
Insertion of Exogenous Genes within the ORF1a Coding Region of Porcine Astrovirus

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Antibodies
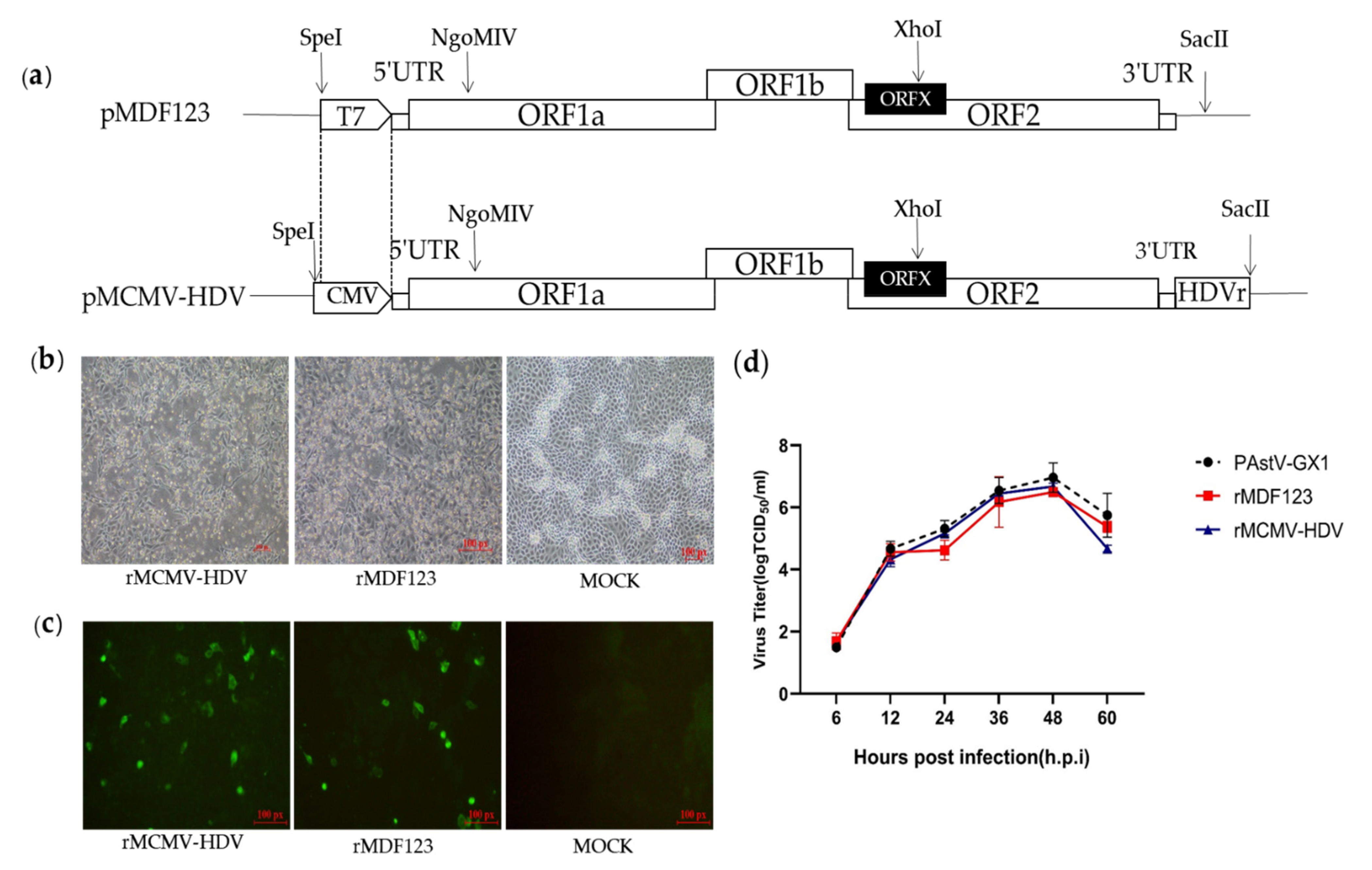
2.2. Construction of DNA-Launched Infectious Clone of a PAstV Type 1 Strain
2.3. Transposon-Mediated 15 bp Random Insertion
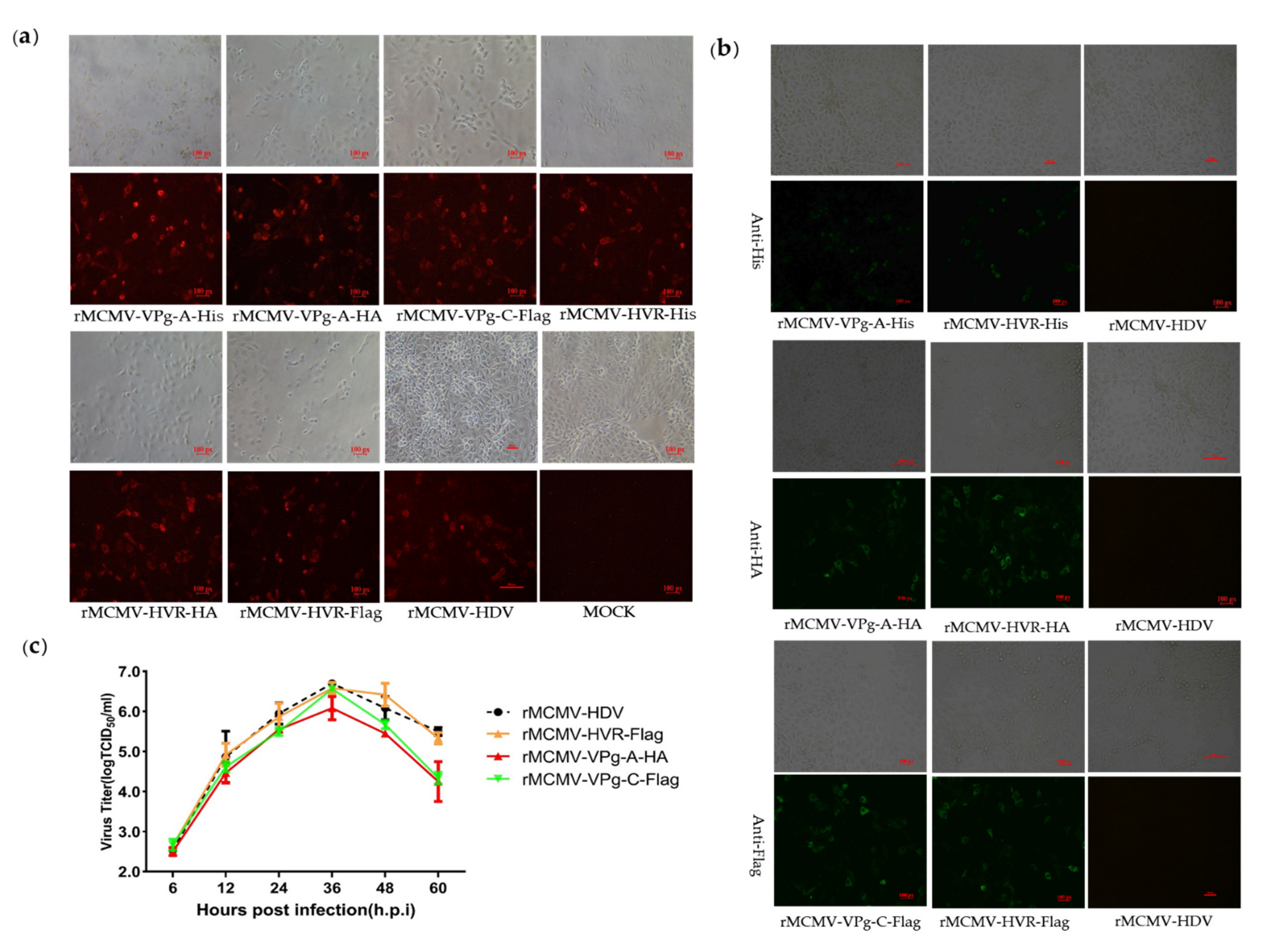
2.4. Construction of Tags or Reporter Recombinant PAstV Infectious Clones
2.5. In Vitro Transcription and Transfection
2.6. RT-PCR and Sequencing
2.7. Indirect Immunofluorescence Assay (IFA)
2.8. Multi-Step Growth Curve
3. Results
3.1. Construction and Recovery of a DNA-Launched Infectious cDNA Clone of PAstV
3.2. Identification of 15 bp Insertion Sites in the PAstV ORF1a Protein
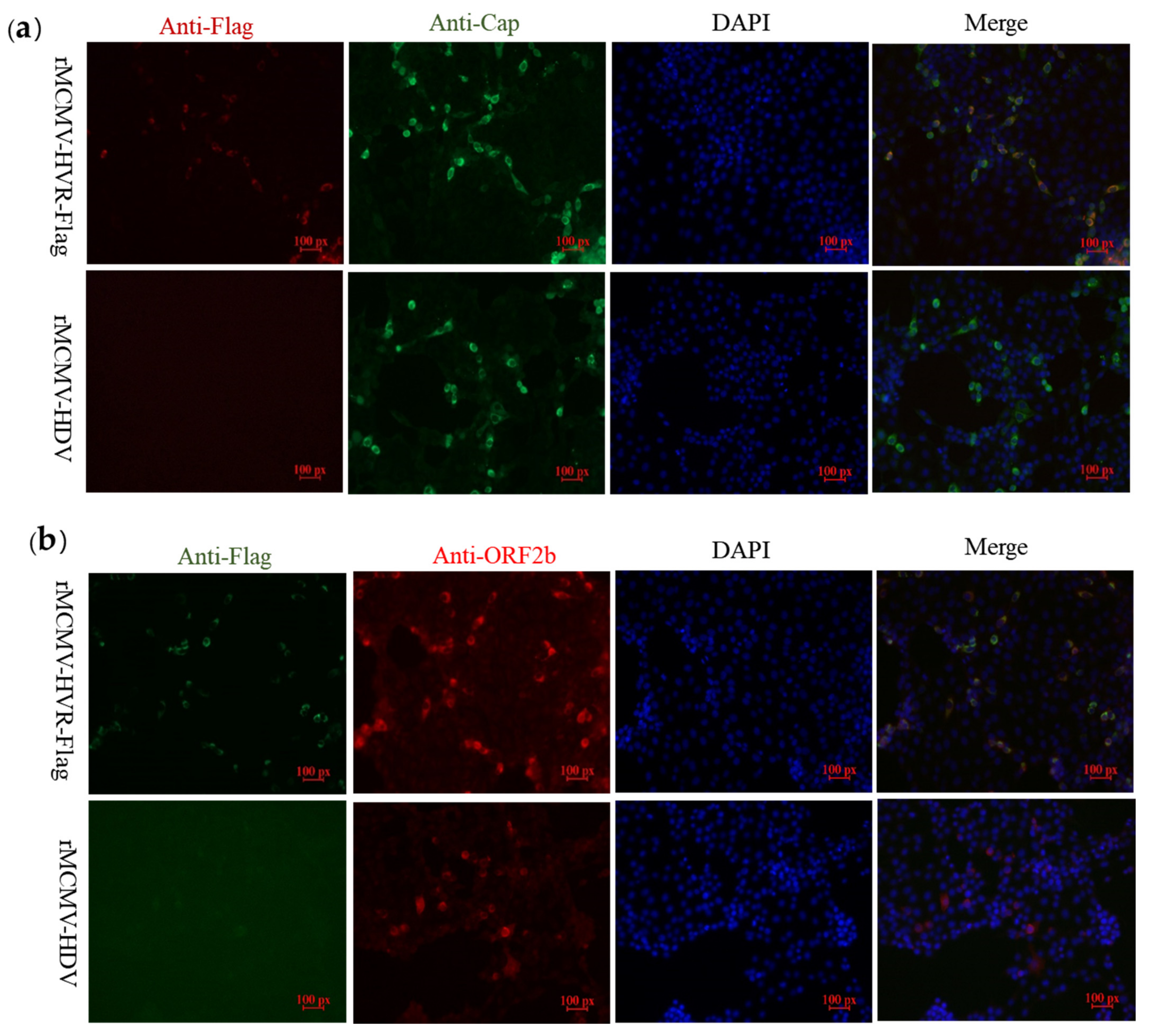
3.3. Recombinant Viruses Harboring Epitope Tags Allow Visualization of the ORF1a Protein
3.4. ORF1a Protein Harboring Epitope Tags Allow Investigation of the Interactions with ORF2b Protein and Cap Protein
3.5. Replicons Harboring iLOV in the HVR Allow Visualization of PAstV Replication
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bosch, A.; Pintó, R.; Guix, S. Human Astroviruses. Clin. Microbiol. Rev. 2014, 27, 1048. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Ji, C.; Liu, T.; Zhang, W.; Fang, Q.; Dong, Q.; Li, M.; Wang, H.; Chen, Y.; Ouyang, K.; et al. Identification of a novel protein in porcine astrovirus that is important for virus replication. Vet. Microbiol. 2021, 255, 108984. [Google Scholar] [CrossRef] [PubMed]
- Lulla, V.; Firth, A.E. A hidden gene in astroviruses encodes a viroporin. Nat. Commun. 2020, 11, 4070. [Google Scholar] [CrossRef] [PubMed]
- Benedictis, P.D.; Schultz-Cherry, S.; Burnham, A.; Cattoli, G. Astrovirus infections in humans and animals-molecular biology, genetic diversity, and interspecies transmissions. Infect. Genet. Evol. 2011, 11, 1529–1544. [Google Scholar] [CrossRef]
