
Direct RNA Sequencing Reveals SARS-CoV-2 m6A Sites and Possible Differential DRACH Motif Methylation among Variants

 , , , , and
, , , , and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Cultures and SARS-CoV-2 Infection
2.2. RNA Isolation
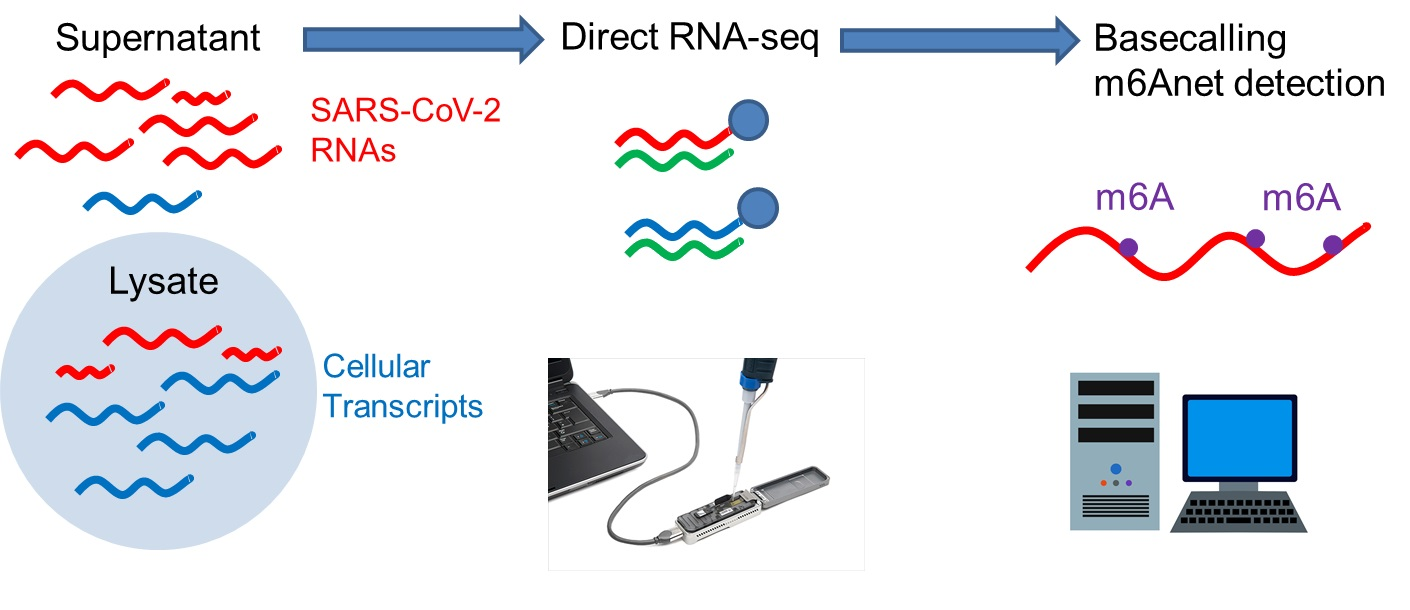
2.3. Direct RNA Sequencing
2.4. Assembly
2.5. Methylation Analysis
2.6. DRACH Motif Comparison
3. Results
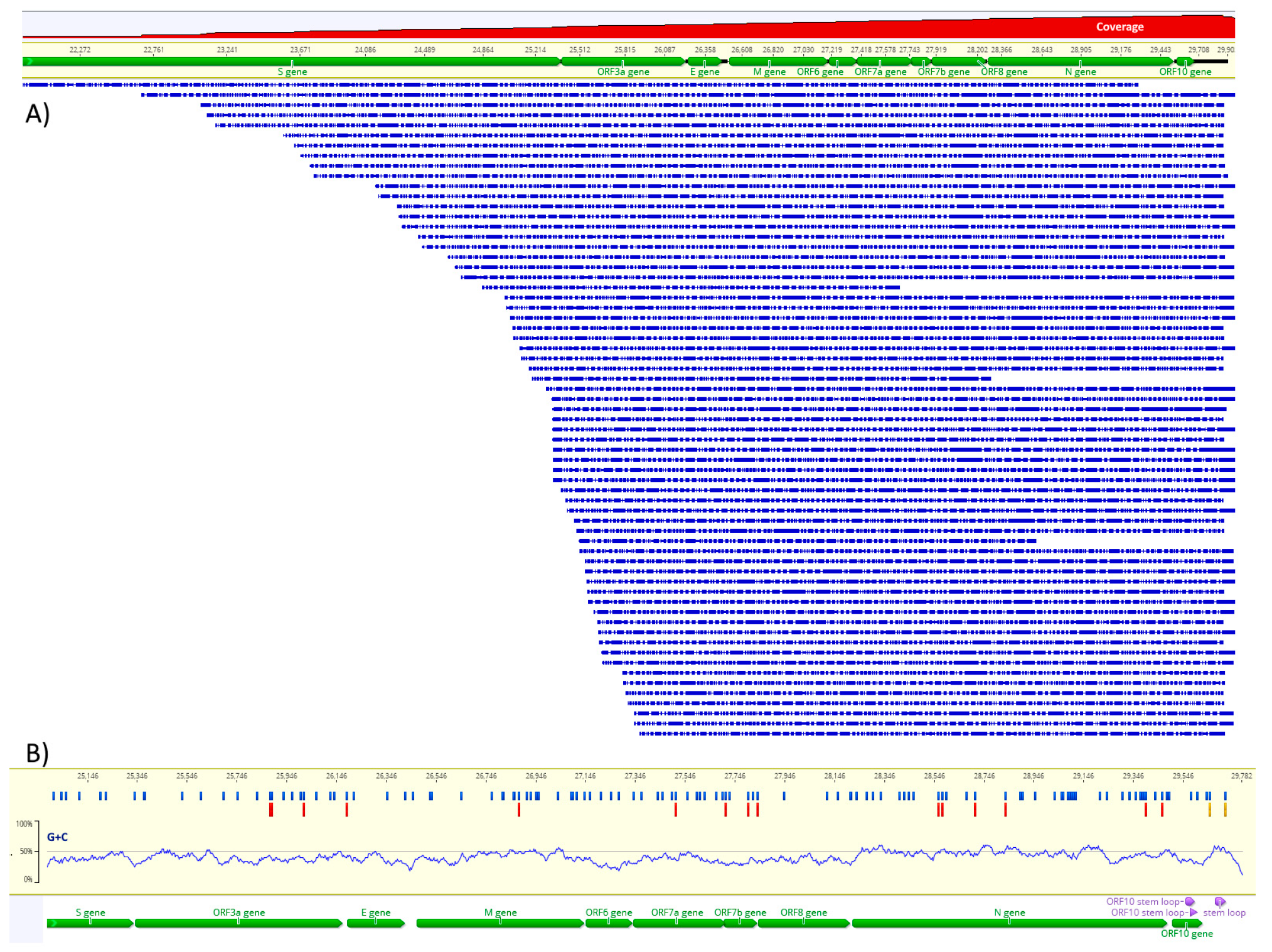
3.1. Direct RNA Sequencing and Assembly
3.2. Detection of m6A
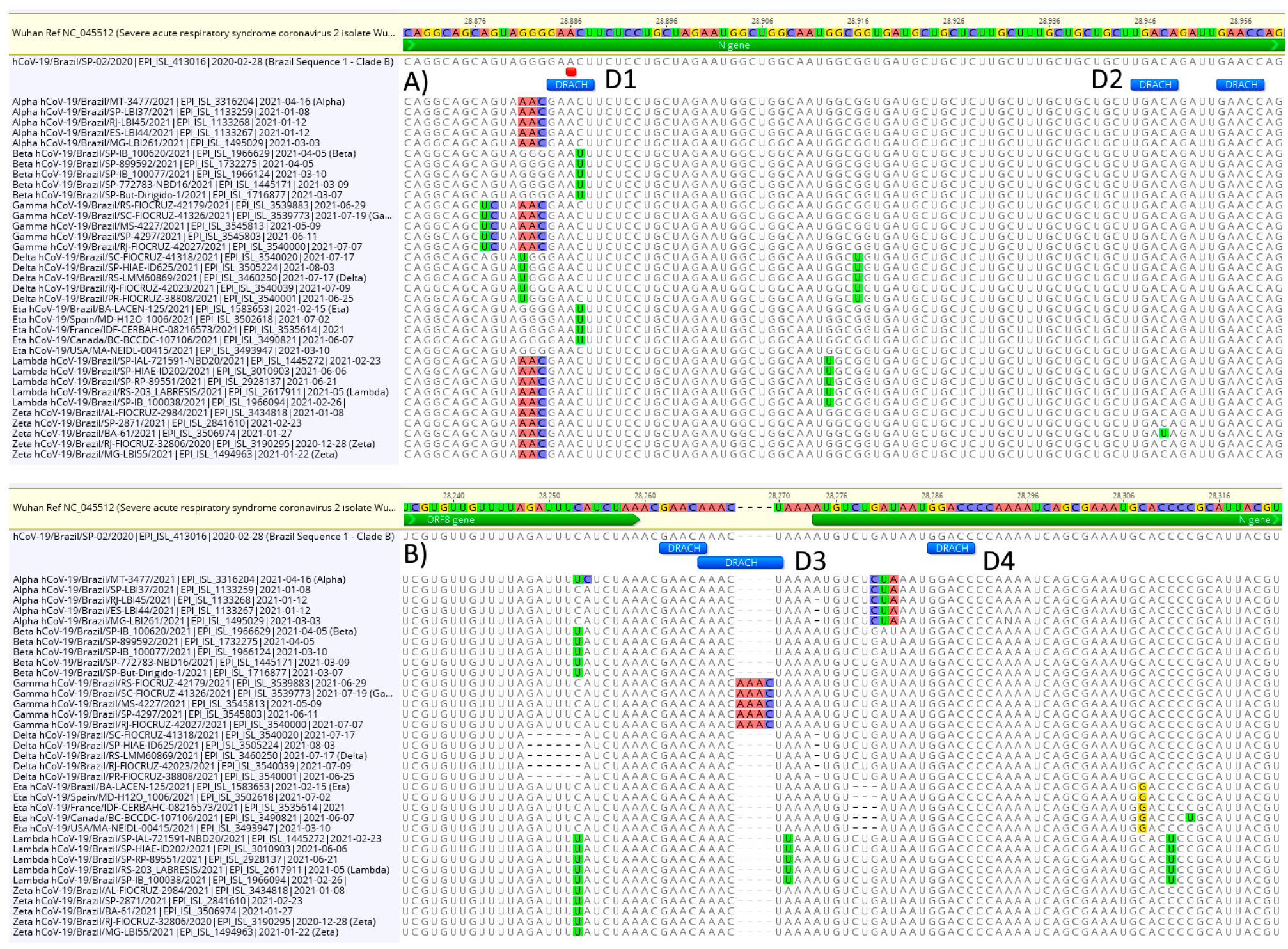
3.3. DRACH Motif Analysis
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hogle, J.M. Poliovirus Cell Entry: Common Structural Themes in Viral Cell Entry Pathways. Annu. Rev. Microbiol. 2002, 56, 677–702. [Google Scholar] [CrossRef] [Green Version]
- Guerra, F.M.; Bolotin, S.; Lim, G.; Heffernan, J.; Deeks, S.L.; Li, Y.; Crowcroft, N.S. The basic reproduction number (R0) of measles: A systematic review. Lancet Infect. Dis. 2017, 17, e420–e428. [Google Scholar] [CrossRef]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primer 2018, 4, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Kundu, S.; Alam, S.S.; Hossan, T.; Kamal, M.A.; Hassan, R. Prevalence and characteristics of fever in adult and paediatric patients with coronavirus disease 2019 (COVID-19): A systematic review and meta-analysis of 17515 patients. PLoS ONE 2021, 16, e0249788. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Z.; Wang, G.; Lau, J.Y.-N.; Zhang, K.; Li, W. COVID-19 in early 2021: Current status and looking forward. Signal Transduct. Target. Ther. 2021, 6, 1–14. [Google Scholar] [CrossRef]
