
Binding of a Pocket Factor to Hepatitis B Virus Capsids Changes the Rotamer Conformation of Phenylalanine 97

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Purification of HBc-CLPs
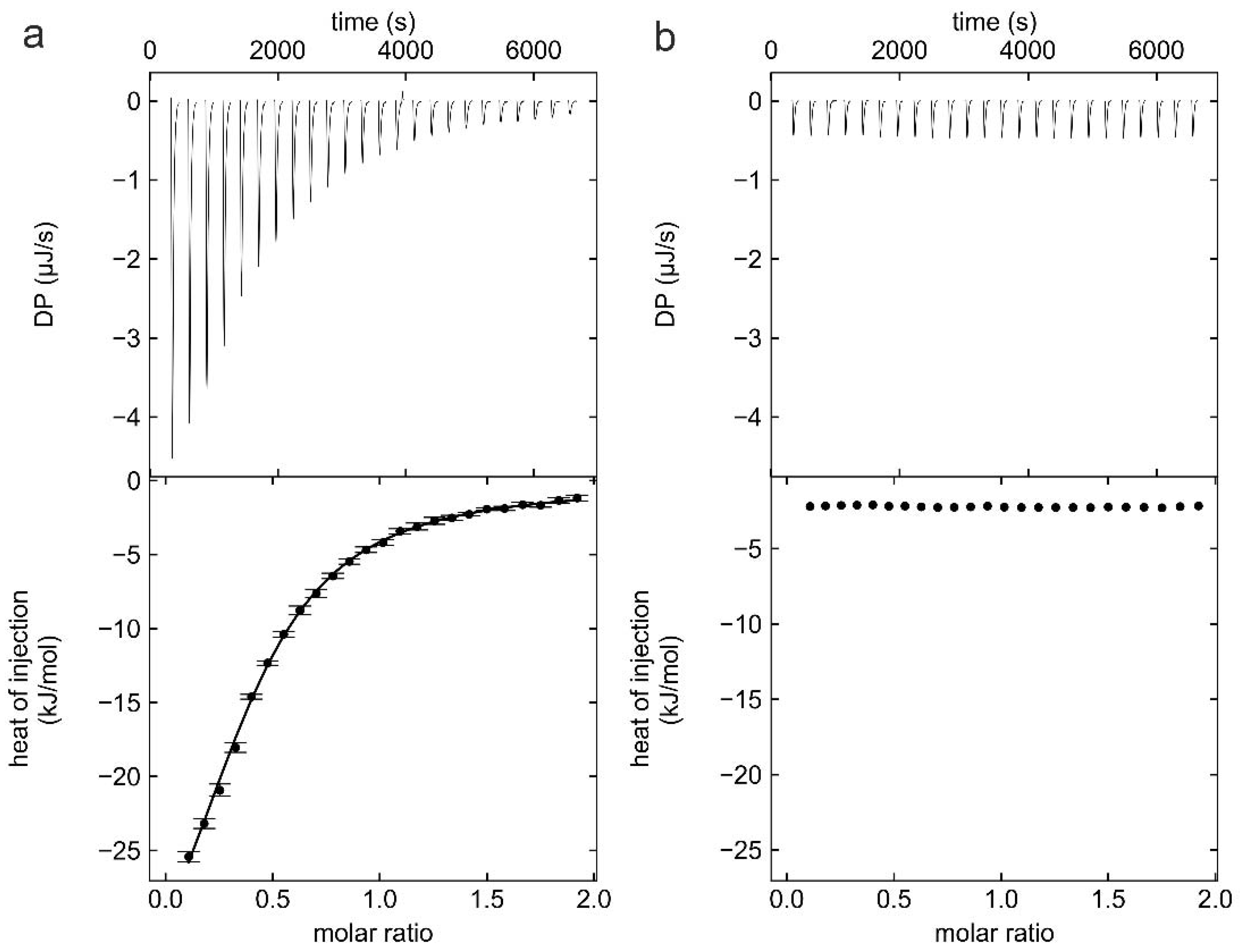
2.2. Isothermal Titration Calorimetry (ITC)
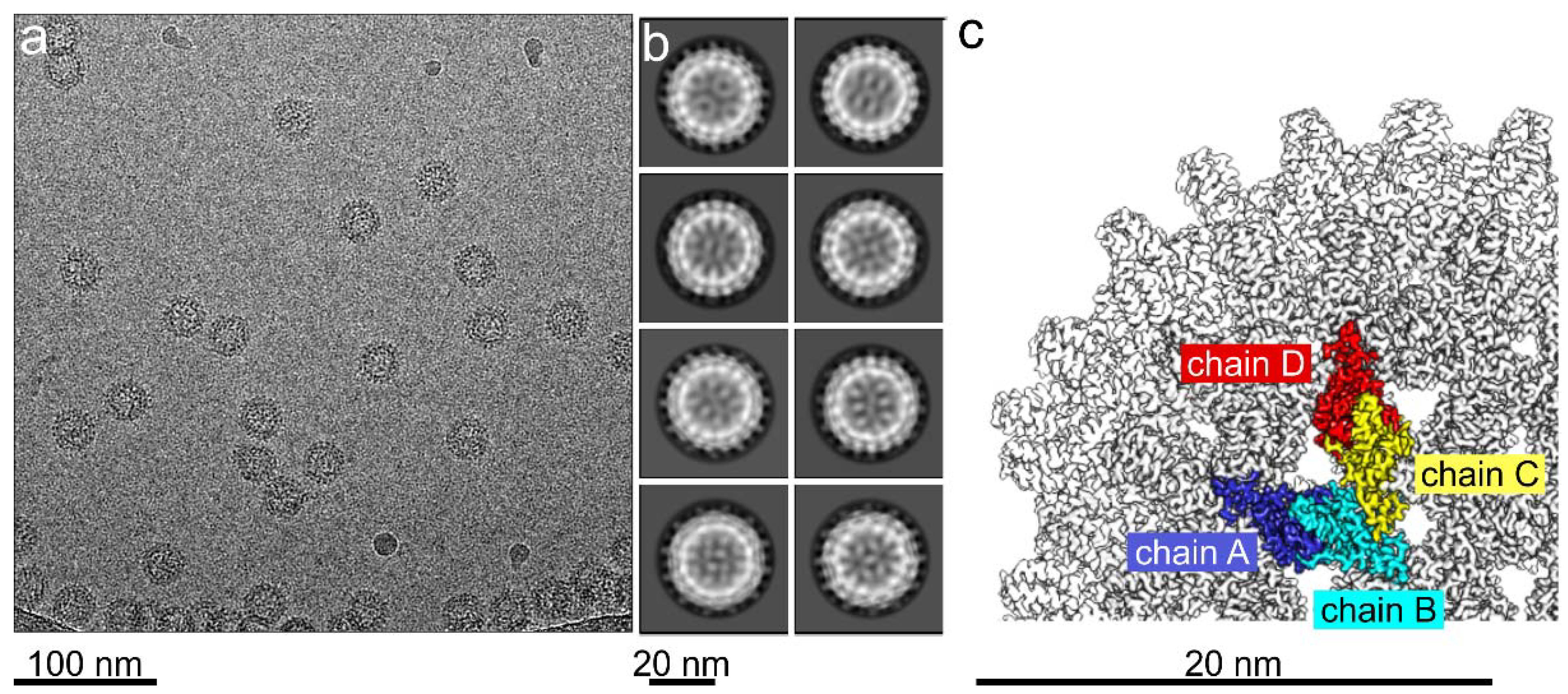
2.3. Sample Preparation for Electron Cryo-Microscopy
2.4. Electron Cryo-Microscopy
2.5. Image Processing
2.6. Modelling of Cryo-EM Maps and their Validation
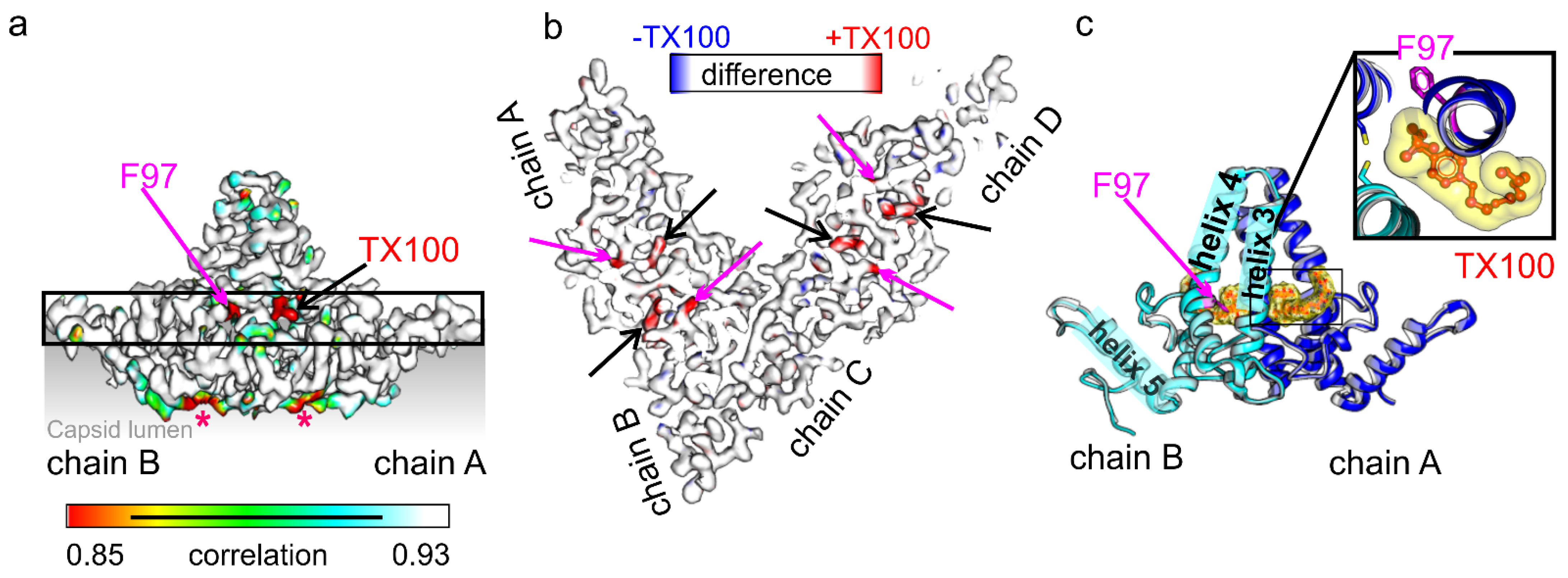
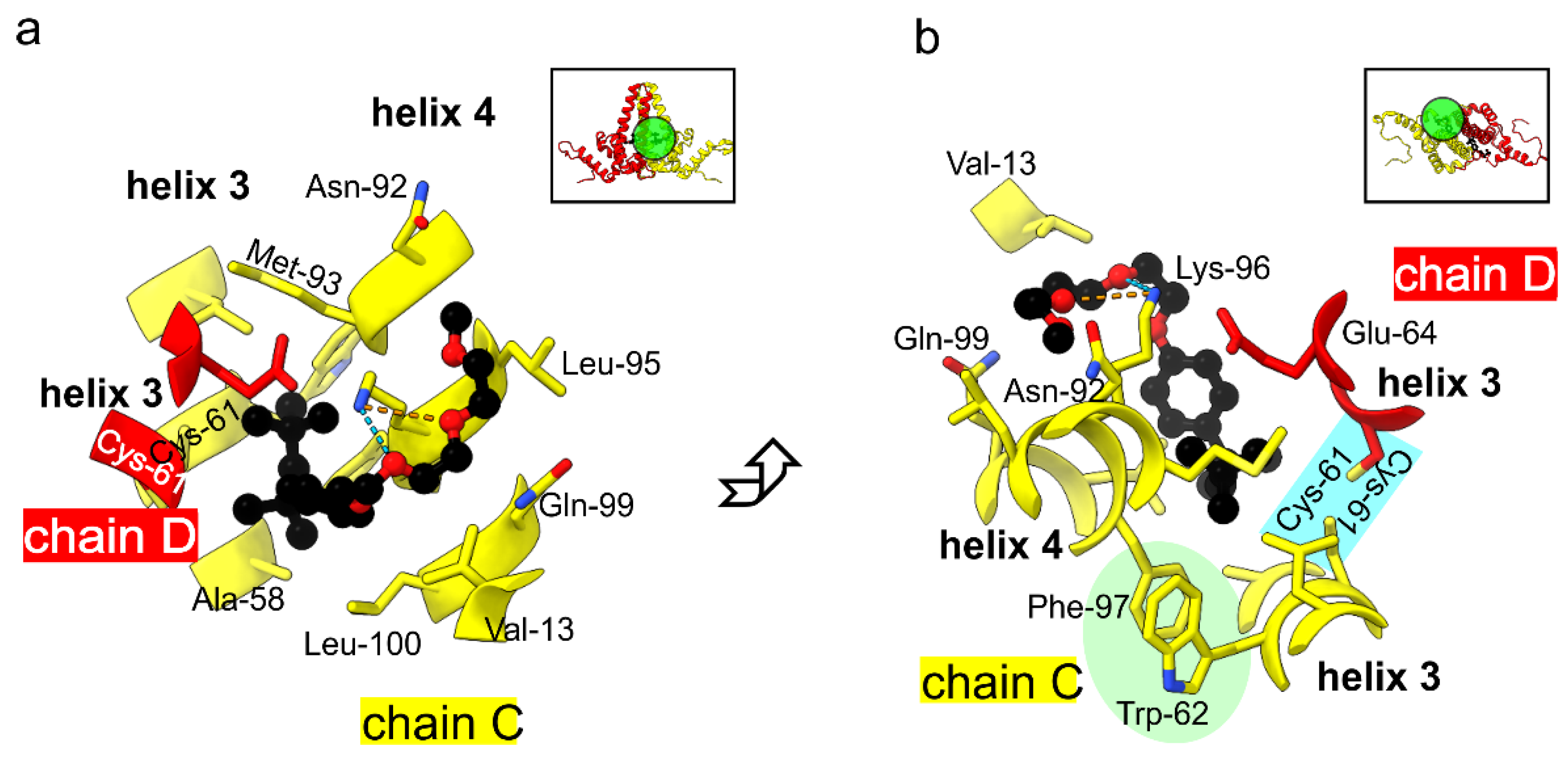
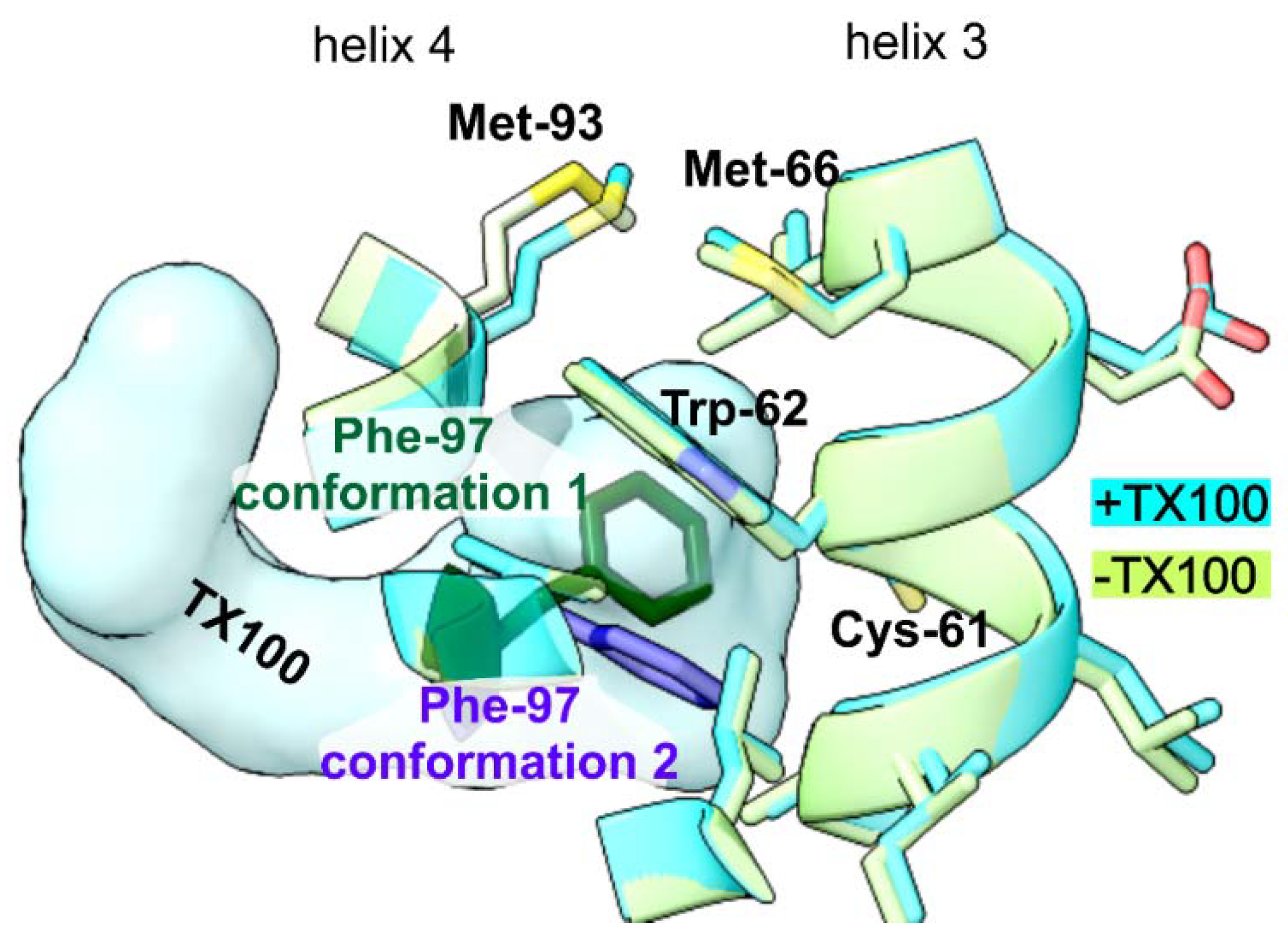
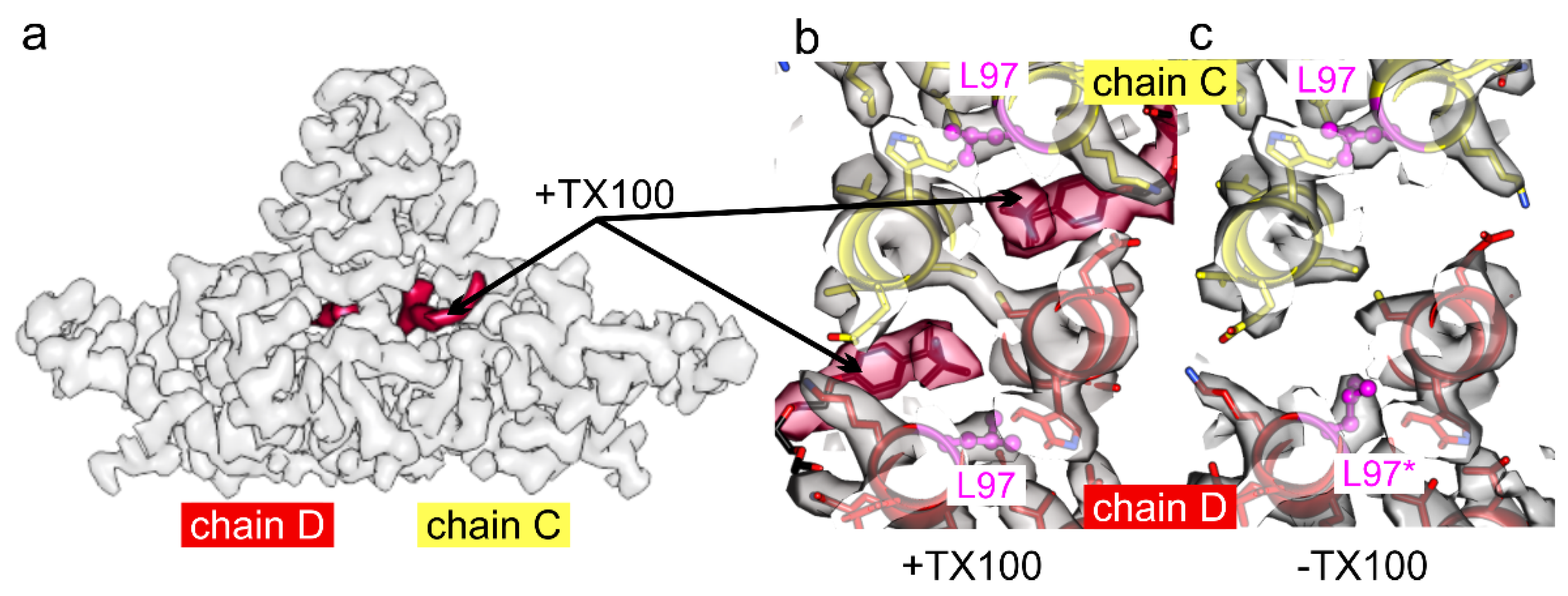
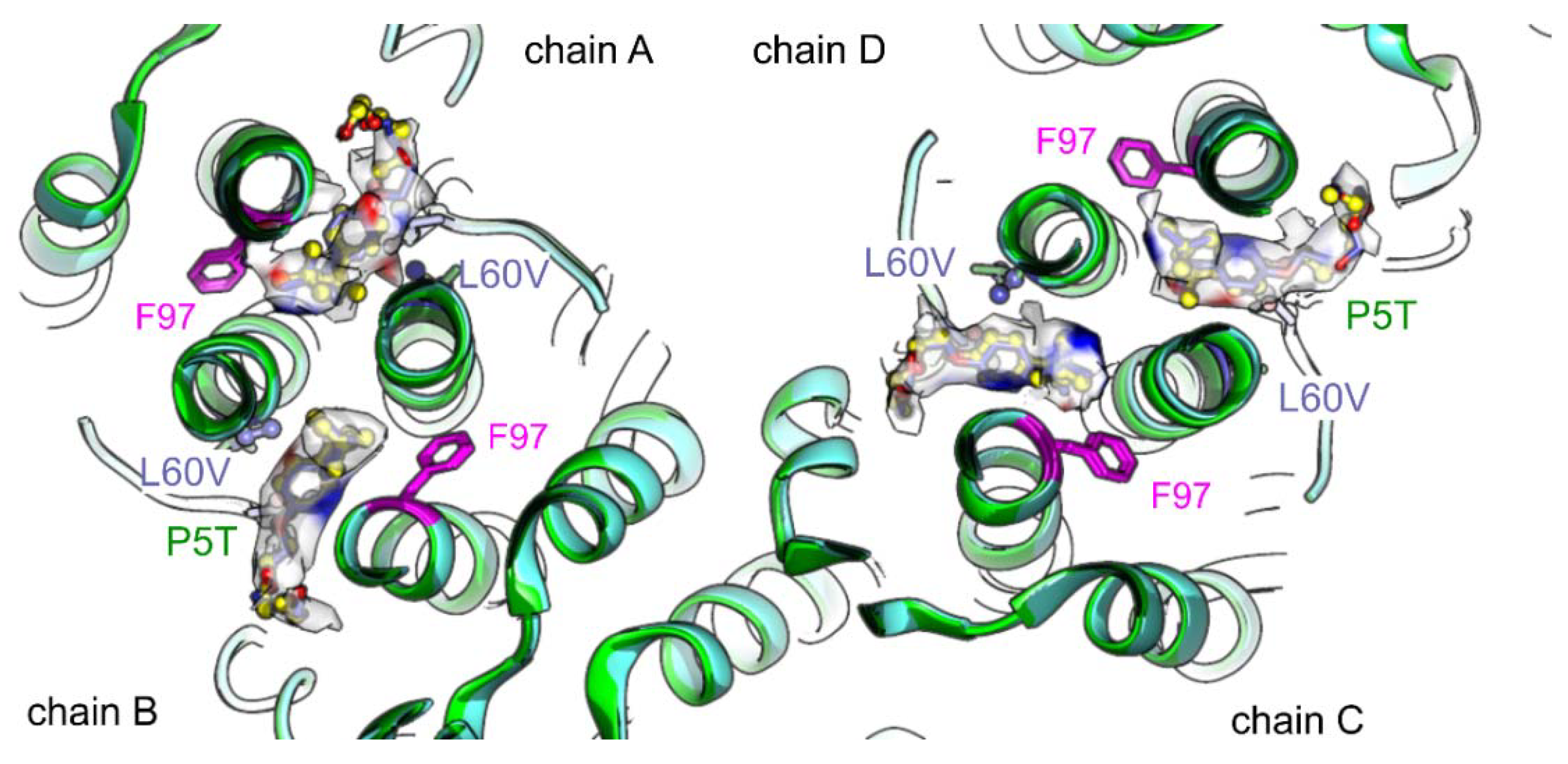
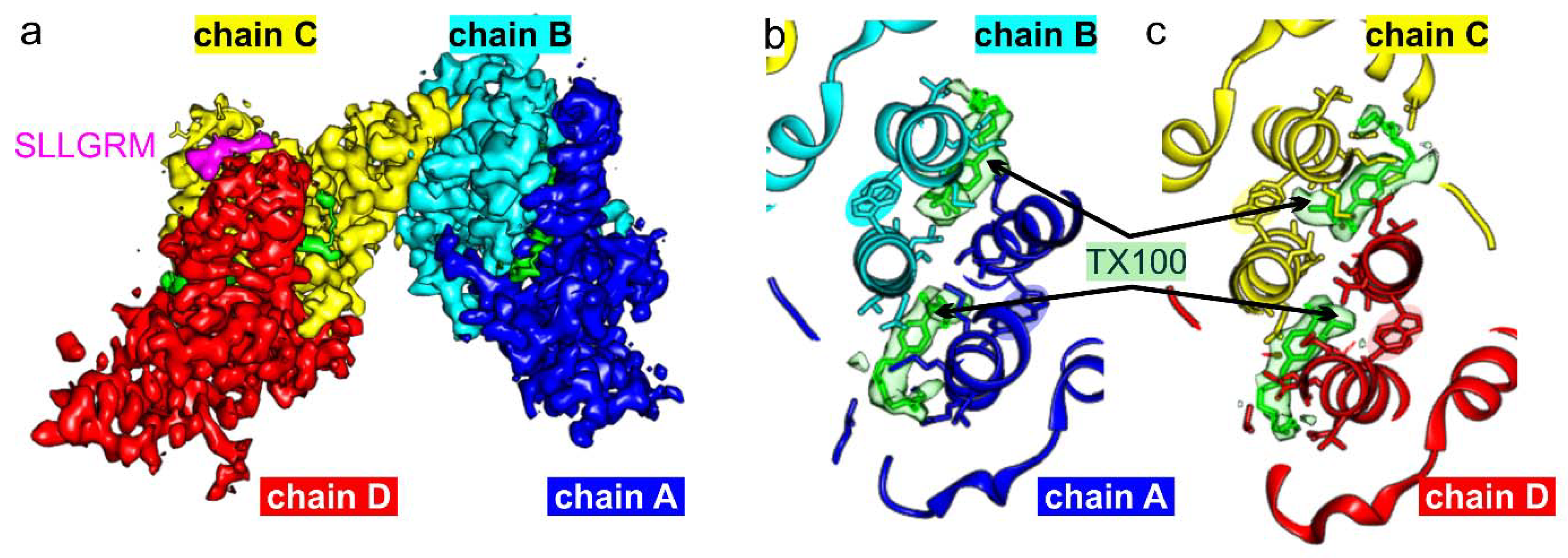
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lauber, C.; Seitz, S.; Mattei, S.; Suh, A.; Beck, J.; Herstein, J.; Borold, J.; Salzburger, W.; Kaderali, L.; Briggs, J.A.G.; et al. Deciphering the origin and evolution of hepatitis b viruses by means of a family of non-enveloped fish viruses. Cell Host Microbe 2017, 22, 387–399.e386. [Google Scholar] [CrossRef] [Green Version]
- Krause-Kyora, B.; Susat, J.; Key, F.M.; Kuhnert, D.; Bosse, E.; Immel, A.; Rinne, C.; Kornell, S.C.; Yepes, D.; Franzenburg, S.; et al. Neolithic and medieval virus genomes reveal complex evolution of hepatitis b. eLife 2018, 7, e36666. [Google Scholar] [CrossRef] [PubMed]
- Alter, M.J. Epidemiology of hepatitis b in europe and worldwide. J. Hepatol. 2003, 39 (Suppl. 1), S64–S69. [Google Scholar] [CrossRef]
- Heermann, K.H.; Goldmann, U.; Schwartz, W.; Seyffarth, T.; Baumgarten, H.; Gerlich, W.H. Large surface proteins of hepatitis b virus containing the pre-s sequence. J. Virol. 1984, 52, 396–402. [Google Scholar] [CrossRef] [Green Version]
- Dane, D.S.; Cameron, C.H.; Briggs, M. Virus-like particles in serum of patients with australia-antigen-associated hepatitis. Lancet 1970, 295, 695–698. [Google Scholar] [CrossRef]
- Mason, W.S.; Aldrich, C.; Summers, J.; Taylor, J.M. Asymmetric replication of duck hepatitis b virus DNA in liver cells: Free minus-strand DNA. Proc. Natl. Acad. Sci. USA 1982, 79, 3997–4001. [Google Scholar] [CrossRef] [Green Version]
