
Mutational Hotspot in the SARS-CoV-2 Spike Protein N-Terminal Domain Conferring Immune Escape Potential


Abstract
:1. Introduction
2. Methods
2.1. Identification of Emerging Mutations in DDM Database
2.2. Analysis of Publicly Available SARS-CoV-2 Sequences
2.2.1. Phylogenetic and Protein Data
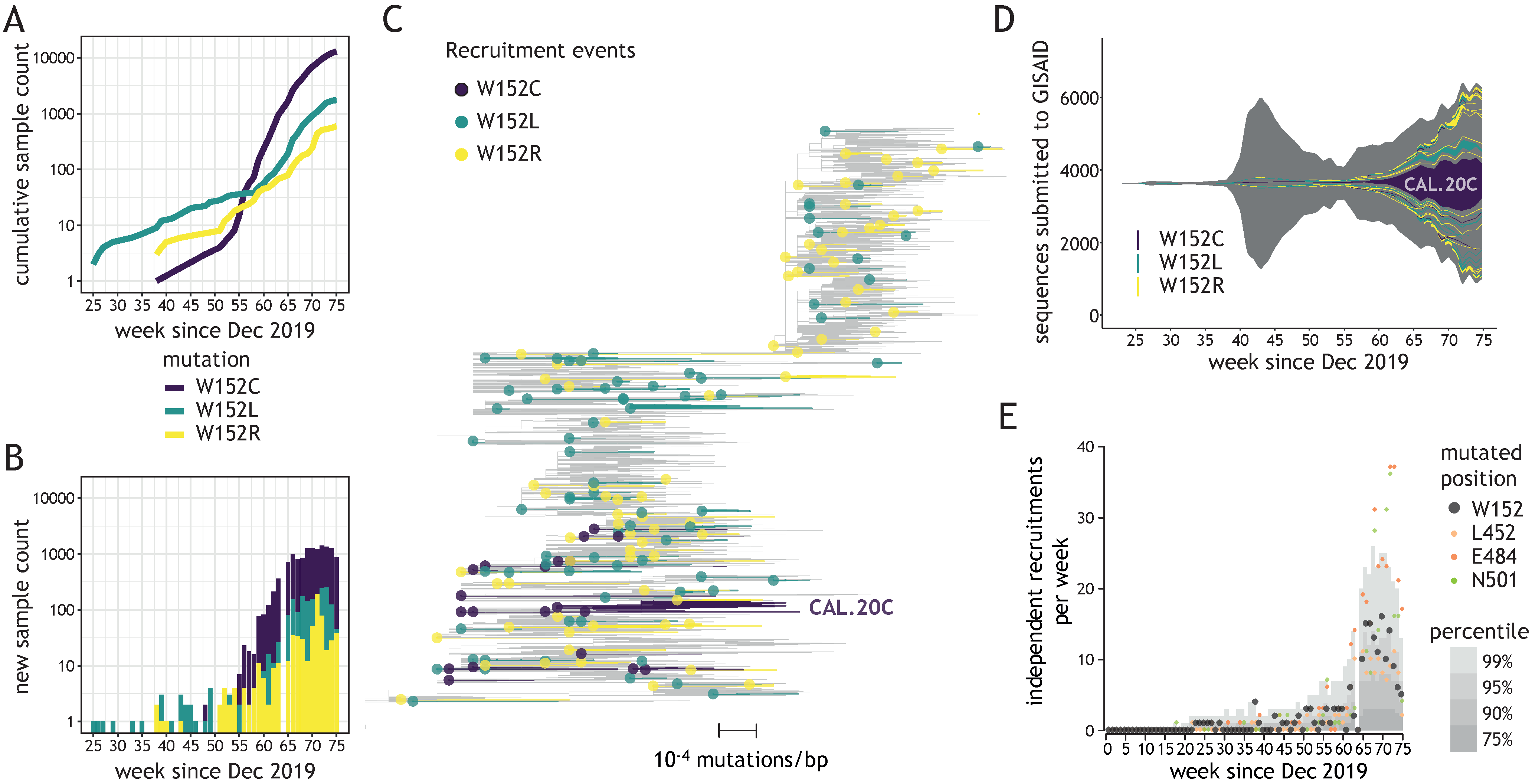
2.2.2. Identification of Independent W152 Recruitments
2.3. Structure-Based Prediction of the Impact of the Mutations on Spike Interactions
3. Results
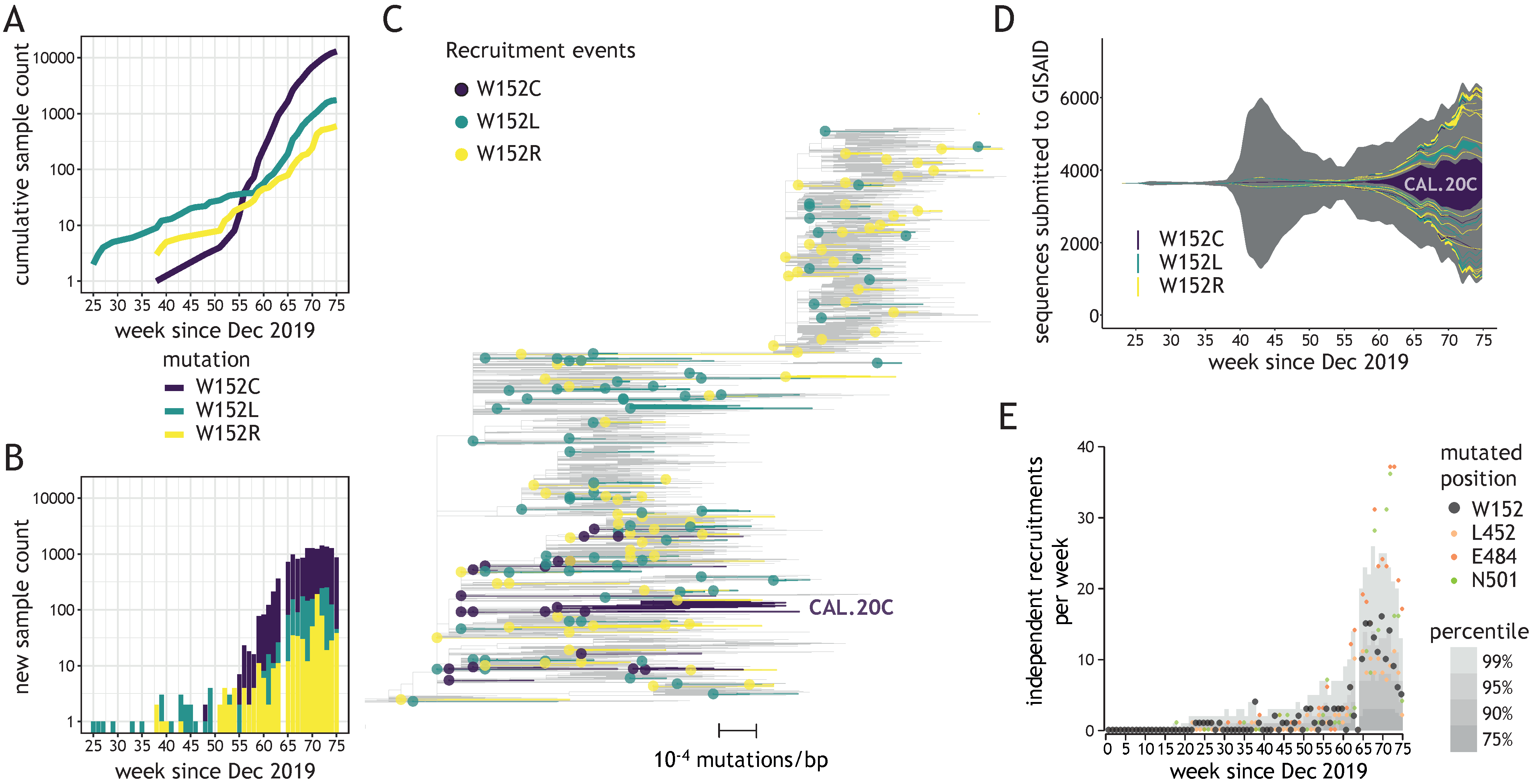
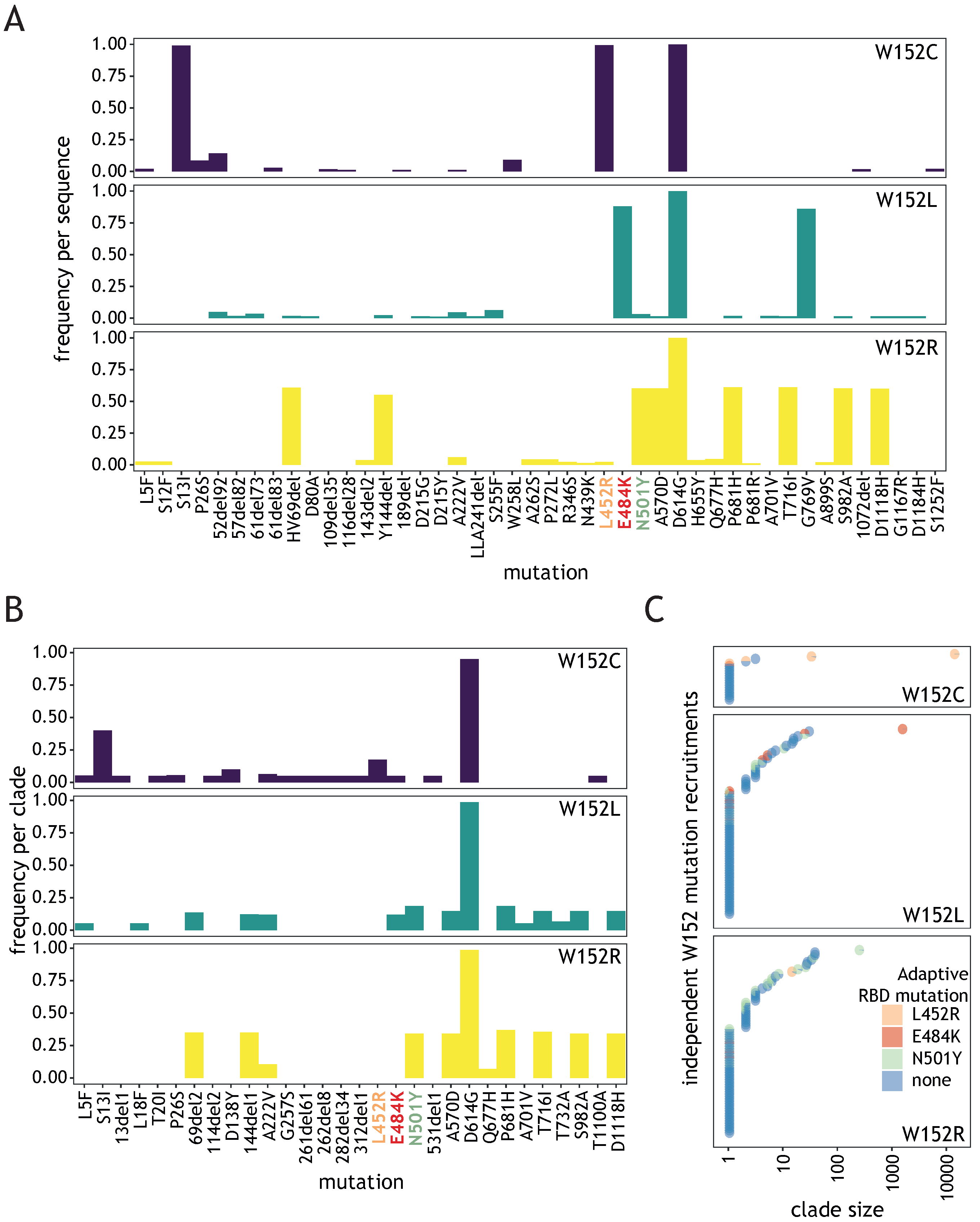
3.1. Tryptophan at Position 152 Is a Mutational Hotspot
3.2. Diverse W152 Mutations Were Recruited Independently in Multiple Phylogenetic Contexts
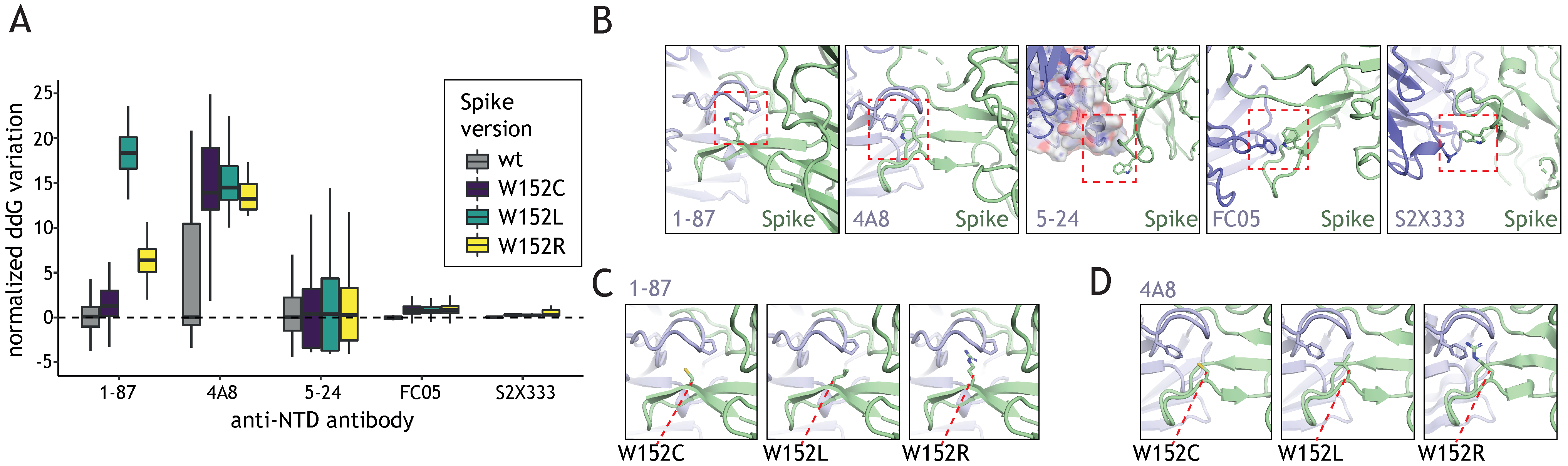
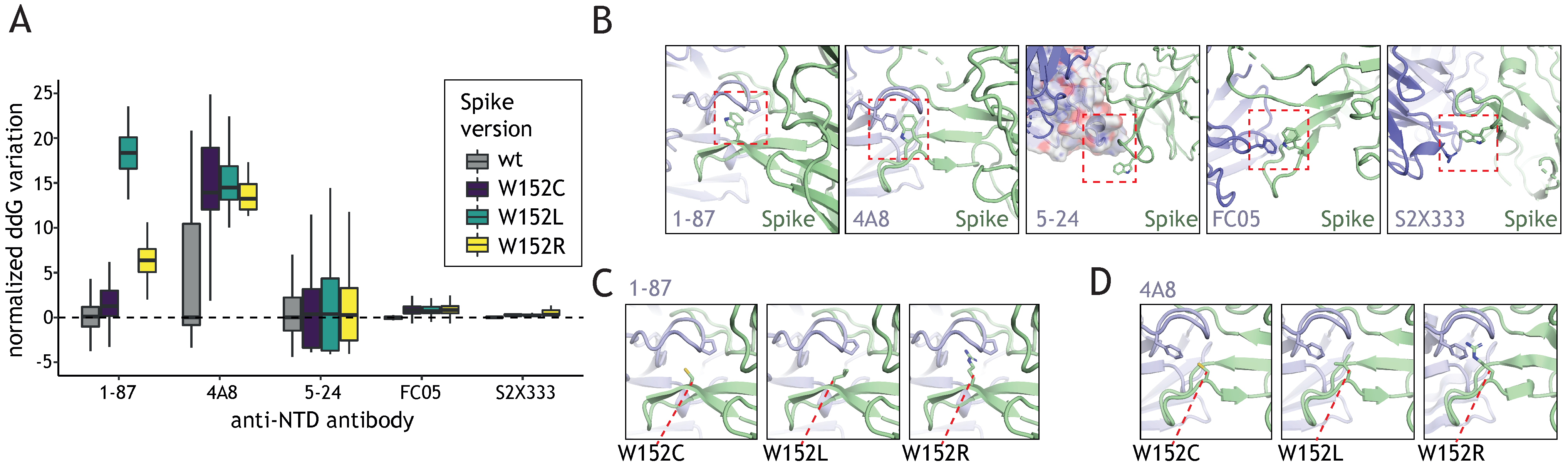
3.3. W152 Is an Important Interaction Point for Neutralizing Antibodies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Duffy, S. Why Are RNA Virus Mutation Rates so Damn High? PLoS Biol. 2018, 16, e3000003. [Google Scholar] [CrossRef] [Green Version]
- van Dorp, L.; Richard, D.; Tan, C.C.S.; Shaw, L.P.; Acman, M.; Balloux, F. No Evidence for Increased Transmissibility from Recurrent Mutations in SARS-CoV-2. Nat. Commun. 2020, 11, 5986. [Google Scholar] [CrossRef]
- Seeholzer, A.; Frey, E.; Obermayer, B. Periodic versus Intermittent Adaptive Cycles in Quasispecies Coevolution. Phys. Rev. Lett. 2014, 113, 128101. [Google Scholar] [CrossRef] [Green Version]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain That Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.e9. [Google Scholar] [CrossRef]