- Fernández-Correa, I.; Truchado, D.A.; Gomez, A.D.; Doménech, A.; Pérez-Tris, J.; Schmidt-Chanasit, J.; Cadar, D.; Benítez, L. A novel group of avian astroviruses from Neotropical passerine birds broaden the diversity and host range of Astroviridae. Sci. Rep. 2019, 9, 9513. [Google Scholar] [CrossRef]
- González, J.M.; Pénzes, Z.; Almazán, F.; Calvo, E.; Enjuanes, L. Stabilization of a Full-Length Infectious cDNA Clone of Transmissible Gastroenteritis Coronavirus by Insertion of an Intron. J. Virol. 2002, 76, 4655–4661. [Google Scholar] [CrossRef] [Green Version]
- Appleton, H.; Higgins, P.G. Viruses and gastroenteritis in infants. Lancet 1975, 305, 7919. [Google Scholar] [CrossRef]
- Qin, Y.; Fang, Q.; Liu, H.; Ji, C.; Chen, Y.; Ouyang, K.; Wei, Z.; Huang, W. Construction of a reverse genetic system for porcine astrovirus. Arch. Virol. 2018, 163, 1511–1518. [Google Scholar] [CrossRef]
- Wohlgemuth, N.; Honce, R.; Schultz-Cherry, S. Astrovirus evolution and emergence. Infect. Genet. Evol. 2019, 69, 30–37. [Google Scholar] [CrossRef]
- Ulloa, J.C.; Gutiérrez, M. Genomic analysis of two ORF2 segments of new porcine astrovirus isolates and their close relationship with human astroviruses. Can. J. Microbiol. 2010, 56, 569. [Google Scholar] [CrossRef]
- Rivera, R.; Nollens, H.H.; Venn-Watson, S.; Gulland, F.; Wellehan, J. Characterization of phylogenetically diverse astro-viruses of marine mammals. J. Gen. Virol. 2010, 91, 166. [Google Scholar] [CrossRef]
- Chu, D.K.W.; Chin, A.W.; Smith, G.J.; Chan, K.-H.; Guan, Y.; Peiris, J.S.M.; Poon, L.L.M. Detection of novel astroviruses in urban brown rats and previously known astroviruses in humans. J. Gen. Virol. 2010, 91, 2457–2462. [Google Scholar] [CrossRef]
- Kirkwood, C.D.; Clark, R.; Bogdanovic-Sakran, N.; Ruth, A.O. A 5-year study of the prevalence and genetic diversity of human caliciviruses associated with sporadic cases of acute gastroenteritis in young children admitted to hospital in Mel-bourne, Australia (1998–2002). Med. Klin. 2005, 77, 96–101. [Google Scholar] [CrossRef]