- Summers, J.; Mason, W.S. Replication of the genome of a hepatitis b--like virus by reverse transcription of an rna intermediate. Cell 1982, 29, 403–415. [Google Scholar] [CrossRef]
- Ning, X.; Nguyen, D.; Mentzer, L.; Adams, C.; Lee, H.; Ashley, R.; Hafenstein, S.; Hu, J. Secretion of genome-free hepatitis b virus--single strand blocking model for virion morphogenesis of para-retrovirus. PLoS Pathog. 2011, 7, e1002255. [Google Scholar] [CrossRef]
- Ning, X.; Luckenbaugh, L.; Liu, K.; Bruss, V.; Sureau, C.; Hu, J. Common and distinct capsid and surface protein requirements for secretion of complete and genome-free hepatitis b virions. J. Virol. 2018, 92, e00272-00218. [Google Scholar] [CrossRef] [Green Version]
- Makbul, C.; Khayenko, V.; Maric, H.M.; Bottcher, B. Conformational plasticity of hepatitis b core protein spikes promotes peptide binding independent of the secretion phenotype. Microorganisms 2021, 9, 956. [Google Scholar] [CrossRef]
- Makbul, C.; Nassal, M.; Bottcher, B. Slowly folding surface extension in the prototypic avian hepatitis b virus capsid governs stability. eLife 2020, 9, e57277. [Google Scholar] [CrossRef] [PubMed]
- Heger-Stevic, J.; Zimmermann, P.; Lecoq, L.; Böttcher, B.; Nassal, M. Hepatitis b virus core protein phosphorylation: Identification of the srpk1 target sites and impact of their occupancy on rna binding and capsid structure. PLoS Pathog. 2018, 14, e1007488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böttcher, B.; Nassal, M. Structure of mutant hepatitis b core protein capsids with premature secretion phenotype. J. Mol. Biol. 2018, 430, 4941–4954. [Google Scholar] [CrossRef] [PubMed]
- Selzer, L.; Kant, R.; Wang, J.C.Y.; Bothner, B.; Zlotnick, A. Hepatitis b virus core protein phosphorylation sites affect capsid stability and transient exposure of the c-terminal domain. J. Biol. Chem. 2015, 290, 28584. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Jin, L.; Jih, J.; Shih, C.; Zhou, Z.H. 3.5a cryoem structure of hepatitis b virus core assembled from full-length core protein. PLoS ONE 2013, 8, e69729. [Google Scholar] [CrossRef] [Green Version]
