
Genome-Wide Analysis of Alternative Splicing during Host-Virus Interactions in Chicken

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Sources
2.2. Read Alignment to the Reference Chicken Genome and Gene Expression Estimation
2.3. Putative Transcript Assembly and AS Event Identification
2.4. Alternative Splicing Landscapes and Differential Splicing Analysis
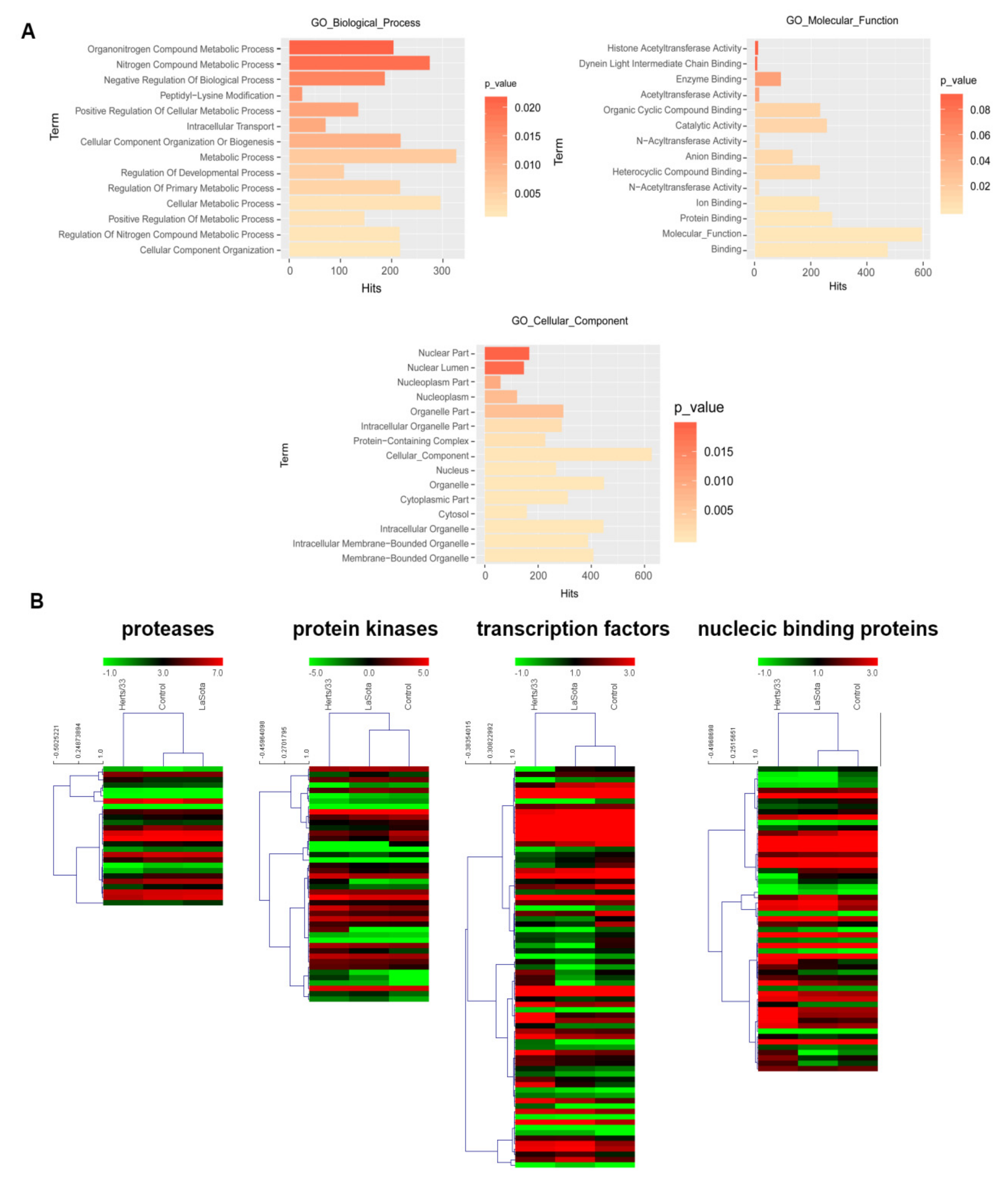
2.5. Functional Enrichment, Clustering, and Isoform-Level Differential Expression Analysis