- Caracciolo, S.; Minini, C.; Colombrita, D.; Foresti, I.; Caruso, A. Detection of sporadic cases of Norovirus infection in hospitalized children in Italy. New Microbiol. 2007, 30, 49–52. [Google Scholar]
- Fang, Q.; Wang, C.; Liu, H.; Wu, Q.; Liang, S.; Cen, M.; Dong, Q.; Wei, Y.; Chen, Y.; Ouyang, K.; et al. Pathogenic Characteristics of a Porcine Astrovirus Strain Isolated in China. Viruses 2019, 11, 1156. [Google Scholar] [CrossRef] [Green Version]
- Vannucci, F.A.; Linhares, D.C.L.; de Barcellos, D.E.S.N.; Lam, H.C.; Collins, J.; Marthaler, D. Identification and Complete Genome of Seneca Valley Virus in Vesicular Fluid and Sera of Pigs Affected with Idiopathic Vesicular Disease, Brazil. Transbound. Emerg. Dis. 2015, 62, 589–593. [Google Scholar] [CrossRef]
- Vu, D.L.; Cordey, S.; Brito, F.; Kaiser, L. Novel human astroviruses: Novel human diseases? J. Clin. Virol. 2016, 82, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Schlottau, K.; Schulze, C.; Bilk, S.; Hanke, D.; Höper, D.; Beer, M.; Hoffmann, B. Detection of a Novel Bovine Astrovirus in a Cow with Encephalitis. Transbound. Emerg. Dis. 2016, 63, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Seuberlich, T.; Wüthrich, D.; Selimovichamza, S.; Drögemüller, C.; Oevermann, A.; Bruggmann, R.; Bouzalas, I. Identification of a second encephalitis-associated astrovirus in cattle. Emerg. Microbes Infect. 2016, 5, e5. [Google Scholar] [CrossRef] [Green Version]
- Deiss, R.; Selimovic-Hamza, S.; Seuberlich, T.; Meylan, M. Neurologic Clinical Signs in Cattle with Astrovirus-Associated Encephalitis. J. Vet. Intern. Med. 2017, 31, 1209–1214. [Google Scholar] [CrossRef] [Green Version]
- Truchet, L.; Walland, J.; Wüthrich, D.; Boujon, C.L.; Posthaus, H.; Bruggmann, R.; Schüpbach-Regula, G.; Oevermann, A.; Seuberlich, T. Neuropathological survey reveals underestimation of the prevalence of neuroinfectious diseases in cattle in Switzerland. Vet. Microbiol. 2017, 208, 137–145. [Google Scholar] [CrossRef]