- Wynne, S.A.; Crowther, R.A.; Leslie, A.G. The crystal structure of the human hepatitis b virus capsid. Mol. Cell 1999, 3, 771–780. [Google Scholar] [CrossRef]
- Conway, J.F.; Chenng, N.; Zlotnick, A.; Wingfield, P.T.; Stahl, S.J.; Steven, A.C. Visualization of a4-helix bundle in the hepatitis b virus capsid by cryo-electron microscopy. Nature 1997, 368, 91–94. [Google Scholar] [CrossRef]
- Böttcher, B.; Wynne, S.A.; Crowther, R.A. Determination of the fold of the core protein of hepatitis b virus by electron cryomicroscopy. Nature 1997, 386, 88–91. [Google Scholar] [CrossRef]
- Roseman, A.M.; Berriman, J.A.; Wynne, S.A.; Butler, P.J.; Crowther, R.A. A structural model for maturation of the hepatitis b virus core. Proc. Natl. Acad. Sci. USA 2005, 102, 15821–15826. [Google Scholar] [CrossRef] [Green Version]
- Chua, P.K.; Tang, F.M.; Huang, J.Y.; Suen, C.S.; Shih, C. Testing the balanced electrostatic interaction hypothesis of hepatitis b virus DNA synthesis by using an in vivo charge rebalance approach. J. Virol. 2010, 84, 2340–2351. [Google Scholar] [CrossRef] [Green Version]
- Le Pogam, S.; Chua, P.K.; Newman, M.; Shih, C. Exposure of rna templates and encapsidation of spliced viral rna are influenced by the arginine-rich domain of human hepatitis b virus core antigen (hbcag 165-173). J. Virol. 2005, 79, 1871–1887. [Google Scholar] [CrossRef] [Green Version]
- de Rocquigny, H.; Rat, V.; Pastor, F.; Darlix, J.L.; Hourioux, C.; Roingeard, P. Phosphorylation of the arginine-rich c-terminal domains of the hepatitis b virus (hbv) core protein as a fine regulator of the interaction between hbc and nucleic acid. Viruses 2020, 12, 738. [Google Scholar] [CrossRef]
- Zhao, Q.; Hu, Z.; Cheng, J.; Wu, S.; Luo, Y.; Chang, J.; Hu, J.; Guo, J.T. Hepatitis b virus core protein dephosphorylation occurs during pregenomic rna encapsidation. J. Virol. 2018, 92, e02139-17. [Google Scholar] [CrossRef] [Green Version]