- Baltimore, D. Expression of animal virus genomes. Bacteriol. Rev. 1971, 35, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Dill, J.A.; Camus, A.C.; Leary, J.H.; Di Giallonardo, F.; Holmes, E.C.; Ng, T.F.F. Distinct Viral Lineages from Fish and Amphibians Reveal the Complex Evolutionary History of Hepadnaviruses. J. Virol. 2016, 90, 7920–7933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Xia, S.; Ying, T.; Lu, L. A novel coronavirus (2019-nCoV) causing pneumonia-associated respiratory syndrome. Cell. Mol. Immunol. 2020, 17, 554. [Google Scholar] [CrossRef] [Green Version]
- Hanff, T.C.; Mohareb, A.M.; Giri, J.; Cohen, J.B.; Chirinos, J.A. Thrombosis in COVID-19. Am. J. Hematol. 2020, 95, 1578–1589. [Google Scholar] [CrossRef]
- Almeida, J.D.; Berry, D.M.; Cunningham, C.H.; Hamre, D.; Hofstad, M.S.; Mallucci, L.; McInstosh, D.A.; Tyrrell, D.A.J. Virology: Coronaviruses. Nature 1968, 220, 650. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Brocard, M.; Ruggieri, A.; Locker, N. m6A RNA methylation, a new hallmark in virus-host interactions. J. Gen. Virol. 2017, 98, 2207–2214. [Google Scholar] [CrossRef]
- Gokhale, N.S.; McIntyre, A.B.R.; McFadden, M.J.; Roder, A.E.; Kennedy, E.M.; Gandara, J.A.; Hopcraft, S.E.; Quicke, K.M.; Vazquez, C.; Willer, J.; et al. N6-Methyladenosine in Flaviviridae Viral RNA Genomes Regulates Infection. Cell Host Microbe 2016, 20, 654–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baquero-Perez, B.; Geers, D.; Díez, J. From A to m6A: The Emerging Viral Epitranscriptome. Viruses 2021, 13, 1049. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Hui, H.; Bray, B.; Gonzalez, G.M.; Zeller, M.; Anderson, K.G.; Knight, R.; Smith, D.; Wang, Y.; Carlin, A.F.; et al. METTL3 regulates viral m6A RNA modification and host cell innate immune responses during SARS-CoV-2 infection. Cell Rep. 2021, 35, 109091. [Google Scholar] [CrossRef]
- Liu, J.; Xu, Y.-P.; Li, K.; Ye, Q.; Zhou, H.-Y.; Sun, H.; Li, X.; Yu, L.; Deng, Y.-Q.; Li, R.-T.; et al. The m6A methylome of SARS-CoV-2 in host cells. Cell Res. 2021, 31, 404–414. [Google Scholar] [CrossRef]
- Gatsiou, A.; Stellos, K. Dawn of Epitranscriptomic Medicine. Circ. Genomic Precis. Med. 2018, 11, e001927. [Google Scholar] [CrossRef] [PubMed]
- Nazario-Toole, A.E.; Xia, H.; Gibbons, T.F. Whole-genome Sequencing of SARS-CoV-2: Using Phylogeny and Structural Modeling to Contextualize Local Viral Evolution. Mil. Med. 2021, usab031. [Google Scholar] [CrossRef] [PubMed]
- Kietrys, A.M.; Velema, W.A.; Kool, E.T. Fingerprints of Modified RNA Bases from Deep Sequencing Profiles. J. Am. Chem. Soc. 2017, 139, 17074–17081. [Google Scholar] [CrossRef] [Green Version]
- Jenjaroenpun, P.; Wongsurawat, T.; Wadley, T.D.; Wassenaar, T.M.; Liu, J.; Dai, Q.; Wanchai, V.; Akel, N.S.; Jamshidi-Parsian, A.; Franco, A.T.; et al. Decoding the epitranscriptional landscape from native RNA sequences. Nucleic Acids Res. 2021, 49, e7. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.A.; Adikari, T.N.; Ferguson, J.M.; Hammond, J.M.; Stevanovski, I.; Beukers, A.G.; Naing, Z.; Yeang, M.; Verich, A.; Gamaarachchi, H.; et al. Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nat. Commun. 2020, 11, 6272. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef] [PubMed]