- Bailey, A.; Paulo, A.; Melissa, H.; Chen, Q.; Zheng, Y.; Yang, C.; Honorato, G.; Matias, F.F.; Phil, G.; Kent, S. Porcine Astrovirus Type 3 in Central Nervous System of Swine with Polioencephalomyelitis. Emerg. Infect. Dis. 2017, 23, 2097–2100. [Google Scholar]
- Pfaff, F.; Schlottau, K.; Scholes, S.; Courtenay, A.; Hoffmann, B.; Höper, D.; Beer, M. A novel astrovirus associated with encephalitis and ganglionitis in domestic sheep. Transbound. Emerg. Dis. 2017, 64, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, A.; Hause, B.M. Detection and characterization of a novel genotype of porcine astrovirus 4 from nasal swabs from pigs with acute respiratory disease. Arch. Virol. 2016, 161, 2575–2579. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Tian, J.; Yang, J.; Jiang, X.; Wang, H.; Chen, H.; Yi, T.; Diao, Y. Novel Goose Astrovirus Associated Gout in Gosling, China. Vet. Microbiol. 2018, 220, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Almazan, F.; Gonzalez, J.M.; Pénzes, Z.; Izeta, A.; Calvo, E.; Plana-Durán, J.; Enjuanes, L. Engineering the largest RNA virus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA 2000, 97, 5516–5521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.-J.; Yun, S.-I.; Kang, S.-Y.; Lee, Y.-M. Identification of 5′ and 3′ cis-Acting Elements of the Porcine Reproductive and Respiratory Syndrome Virus: Acquisition of Novel 5′ AU-Rich Sequences Restored Replication of a 5′-Proximal 7-Nucleotide Deletion Mutant. J. Virol. 2006, 80, 723–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geigenmüller, U.; Ginzton, N.H.; Matsui, S.M. Construction of a genome-length cDNA clone for human astrovirus serotype 1 and synthesis of infectious RNA transcripts. J. Virol. 1997, 71, 1713–1717. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Lan, J.; Li, H.; Chen, J.; Yang, Y.; Lin, S.; Xie, Z.; Jian, S. A novel method to rescue and culture duck Astrovirus type 1 in vitro. Virol. J. 2019, 16, 112. [Google Scholar] [CrossRef] [Green Version]