- Dryden, K.A.; Wieland, S.F.; Whitten-Bauer, C.; Gerin, J.L.; Chisari, F.V.; Yeager, M. Native hepatitis b virions and capsids visualized by electron cryomicroscopy. Mol. Cell 2006, 22, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Seitz, S.; Urban, S.; Antoni, C.; Böttcher, B. Cryo-electron microscopy of hepatitis b virions reveals variability in envelope capsid interactions. EMBO J. 2007, 26, 4160–4167. [Google Scholar] [CrossRef]
- Selzer, L.; Katen, S.P.; Zlotnick, A. The hepatitis b virus core protein intradimer interface modulates capsid assembly and stability. Biochemistry 2014, 53, 5496–5504. [Google Scholar] [CrossRef] [PubMed]
- Hadden, J.A.; Perilla, J.R.; Schlicksup, C.J.; Venkatakrishnan, B.; Zlotnick, A.; Schulten, K. All-atom molecular dynamics of the hbv capsid reveals insights into biological function and cryo-em resolution limits. eLife 2018, 7, e32478. [Google Scholar] [CrossRef]
- Perez-Segura, C.; Goh, B.C.; Hadden-Perilla, J.A. All-atom md simulations of the hbv capsid complexed with at130 reveal secondary and tertiary structural changes and mechanisms of allostery. Viruses 2021, 13, 564. [Google Scholar] [CrossRef] [PubMed]
- Freund, S.M.; Johnson, C.M.; Jaulent, A.M.; Ferguson, N. Moving towards high-resolution descriptions of the molecular interactions and structural rearrangements of the human hepatitis b core protein. J. Mol. Biol. 2008, 384, 1301–1313. [Google Scholar] [CrossRef]
- Muhamad, A.; Ho, K.L.; Rahman, M.B.A.; Tejo, B.A.; Uhrin, D.; Tan, W.S. Hepatitis b virus peptide inhibitors: Solution structures and interactions with the viral capsid. Org. Biomol. Chem. 2015, 13, 7780–7789. [Google Scholar] [CrossRef] [Green Version]
- Ponsel, D.; Bruss, V. Mapping of amino acid side chains on the surface of hepatitis b virus capsids required for envelopment and virion formation. J. Virol. 2003, 77, 416–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, T.T.; Sahu, G.K.; Whitehead, W.E.; Greenberg, R.; Shih, C. The mechanism of an immature secretion phenotype of a highly frequent naturally occurring missense mutation at codon 97 of human hepatitis b virus core antigen. J. Virol. 