; Costa, Á.; Duarte, D.; Pessoa, K.; Mejía, M.; et al. COVID-19 in Amazonas, Brazil, Was Driven by the Persistence of Endemic Lineages and P.1 Emergence. Nat. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, L.G.P.; Lamarca, A.L.; Fracisco Junior, R.S.; Cavalcante, L.; Gerber, A.L.; Guimarães, A.P.C.; Machado, D.T.; Alves, C.; Mariani, D.; Cruz, T.F.; et al. Genomic Surveillance of SARS-CoV-2 in the State of Rio de Janeiro, Brazil: Technical Briefing. Available online: https://virological.org/t/genomic-surveillance-of-sars-cov-2-in-the-state-of-rio-de-janeiro-brazil-technical-briefing/683 (accessed on 14 July 2021).


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francisco Junior, R.d.S.; Lamarca, A.P.; de Almeida, L.G.P.; Cavalcante, L.; Machado, D.T.; Martins, Y.; Brustolini, O.; Gerber, A.L.; Guimarães, A.P.d.C.; Gonçalves, R.B.; et al. Turnover of SARS-CoV-2 Lineages Shaped the Pandemic and Enabled the Emergence of New Variants in the State of Rio de Janeiro, Brazil. Viruses 2021, 13, 2013. https://doi.org/10.3390/v13102013
Francisco Junior RdS, Lamarca AP, de Almeida LGP, Cavalcante L, Machado DT, Martins Y, Brustolini O, Gerber AL, Guimarães APdC, Gonçalves RB, et al. Turnover of SARS-CoV-2 Lineages Shaped the Pandemic and Enabled the Emergence of New Variants in the State of Rio de Janeiro, Brazil. Viruses. 2021; 13(10):2013. https://doi.org/10.3390/v13102013
Chicago/Turabian StyleFrancisco Junior, Ronaldo da Silva, Alessandra P Lamarca, Luiz G P de Almeida, Liliane Cavalcante, Douglas Terra Machado, Yasmmin Martins, Otávio Brustolini, Alexandra L Gerber, Ana Paula de C Guimarães, Reinaldo Bellini Gonçalves, and et al. 2021. "Turnover of SARS-CoV-2 Lineages Shaped the Pandemic and Enabled the Emergence of New Variants in the State of Rio de Janeiro, Brazil" Viruses 13, no. 10: 2013. https://doi.org/10.3390/v13102013
APA StyleFrancisco Junior, R. d. S., Lamarca, A. P., de Almeida, L. G. P., Cavalcante, L., Machado, D. T., Martins, Y., Brustolini, O., Gerber, A. L., Guimarães, A. P. d. C., Gonçalves, R. B., Alves, C., Mariani, D., Cruz, T. F., de Souza, I. V., de Carvalho, E. M., Ribeiro, M. S., Carvalho, S., da Silva, F. D., Garcia, M. H. d. O., ... de Vasconcelos, A. T. R. (2021). Turnover of SARS-CoV-2 Lineages Shaped the Pandemic and Enabled the Emergence of New Variants in the State of Rio de Janeiro, Brazil. Viruses, 13(10), 2013. https://doi.org/10.3390/v13102013