- Crivat, G.; Taraska, J.W. Imaging proteins inside cells with fluorescent tags. Trends Biotechnol. 2012, 30, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Moradpour, D.; Evans, M.J.; Gosert, R.; Yuan, Z.; Blum, H.E.; Goff, S.P.; Lindenbach, B.D.; Rice, C.M. Insertion of Green Fluorescent Protein into Nonstructural Protein 5A Allows Direct Visualization of Functional Hepatitis C Virus Replication Complexes. Hepatology 2004, 38, 343. [Google Scholar] [CrossRef]
- Szkolnicka, D.; Pollán, A.; Da Silva, N.; Oechslin, N.; Gouttenoire, J.; Moradpour, D. Recombinant Hepatitis E Viruses Harboring Tags in the ORF1 Protein. J. Virol. 2019, 93, e00459-19. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Yang, F.; Zhang, Y.; Liu, H.; Jin, Y.; Cao, W.; Zhu, Z.; Zheng, H.; Yin, H. The rescue and evaluation of FLAG and HIS epitope-tagged Asia 1 type foot-and-mouth disease viruses. Virus Res. 2016, 213, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Abente, E.J.; Sosnovtsev, S.V.; Bok, K.; Green, K.Y. Visualization of feline calicivirus replication in real-time with re-combinant viruses engineered to express fluorescent reporter proteins. Virology 2010, 400, 18–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guix, S.; Caballero, S.; Bosch, A.; Pintó, R.M. Human astrovirus C-terminal nsP1a protein is involved in RNA replication. Virology 2005, 333, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, M.; Rajput, C.; Hinde, J.L.; Wu, Q.; Lei, J.; Ishikawa, T.; Bentley, J.K.; Hershenson, M.B. Construction of a recom-binant rhinovirus accommodating fluorescent marker expression. Influenza Other. Respir. Viruses 2018, 12, 717–727. [Google Scholar] [CrossRef]