- McCallum, M.; Bassi, J.; De Marco, A.; Chen, A.; Walls, A.C.; Di Iulio, J.; Tortorici, M.A.; Navarro, M.-J.; Silacci-Fregni, C.; Saliba, C.; et al. SARS-CoV-2 Immune Evasion by the B.1.427/B.1.429 Variant of Concern. Science 2021, 373, 648–654. [Google Scholar] [CrossRef]
- Vilar, S.; Isom, D.G. One Year of SARS-CoV-2: How Much Has the Virus Changed? Biology 2021, 10, 91. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Krammer, F. SARS-CoV-2 Vaccines in Development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G Variant Exhibits Efficient Replication Ex Vivo and Transmission in Vivo. Science 2020, 370, eabe8499. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef]
- Luan, B.; Wang, H.; Huynh, T. Enhanced Binding of the N501Y-mutated SARS-CoV-2 Spike Protein to the Human ACE2 Receptor: Insights from Molecular Dynamics Simulations. FEBS Lett. 2021, 14076, 1873–3468. [Google Scholar] [CrossRef]
- Laffeber, C.; de Koning, K.; Kanaar, R.; Lebbink, J.H.G. Experimental Evidence for Enhanced Receptor Binding by Rapidly Spreading SARS-CoV-2 Variants. J. Mol. Biol. 2021, 433, 167058. [Google Scholar] [CrossRef]
- Tian, F.; Tong, B.; Sun, L.; Shi, S.; Zheng, B.; Wang, Z.; Dong, X.; Zheng, P. N501Y Mutation of Spike Protein in SARS-CoV-2 Strengthens Its Binding to Receptor ACE2. eLife 2021, 10, e69091. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C.; et al. The N501Y Spike Substitution Enhances SARS-CoV-2 Transmission. BioRxiv 2021. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Dingens, A.S.; Bloom, J.D. Complete Map of SARS-CoV-2 RBD Mutations That Escape the Monoclonal Antibody LY-CoV555 and Its Cocktail with LY-CoV016. Cell Rep. Med. 2021, 2, 100255. [Google Scholar] [CrossRef]