- Miladi, M.; Fuchs, J.; Maier, W.; Weigang, S.; i Pedrosa, N.D.; Weiss, L.; Lother, A.; Nekrutenko, A.; Ruzsics, Z.; Panning, M.; et al. The Landscape of SARS-CoV-2 RNA Modifications. bioRxiv 2020. [Google Scholar] [CrossRef]
- Taiaroa, G.; Rawlinson, D.; Featherstone, L.; Pitt, M.; Caly, L.; Druce, J.; Purcell, D.; Harty, L.; Tran, T.; Roberts, J.; et al. Direct RNA sequencing and early evolution of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Vacca, D.; Fiannaca, A.; Tramuto, F.; Cancila, V.; Paglia, L.L.; Mazzucco, W.; Gulino, A.; Rosa, M.L.; Maida, C.M.; Morello, G.; et al. Direct RNA nanopore sequencing of SARS-CoV-2 extracted from critical material from swabs. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bayoumi, M.; Munir, M. Evolutionary conservation of the DRACH signatures of potential N6-methyladenosine (m6A) sites among influenza A viruses. Sci. Rep. 2021, 11, 4548. [Google Scholar] [CrossRef]
- Araujo, D.B.; Machado, R.R.G.; Amgarten, D.E.; Malta, F.d.M.; de Araujo, G.G.; Monteiro, C.O.; Candido, E.D.; Soares, C.P.; de Menezes, F.G.; Pires, A.C.C.; et al. SARS-CoV-2 isolation from the first reported patients in Brazil and establishment of a coordinated task network. Mem. Inst. Oswaldo Cruz 2020, 115, e200342. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinforma. Oxf. Engl. 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendra, C.; Pratanwanich, P.N.; Wan, Y.K.; Goh, W.S.S.; Thiery, A.; Göke, J. Detection of m6A from direct RNA sequencing using a Multiple Instance Learning framework. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hendra, C.; Wan, Y.K. GoekeLab/m6anet: Pre-Release; Zenodo: Genève, Switzerland, 2021. [Google Scholar]