- Seago, J.; Juleff, N.; Moffat, K.; Berryman, S.; Christie, J.; Charleston, B.; Jackson, T. An infectious recombinant foot-and-mouth disease virus expressing a fluorescent marker protein. J. Gen. Virol. 2013, 94, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Di, H.; Morantz, E.K.; Sadhwani, H.; Madden, J.C.; Brinton, M.A. Insertion position as well as the inserted TRS and gene sequences differentially affect the retention of foreign gene expression by simian hemorrhagic fever virus (SHFV). Virology 2018, 525, 150–160. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Predicted Domain | Site of Insertion (nt) | Nucleotide Sequence of 15 nt Insertion |
|---|---|---|
| CC | 1902/1903 | CTAATtgcggccgcactaatGAGAT |
| VPg-A | 2016/2017 | GTGGGtgcggccgcagtgggCGCAC |
| VPg-B | 2033/2034 | CGGACtgcggccgcacggacCAATC |
| VPg-C | 2199/2200 | CGCAATtgcggccgcagcaatGAGGA |
| HVR | 2261/2262 | GCCCATtgcggccgcacccatCCCTG |
| Viral Protein | Site of Insertion (nt) | His | Flag | HA |
|---|---|---|---|---|
| CC | 1902/1903 | ⚪ | × | ⚪ |
| VPg-A | 2016/2017 | √ | × | √ |
| VPg-B | 2033/2034 | × | × | × |
| VPg-C | 2199/2200 | ⚪ | √ | ⚪ |
| HVR | 2261/2262 | √ | √ | √ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, Y.; Liu, T.; Qin, Y.; Dong, Q.; Chen, Y.; Ouyang, K.; Wei, Z.; Huang, W. Insertion of Exogenous Genes within the ORF1a Coding Region of Porcine Astrovirus. Viruses 2021, 13, 2119. https://doi.org/10.3390/v13112119
Du Y, Liu T, Qin Y, Dong Q, Chen Y, Ouyang K, Wei Z, Huang W. Insertion of Exogenous Genes within the ORF1a Coding Region of Porcine Astrovirus. Viruses. 2021; 13(11):2119. https://doi.org/10.3390/v13112119
Chicago/Turabian StyleDu, Yanjie, Teng Liu, Yifeng Qin, Qinting Dong, Ying Chen, Kang Ouyang, Zuzhang Wei, and Weijian Huang. 2021. "Insertion of Exogenous Genes within the ORF1a Coding Region of Porcine Astrovirus" Viruses 13, no. 11: 2119. https://doi.org/10.3390/v13112119
APA StyleDu, Y., Liu, T., Qin, Y., Dong, Q., Chen, Y., Ouyang, K., Wei, Z., & Huang, W. (2021). Insertion of Exogenous Genes within the ORF1a Coding Region of Porcine Astrovirus. Viruses, 13(11), 2119. https://doi.org/10.3390/v13112119