- Zhou, D.; Dejnirattisai, W.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Evidence of Escape of SARS-CoV-2 Variant B.1.351 from Natural and Vaccine-Induced Sera. Cell 2021, 9, 2348–2361.e6. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, Infectivity, and Neutralization of a Spike L452R SARS-CoV-2 Variant. Cell 2021, 184, 3426–3437.e8. [Google Scholar] [CrossRef]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.-M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Identification of SARS-CoV-2 Spike Mutations That Attenuate Monoclonal and Serum Antibody Neutralization. Cell Host Microbe 2021, 29, 477–488.e4. [Google Scholar] [CrossRef]
- Brisson, D. Negative Frequency-Dependent Selection Is Frequently Confounding. Front. Ecol. Evol. 2018, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.-M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Landscape Analysis of Escape Variants Identifies SARS-CoV-2 Spike Mutations That Attenuate Monoclonal and Serum Antibody Neutralization. BioRxiv 2020. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Addetia, A.; Hannon, W.W.; Choudhary, M.C.; Dingens, A.S.; Li, J.Z.; Bloom, J.D. Prospective Mapping of Viral Mutations That Escape Antibodies Used to Treat COVID-19. Science 2021, 371, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.-H.; Michailidis, E.; et al. Escape from Neutralizing Antibodies by SARS-CoV-2 Spike Protein Variants. eLife 2020, 9, e61312. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gao, K.; Wang, R.; Wei, G.-W. Prediction and Mitigation of Mutation Threats to COVID-19 Vaccines and Antibody Therapies. Chem. Sci. 2021, 12, 6929–6948. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Beltran, W.F.; Lam, E.C.; Denis, K.S.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 Variants Escape Neutralization by Vaccine-Induced Humoral Immunity. Cell 2021, 184, 2372–2383.e9. [Google Scholar] [CrossRef]
- Gobeil, S.M.-C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Stalls, V.; Kopp, M.F.; Manne, K.; Li, D.; Wiehe, K.; et al. Effect of Natural Mutations of SARS-CoV-2 on Spike Structure, Conformation, and Antigenicity. Science 2021, 373, eabi6226. [Google Scholar] [CrossRef]
- Piccoli, L.; Park, Y.-J.; Tortorici, M.A.; Czudnochowski, N.; Walls, A.C.; Beltramello, M.; Silacci-Fregni, C.; Pinto, D.; Rosen, L.E.; Bowen, J.E.; et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell 2020, 183, 1024–1042.e21. [Google Scholar] [CrossRef]
- Chi, X.; Yan, R.; Zhang, J.; Zhang, G.; Zhang, Y.; Hao, M.; Zhang, Z.; Fan, P.; Dong, Y.; Yang, Y.; et al. A Neutralizing Human Antibody Binds to the N-Terminal Domain of the Spike Protein of SARS-CoV-2. Science 2020, 369, 650–655. [Google Scholar] [CrossRef]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.-W.; Sahi, V.; Figueroa, A.; et al. Potent Neutralizing Antibodies against Multiple Epitopes on SARS-CoV-2 Spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef]