1999, 73, 5731–5740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, T.T.; Tai, P.C.; Shih, C. Subtype-independent immature secretion and subtype-dependent replication deficiency of a highly frequent, naturally occurring mutation of human hepatitis b virus core antigen. J. Virol. 1999, 73, 10122–10128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Pogam, S.; Yuan, T.T.; Sahu, G.K.; Chatterjee, S.; Shih, C. Low-level secretion of human hepatitis b virus virions caused by two independent, naturally occurring mutations (p5t and l60v) in the capsid protein. J. Virol. 2000, 74, 9099–9105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecoq, L.; Wang, S.; Dujardin, M.; Zimmermann, P.; Schuster, L.; Fogeron, M.L.; Briday, M.; Schledorn, M.; Wiegand, T.; Cole, L.; et al. A pocket-factor-triggered conformational switch in the hepatitis b virus capsid. Proc. Natl. Acad. Sci. USA 2021, 118, e2022464118. [Google Scholar]
- Brautigam, C.A.; Zhao, H.; Vargas, C.; Keller, S.; Schuck, P. Integration and global analysis of isothermal titration calorimetry data for studying macromolecular interactions. Nat. Protoc. 2016, 11, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Lenhart, J.; Flegler, V.J.; Makbul, C.; Rasmussen, T.; Böttcher, B. Capabilities of the falcon III detector for single-particle structure determination. Ultramicroscopy 2019, 203, 145–154. [Google Scholar] [CrossRef]
- Sader, K.; Matadeen, R.; Castro Hartmann, P.; Halsan, T.; Schlichten, C. Industrial cryo-em facility setup and management. Acta Crystallogr. D Struct. Biol. 2020, 76, 313–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, S.Q.; Palovcak, E.; Armache, J.P.; Verba, K.A.; Cheng, Y.; Agard, D.A. Motioncor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 2017, 14, 331–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohou, A.; Grigorieff, N. Ctffind4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015, 192, 216–221. [Google Scholar] [CrossRef]
- Zivanov, J.; Nakane, T.; Scheres, S.H.W. Estimation of high-order aberrations and anisotropic magnification from cryo-em data sets in relion-3.1. IUCrJ 2020, 7, 253–267. [Google Scholar] [CrossRef] [Green Version]