- Chen, Y.; Davidson, N.M.; Wan, Y.K.; Patel, H.; Yao, F.; Low, H.M.; Hendra, C.; Watten, L.; Sim, A.; Sawyer, C.; et al. A systematic benchmark of Nanopore long read RNA sequencing for transcript level analysis in human cell lines. bioRxiv 2021. [Google Scholar] [CrossRef]
- Meng, Y.; Zhang, Q.; Wang, K.; Zhang, X.; Yang, R.; Bi, K.; Chen, W.; Diao, H. RBM15-mediated N6-methyladenosine modification affects COVID-19 severity by regulating the expression of multitarget genes. Cell Death Dis. 2021, 12, 1–10. [Google Scholar] [CrossRef]
- Ahmed-Belkacem, R.; Sutto-Ortiz, P.; Guiraud, M.; Canard, B.; Vasseur, J.-J.; Decroly, E.; Debart, F. Synthesis of adenine dinucleosides SAM analogs as specific inhibitors of SARS-CoV nsp14 RNA cap guanine-N7-methyltransferase. Eur. J. Med. Chem. 2020, 201, 112557. [Google Scholar] [CrossRef]
- Paramasivam, A. RNA 2′-O-methylation modification and its implication in COVID-19 immunity. Cell Death Discov. 2020, 6, 118. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| # | RNA Id—ORF | Position | Coverage | Probability |
|---|---|---|---|---|
| 1 | EPI_ISL_413016—3a | 25,935 | 65/75 | 0.5472/0.2749 |
| 2 | EPI_ISL_413016—3a | 25,940 | 67/61 | 0.5094/0.5865 |
| 3 | EPI_ISL_413016—3a | 26,070 | 72/83 | 0.6659/0.2497 |
| 4 | EPI_ISL_413016—3a/E | 26,241 | 83/100 | 0.5480/0.3461 |
| 5 | EPI_ISL_413016—M | 26,933 | 162/295 | 0.5102/0.8204 |
| 6 | EPI_ISL_413016—7a | 27,562 | 266/679 | 0.8516/0.3973 |
| 7 | EPI_ISL_413016—7b | 27,764 | 309/849 | 0.8433/0.8946 |
| 8 | EPI_ISL_413016—7b | 27,854 | 318/839 | 0.8761/0.8709 |