2.6. 3′ UTR Analysis
2.7. Spliceosome Analysis
3. Results
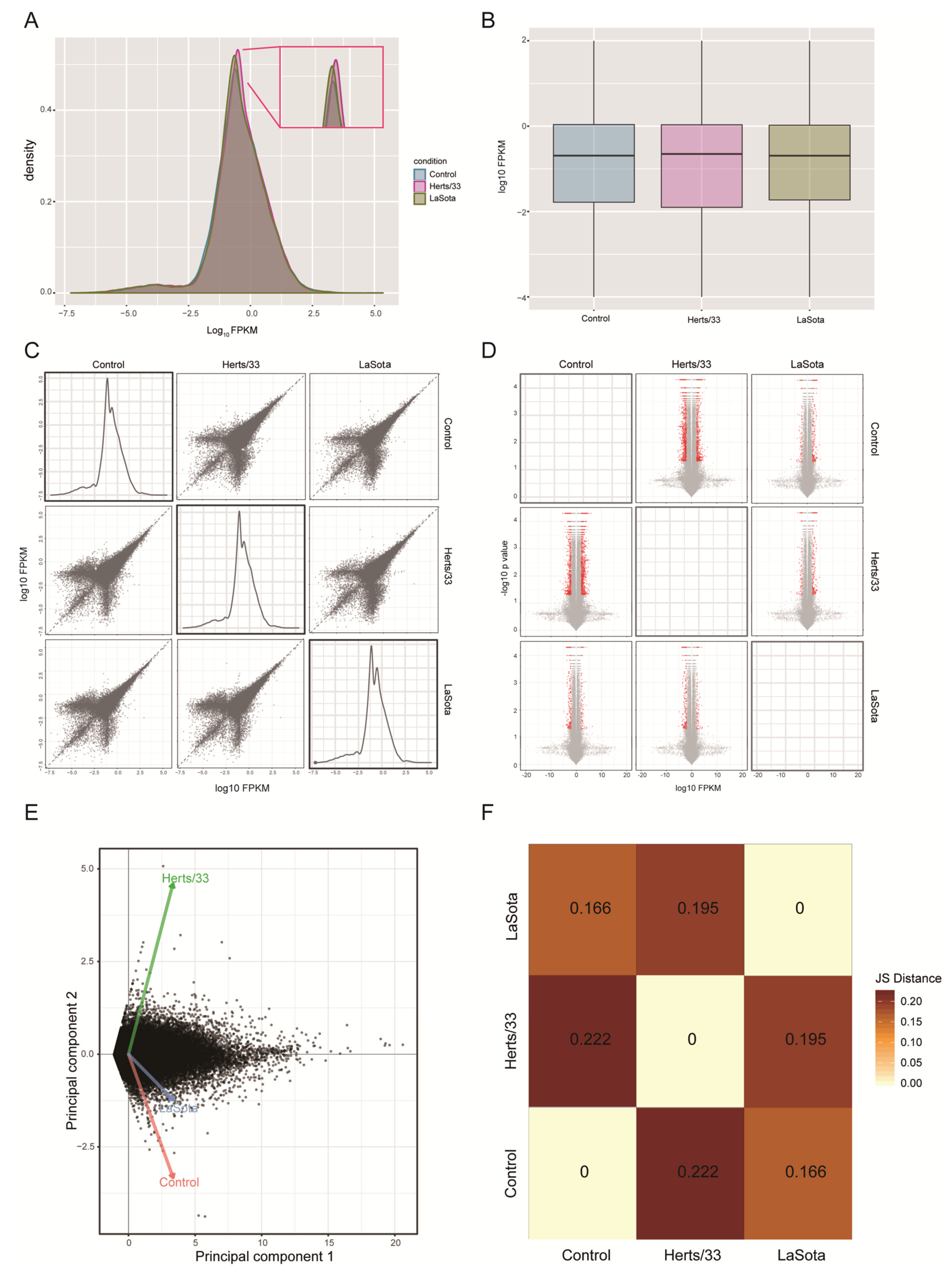
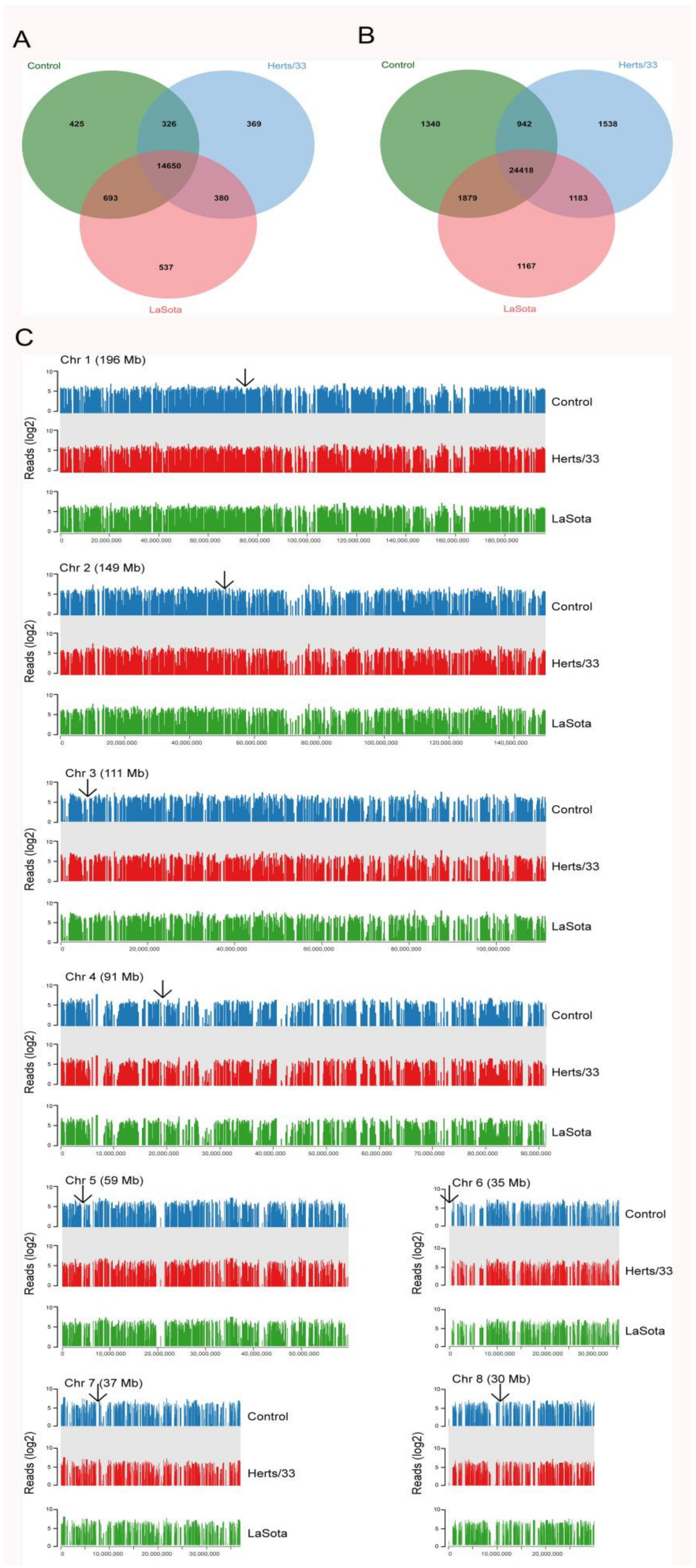
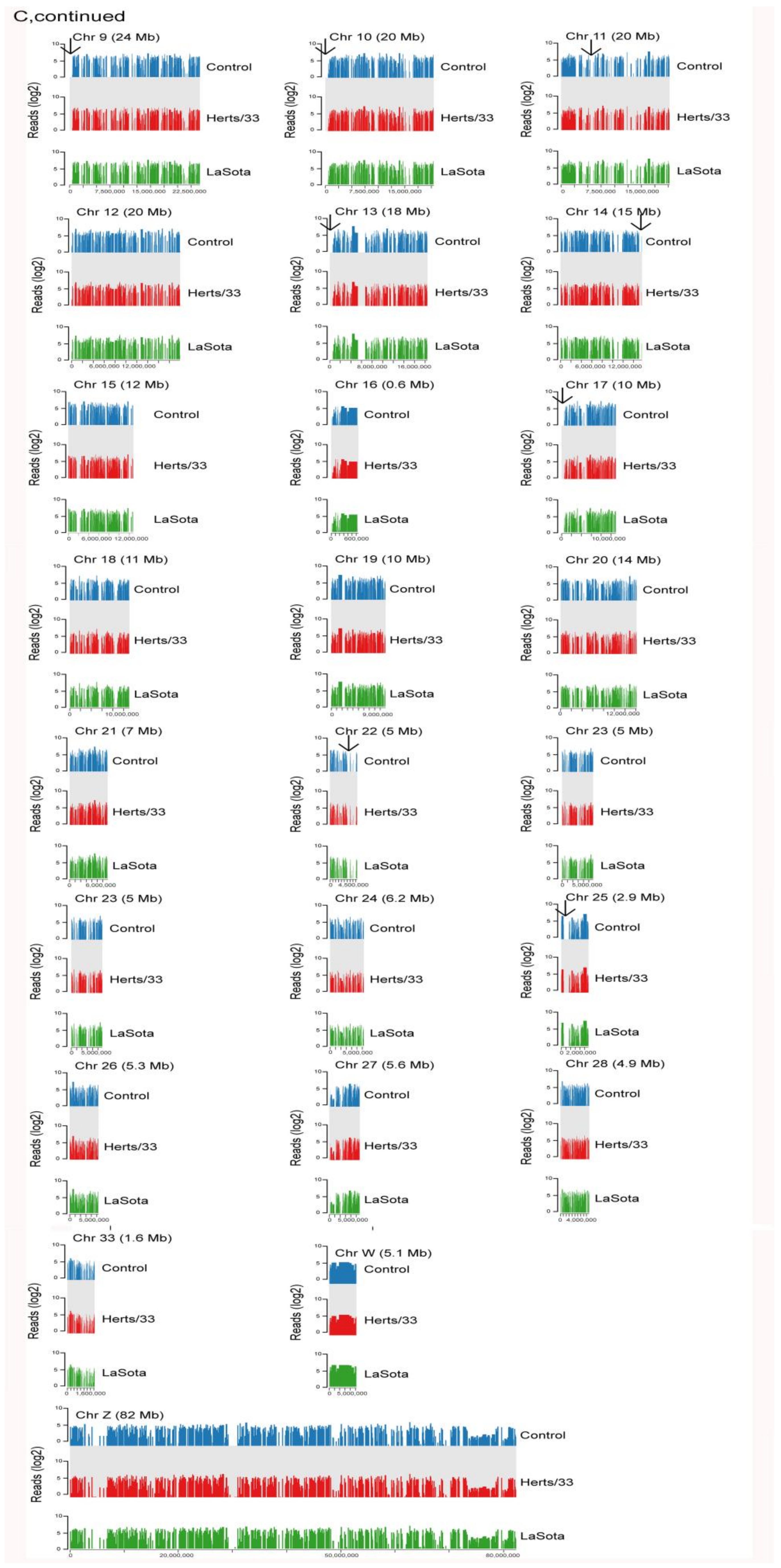
3.1. Mapping Transcriptome Data to the Reference Chicken Genome
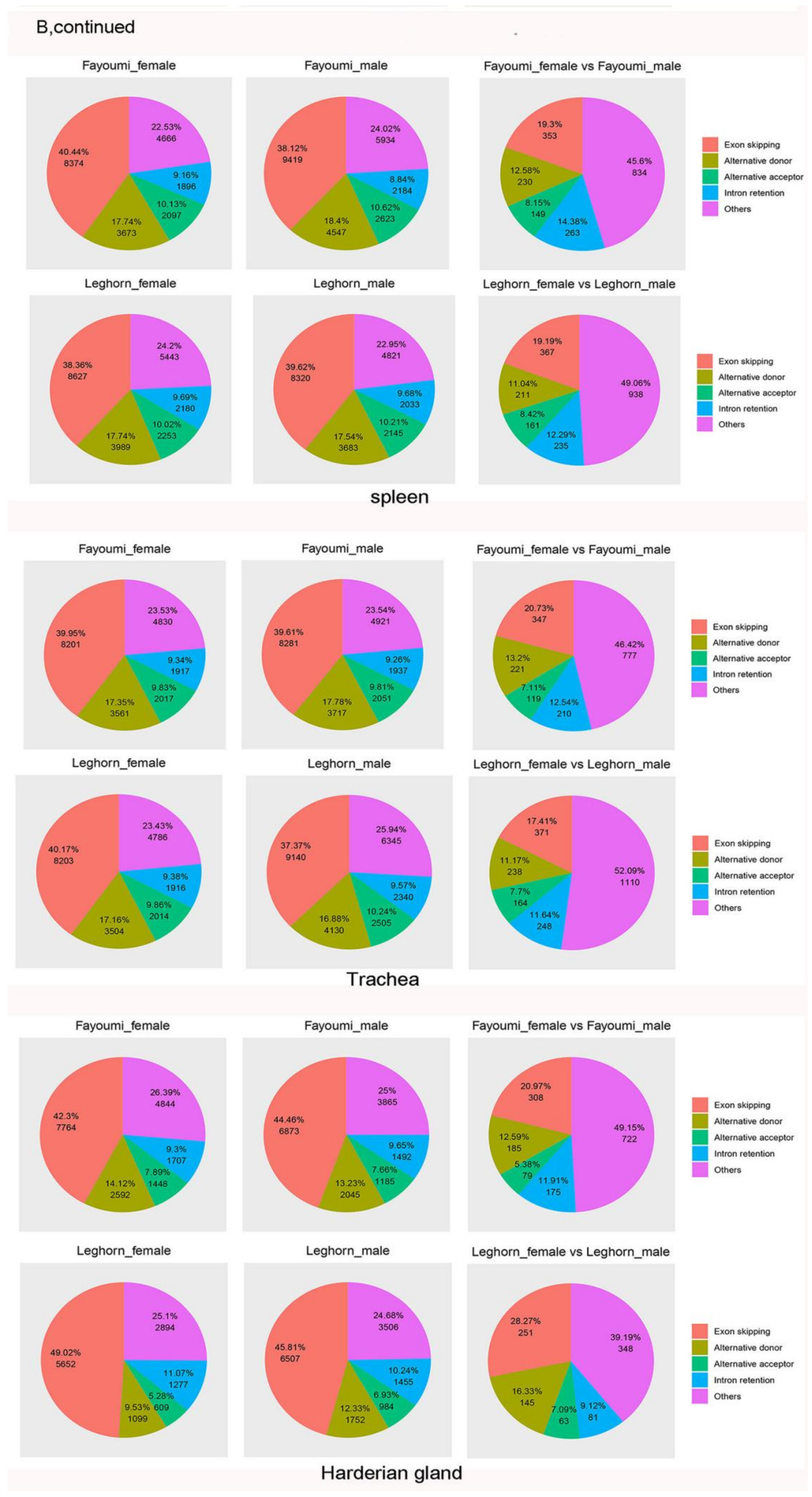
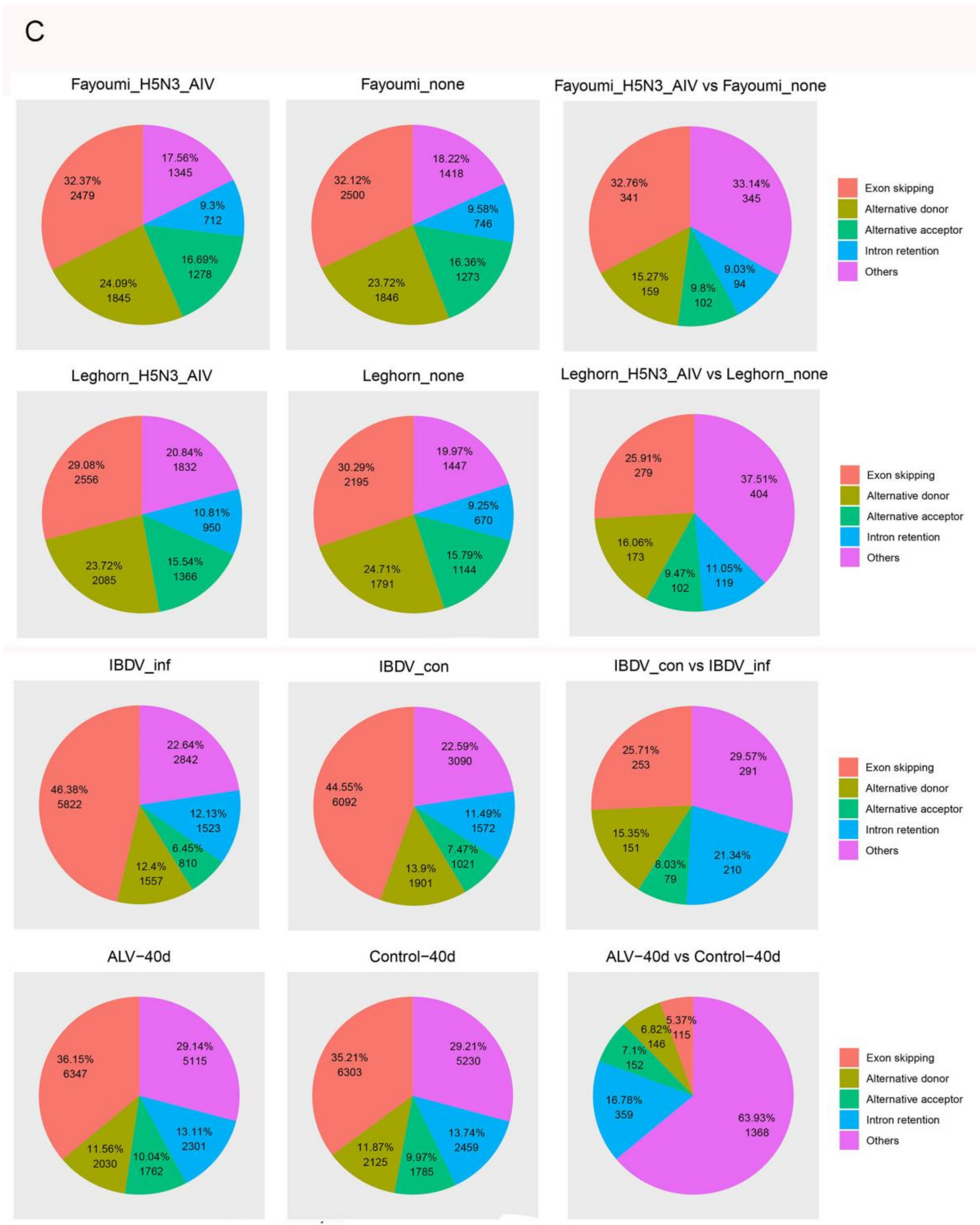
3.2. Alternative Splicing Events of Virus-Infected Chicken
3.3. Virus-Modulated Alternatively Spliced Gene Cluster Analysis