- Scheres, S.H. Relion: Implementation of a bayesian approach to cryo-em structure determination. J. Struct. Biol. 2012, 180, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Russo, C.J.; Henderson, R. Ewald sphere correction using a single side-band image processing algorithm. Ultramicroscopy 2018, 187, 26–33. [Google Scholar] [CrossRef]
- Zivanov, J.; Nakane, T.; Forsberg, B.O.; Kimanius, D.; Hagen, W.J.; Lindahl, E.; Scheres, S.H. New tools for automated high-resolution cryo-em structure determination in relion-3. eLife 2018, 7, e42166. [Google Scholar] [CrossRef]
- Yang, Z.; Lasker, K.; Schneidman-Duhovny, D.; Webb, B.; Huang, C.C.; Pettersen, E.F.; Goddard, T.D.; Meng, E.C.; Sali, A.; Ferrin, T.E. Ucsf chimera, modeller, and imp: An integrated modeling system. J. Struct. Biol. 2012, 179, 269–278. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. Ucsf chimerax: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Terwilliger, T.C.; Sobolev, O.V.; Afonine, P.V.; Adams, P.D. Automated map sharpening by maximization of detail and connectivity. Acta Crystallogr. D Struct. Biol. 2018, 74, 545–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanal, A.; Lohkamp, B.; Emsley, P. Current developments in coot for macromolecular model building of electron cryo-microscopy and crystallographic data. Protein Sci. 2020, 29, 1069–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkoczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using x-rays, neutrons and electrons: Recent developments in phenix. Acta Crystallogr. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Noble, A.J.; Kang, J.Y.; Darst, S.A. Eliminating effects of particle adsorption to the air/water interface in single-particle cryo-electron microscopy: Bacterial rna polymerase and chapso. J. Struct. Biol. X 2019, 1, 100005. [Google Scholar] [CrossRef] [PubMed]
- Flegler, V.J.; Rasmussen, A.; Borbil, K.; Boten, L.; Chen, H.A.; Deinlein, H.; Halang, J.; Hellmanzik, K.; Loffler, J.; Schmidt, V.; et al. Mechanosensitive channel gating by delipidation. Proc. Natl. Acad. Sci. USA 2021, 118, e2107095118. [Google Scholar] [CrossRef]