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-Terminal Domain Antigenic Mapping Reveals a Site of Vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347.e16. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, G.; Guo, Y.; Zhou, T.; Gorman, J.; Lee, M.; Rapp, M.; Reddem, E.R.; Yu, J.; Bahna, F.; Bimela, J.; et al. Potent SARS-CoV-2 Neutralizing Antibodies Directed against Spike N-Terminal Domain Target a Single Supersite. Cell Host Microbe 2021, 29, 819–833.e7. [Google Scholar] [CrossRef]
- Kubik, S.; Marques, A.C.; Xing, X.; Silvery, J.; Bertelli, C.; De Maio, F.; Pournaras, S.; Burr, T.; Duffourd, Y.; Siemens, H.; et al. Recommendations for Accurate Genotyping of SARS-CoV-2 Using Amplicon-Based Sequencing of Clinical Samples. Clin. Microbiol. Infect. 2021, 27, 1036.e1–1036.e8. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Wang, N.; Sun, Y.; Feng, R.; Wang, Y.; Guo, Y.; Zhang, L.; Deng, Y.-Q.; Wang, L.; Cui, Z.; Cao, L.; et al. Structure-Based Development of Human Antibody Cocktails against SARS-CoV-2. Cell Res. 2021, 31, 101–103. [Google Scholar] [CrossRef]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Fleishman, S.J.; Leaver-Fay, A.; Corn, J.E.; Strauch, E.-M.; Khare, S.D.; Koga, N.; Ashworth, J.; Murphy, P.; Richter, F.; Lemmon, G.; et al. RosettaScripts: A Scripting Language Interface to the Rosetta Macromolecular Modeling Suite. PLoS ONE 2011, 6, e20161. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Davis, B.D.; Chen, S.S.; Martinez, J.M.S.; Plummer, J.T.; Vail, E. Emergence of a Novel SARS-CoV-2 Variant in Southern California. PLoS ONE 2011, 6, e20161. [Google Scholar] [CrossRef]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent Deletions in the SARS-CoV-2 Spike Glycoprotein Drive Antibody Escape. Science 2021, 371, 1139–1142. [Google Scholar] [CrossRef]
- Haslwanter, D.; Dieterle, M.E.; Wec, A.Z.; O’Brien, C.M.; Sakharkar, M.; Florez, C.; Tong, K.; Rappazzo, C.G.; Lasso, G.; Vergnolle, O.; et al. A Combination of Receptor-Binding Domain and N-Terminal Domain Neutralizing Antibodies Limits the Generation of SARS-CoV-2 Spike Neutralization-Escape Mutants. mBio 2021, 12, e02473-21. [Google Scholar] [CrossRef]
- Peng, R.; Wu, L.-A.; Wang, Q.; Qi, J.; Gao, G.F. Cell Entry by SARS-CoV-2. Trends Biochem. Sci. 2021, 46, 848–860. [Google Scholar] [CrossRef]
- Puray-Chavez, M.; LaPak, K.M.; Schrank, T.P.; Elliott, J.L.; Bhatt, D.P.; Agajanian, M.J.; Jasuja, R.; Lawson, D.Q.; Davis, K.; Rothlauf, P.W.; et al. Systematic Analysis of SARS-CoV-2 Infection of an ACE2-Negative Human Airway Cell. Cell Rep. 2021, 36, 109364. [Google Scholar] [CrossRef] [PubMed]
- Lusvarghi, S.; Wang, W.; Herrup, R.; Neerukonda, S.N.; Vassell, R.; Bentley, L.; Eakin, A.E.; Erlandson, K.J.; Weiss, C.D. Key Substitutions in the Spike Protein of SARS-CoV-2 Variants Can Predict Resistance to Monoclonal Antibodies, but Other Substitutions Can Modify the Effects. BioRxiv 2021. [Google Scholar] [CrossRef]