3.4. Dynamic Analyses of Alternative Polyadenylation in 3′ UTRs
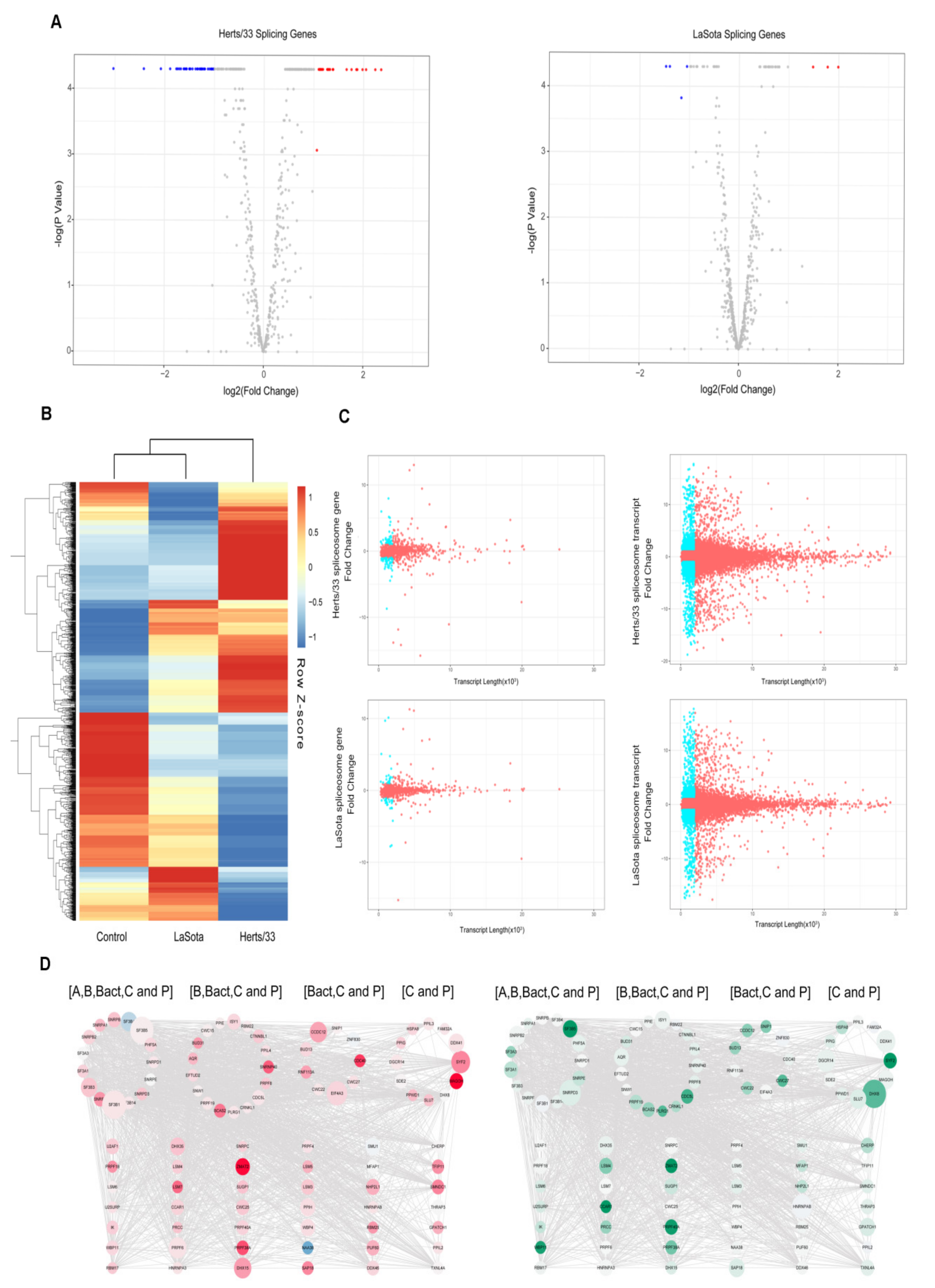
3.5. Spliceosome during Virus Infection
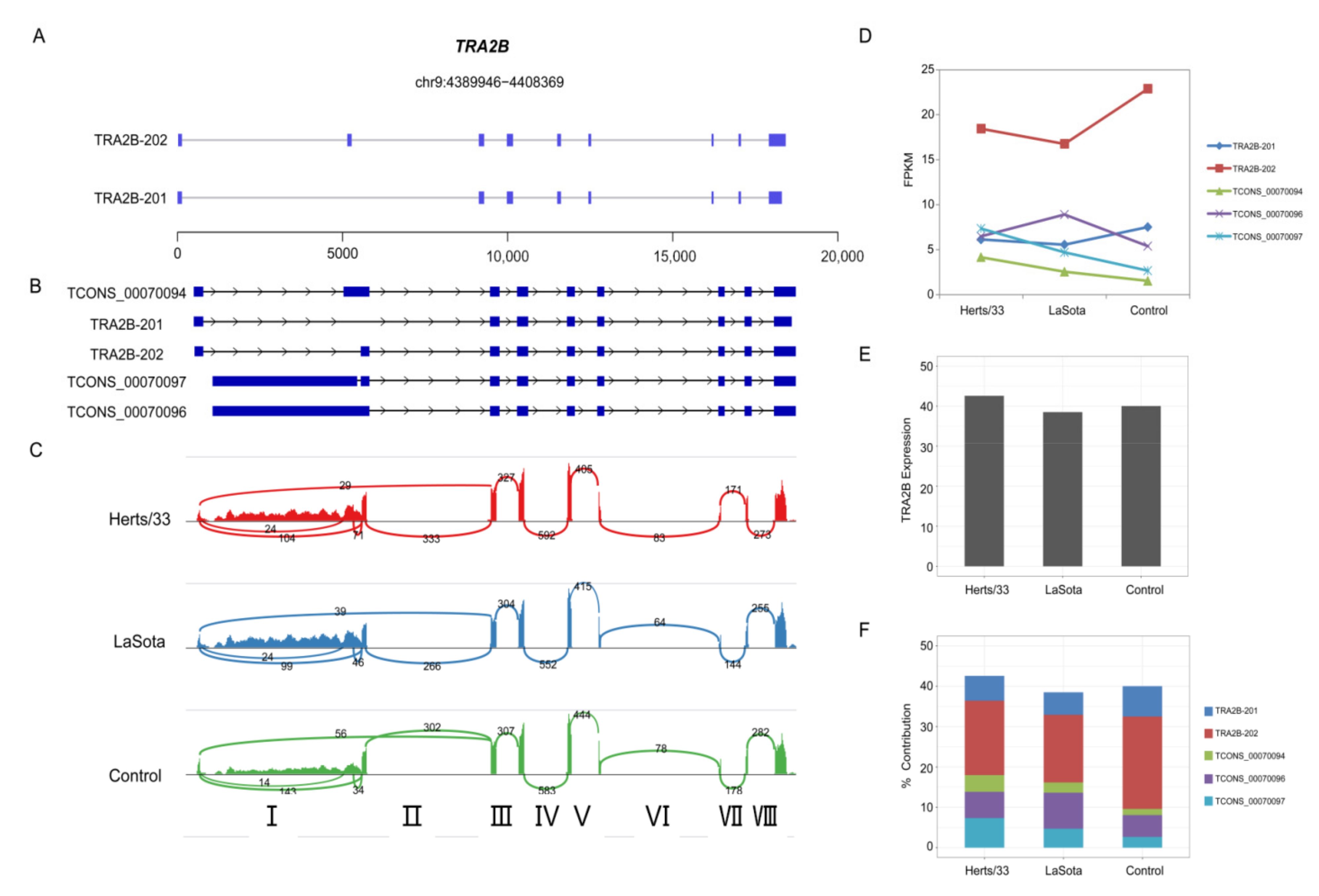
3.6. Virus-Modulated Alternative Splicing of Splicing Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Kalsotra, A.; Cooper, T.A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011, 12, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Will, C.L.; Lührmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef] [Green Version]