- Rosenthal, P.B.; Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 2003, 333, 721–745. [Google Scholar] [CrossRef]
- Weber, D.S.; Warren, J.J. The interaction between methionine and two aromatic amino acids is an abundant and multifunctional motif in proteins. Arch. Biochem. Biophys. 2019, 672, 108053. [Google Scholar] [CrossRef] [PubMed]
- Packianathan, C.; Katen, S.P.; Dann, C.E., 3rd; Zlotnick, A. Conformational changes in the hepatitis b virus core protein are consistent with a role for allostery in virus assembly. J. Virol. 2010, 84, 1607–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Wang, J.C.; Segura, C.P.; Hadden-Perilla, J.A.; Zlotnick, A. The integrity of the intradimer interface of the hepatitis b virus capsid protein dimer regulates capsid self-assembly. ACS Chem. Biol. 2020, 15, 3124–3132. [Google Scholar] [CrossRef]
- Tang, K.F.; Abdullah, M.P.; Yusoff, K.; Tan, W.S. Interactions of hepatitis b core antigen and peptide inhibitors. J. Med. Chem. 2007, 50, 5620–5626. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. Dogsitescorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatakrishnan, B.; Katen, S.P.; Francis, S.; Chirapu, S.; Finn, M.G.; Zlotnick, A. Hepatitis b virus capsids have diverse structural responses to small-molecule ligands bound to the heteroaryldihydropyrimidine pocket. J. Virol. 2016, 90, 3994–4004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlicksup, C.J.; Wang, J.C.; Francis, S.; Venkatakrishnan, B.; Turner, W.W.; VanNieuwenhze, M.; Zlotnick, A. Hepatitis b virus core protein allosteric modulators can distort and disrupt intact capsids. eLife 2018, 7, e31473. [Google Scholar] [CrossRef]
- Chang, S.Y.; Ko, T.P.; Liang, P.H.; Wang, A.H. Catalytic mechanism revealed by the crystal structure of undecaprenyl pyrophosphate synthase in complex with sulfate, magnesium, and triton. J. Biol. Chem. 2003, 278, 29298–29307. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, B.M.; Schmidt, A.; Behrmann, E.; Burger, J.; Mielke, T.; Spahn, C.M.T.; Heck, M.; Scheerer, P. Mechanistic insights into the role of prenyl-binding protein prbp/delta in membrane dissociation of phosphodiesterase 6. Nat. Commun. 2018, 9, 90. [Google Scholar] [CrossRef] [Green Version]