| 9 | EPI_ISL_413016—7b/8 | 27,892 | 357/859 | 0.5770/0.4420 |
| 10 | EPI_ISL_413016—N | 28,616 | 699/812 | 0.8184/0.4929 |
| 11 | EPI_ISL_413016—N | 28,633 | 314/364 | 0.5022/0.4134 |
| 12 | EPI_ISL_413016—N | 28,766 | 794/817 | 0.5403/0.4098 |
| 13 | EPI_ISL_413016—N | 28,886 | 871/835 | 0.7020/0.2212 |
| 14 | EPI_ISL_413016—N | 29,450 | 922/803 | 0.5523/0.2701 |
| 15 | EPI_ISL_413016—N | 29,517 | 887/783 | 0.5705/0.3598 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos, J.H.C.; Maricato, J.T.; Braconi, C.T.; Antoneli, F.; Janini, L.M.R.; Briones, M.R.S. Direct RNA Sequencing Reveals SARS-CoV-2 m6A Sites and Possible Differential DRACH Motif Methylation among Variants. Viruses 2021, 13, 2108. https://doi.org/10.3390/v13112108
Campos JHC, Maricato JT, Braconi CT, Antoneli F, Janini LMR, Briones MRS. Direct RNA Sequencing Reveals SARS-CoV-2 m6A Sites and Possible Differential DRACH Motif Methylation among Variants. Viruses. 2021; 13(11):2108. https://doi.org/10.3390/v13112108
Chicago/Turabian StyleCampos, João H. C., Juliana T. Maricato, Carla T. Braconi, Fernando Antoneli, Luiz Mario R. Janini, and Marcelo R. S. Briones. 2021. "Direct RNA Sequencing Reveals SARS-CoV-2 m6A Sites and Possible Differential DRACH Motif Methylation among Variants" Viruses 13, no. 11: 2108. https://doi.org/10.3390/v13112108
APA StyleCampos, J. H. C., Maricato, J. T., Braconi, C. T., Antoneli, F., Janini, L. M. R., & Briones, M. R. S. (2021). Direct RNA Sequencing Reveals SARS-CoV-2 m6A Sites and Possible Differential DRACH Motif Methylation among Variants. Viruses, 13(11), 2108. https://doi.org/10.3390/v13112108