- Ip, J.D.; Kok, K.-H.; Chan, W.-M.; Chu, A.W.-H.; Wu, W.-L.; Yip, C.C.-Y.; To, W.-K.; Tsang, O.T.-Y.; Leung, W.-S.; Chik, T.S.-H.; et al. Intra-Host Non-Synonymous Diversity at a Neutralizing Antibody Epitope of SARS-CoV-2 Spike Protein N-Terminal Domain. Clin. Microbiol. Infect. 2020, 27, 1350.e1–1350.e5. [Google Scholar] [CrossRef] [PubMed]
- Hastie, K.M.; Li, H.; Bedinger, D.; Schendel, S.L.; Dennison, S.M.; Li, K.; Rayaprolu, V.; Yu, X.; Mann, C.; Zandonatti, M.; et al. Defining Variant-Resistant Epitopes Targeted by SARS-CoV-2 Antibodies: A Global Consortium Study. Science 2021, eabh2315. [Google Scholar] [CrossRef]
- Cerutti, G.; Guo, Y.; Wang, P.; Nair, M.S.; Huang, Y.; Yu, J.; Liu, L.; Katsamba, P.S.; Bahna, F.; Reddem, E.R.; et al. Neutralizing Antibody 5–7 Defines a Distinct Site of Vulnerability in SARS-CoV-2 Spike N-Terminal Domain. BioRxiv 2021. [Google Scholar] [CrossRef]
- Liu, Y.; Arase, N.; Kishikawa, J.; Hirose, M.; Li, S.; Tada, A.; Matsuoka, S.; Arakawa, A.; Akamatsu, K.; Ono, C.; et al. The SARS-CoV-2 Delta Variant Is Poised to Acquire Complete Resistance to Wild-Type Spike Vaccines. BioRxiv 2021. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Omata, M. Detection of R.1 Lineage Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) with Spike Protein W152L/E484K/G769V Mutations in Japan. PLoS Pathog. 2021, 17, e1009619. [Google Scholar] [CrossRef]
- Cavanaugh, A.M.; Fortier, S.; Lewis, P.; Arora, V.; Johnson, M.; George, K.; Tobias, J.; Lunn, S.; Miller, T.; Thoroughman, D.; et al. COVID-19 Outbreak Associated with a SARS-CoV-2 R.1 Lineage Variant in a Skilled Nursing Facility After Vaccination Program—Kentucky, March 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 639–643. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | DDM Frequency (%) | Global Frequency (%) | Country Count | Continent Count | Clade Count | Clades | Lineage Count | Lineages |