- Urban, S.; Neumann-Haefelin, C.; Lampertico, P. Hepatitis d virus in 2021: Virology, immunology and new treatment approaches for a difficult-to-treat disease. Gut 2021, 70, 1782–1794. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, K. Complete and incomplete hepatitis b virus particles: Formation, function, and application. Viruses 2017, 9, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | # of Movies (3) | # of Selected Particles | # of Particles in Final Map | Resolution of Map (1) | B-Factor (2) |
|---|---|---|---|---|---|
| HBc + TX100 | 6750 | 69,257 | 41,478 | 3.1 Š| 121 Ų |
| HBc +TX100 + SLLGRM | 1520 | 19,232 | 11,525 | 3.2 Š| 102 Ų |
| HBc-F97L + TX100 | 5696 | 146,930 | 89,978 | 2.9 Š| 121 Ų |
| HBc-F97L | 2409 | 180,259 | 114,274 | 2.8 Š| 107 Ų |
| HBc-L60V +TX100 | 6675 | 108,843 | 73,540 | 2.8 Š| 106 Ų |
| HBc-P5T + TX100 | 5625 | 16,689 | 130,610 | 2.9 Š| 117 Ų |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makbul, C.; Kraft, C.; Grießmann, M.; Rasmussen, T.; Katzenberger, K.; Lappe, M.; Pfarr, P.; Stoffer, C.; Stöhr, M.; Wandinger, A.-M.; et al. Binding of a Pocket Factor to Hepatitis B Virus Capsids Changes the Rotamer Conformation of Phenylalanine 97. Viruses 2021, 13, 2115. https://doi.org/10.3390/v13112115
Makbul C, Kraft C, Grießmann M, Rasmussen T, Katzenberger K, Lappe M, Pfarr P, Stoffer C, Stöhr M, Wandinger A-M, et al. Binding of a Pocket Factor to Hepatitis B Virus Capsids Changes the Rotamer Conformation of Phenylalanine 97. Viruses. 2021; 13(11):2115. https://doi.org/10.3390/v13112115
Chicago/Turabian StyleMakbul, Cihan, Christian Kraft, Matthias Grießmann, Tim Rasmussen, Kilian Katzenberger, Melina Lappe, Paul Pfarr, Cato Stoffer, Mara Stöhr, Anna-Maria Wandinger, and et al. 2021. "Binding of a Pocket Factor to Hepatitis B Virus Capsids Changes the Rotamer Conformation of Phenylalanine 97" Viruses 13, no. 11: 2115. https://doi.org/10.3390/v13112115