|---|---|---|---|---|---|---|---|---|
| Trp152Leu | 0.82 | 0.06 | 3 | 2 | 5 | 19A, 20A, 20B, 20I/501Y.V1, 20H/501Y.V2 | 9 | B, B.1, B.1.1, B.1.1.7, B.1.29, B.1.351, B.1.416, B.1.420, R.1 |
| Trp152Arg | 0.73 | 0.04 | 3 | 2 | 4 | 19A, 20A, 20D, 20I/501Y.V1 | 7 | B, B.1, B.1.1.1, B.1.111, B.1.1.7, B.1.166, B.1.402 |
| Trp152Cys | 0.02 | 1.56 | 1 | 1 | 1 | 20C | 1 | B.1.429 |
| Trp152Gly | 0.02 | 2 × 10−4 | 1 | 1 | 1 | 20I/501Y.v1 | 1 | B.1.1.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubik, S.; Arrigo, N.; Bonet, J.; Xu, Z. Mutational Hotspot in the SARS-CoV-2 Spike Protein N-Terminal Domain Conferring Immune Escape Potential. Viruses 2021, 13, 2114. https://doi.org/10.3390/v13112114
Kubik S, Arrigo N, Bonet J, Xu Z. Mutational Hotspot in the SARS-CoV-2 Spike Protein N-Terminal Domain Conferring Immune Escape Potential. Viruses. 2021; 13(11):2114. https://doi.org/10.3390/v13112114
Chicago/Turabian StyleKubik, Slawomir, Nils Arrigo, Jaume Bonet, and Zhenyu Xu. 2021. "Mutational Hotspot in the SARS-CoV-2 Spike Protein N-Terminal Domain Conferring Immune Escape Potential" Viruses 13, no. 11: 2114. https://doi.org/10.3390/v13112114
APA StyleKubik, S., Arrigo, N., Bonet, J., & Xu, Z. (2021). Mutational Hotspot in the SARS-CoV-2 Spike Protein N-Terminal Domain Conferring Immune Escape Potential. Viruses, 13(11), 2114. https://doi.org/10.3390/v13112114