- Blencowe, B.J. Alternative splicing: New insights from global analyses. Cell 2006, 126, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [Green Version]
- Lindeboom, R.G.H.; Vermeulen, M.; Lehner, B.; Supek, F. The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 2019, 51, 1645–1651. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Tazi, J.; Bakkour, N.; Stamm, S. Alternative splicing and disease. Biochim. Biophys. Acta 2009, 1792, 14–26. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, M.H.; Wu, X.; Kodani, A.; Fan, J.; Doan, R.; Ozawa, M.; Ma, J.; Yoshida, N.; Reiter, J.F.; et al. Cell-Type-Specific Alternative Splicing Governs Cell Fate in the Developing Cerebral Cortex. Cell 2016, 166, 1147–1162. [Google Scholar] [CrossRef] [Green Version]
- Gabut, M.; Samavarchi-Tehrani, P.; Wang, X.; Slobodeniuc, V.; O’Hanlon, D.; Sung, H.K.; Alvarez, M.; Talukder, S.; Pan, Q.; Mazzoni, E.O.; et al. An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell 2011, 147, 132–146. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Yu, J.; Han, W.; Fan, X.; Qian, H.; Wei, H.; Tsai, Y.H.; Zhao, J.; Zhang, W.; Liu, Q.; et al. A splicing isoform of TEAD4 attenuates the Hippo-YAP signalling to inhibit tumour proliferation. Nat. Commun. 2016, 7, ncomms11840. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.I.; van de Geijn, B.; Raj, A.; Knowles, D.A.; Petti, A.A.; Golan, D.; Gilad, Y.; Pritchard, J.K. RNA splicing is a primary link between genetic variation and disease. Science 2016, 352, 600–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716. [CrossRef]
- Davey, M.G.; Tickle, C. The chicken as a model for embryonic development. Cytogenet. Genome Res. 2007, 117, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Barbosa-Morais, N.L.; Irimia, M.; Pan, Q.; Xiong, H.Y.; Gueroussov, S.; Lee, L.J.; Slobodeniuc, V.; Kutter, C.; Watt, S.; Colak, R.; et al. The evolutionary landscape of alternative splicing in vertebrate species. Science 2012, 338, 1587–1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chacko, E.; Ranganathan, S. Comprehensive splicing graph analysis of alternative splicing patterns in chicken, compared to human and mouse. BMC Genom. 2009, 10, S5. [Google Scholar] [CrossRef] [Green Version]
- Weiss, R.A. The discovery of endogenous retroviruses. Retrovirology 2006, 3, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, S.; Pathak, D.C.; Ramamurthy, N.; Maity, H.K.; Chellappa, M.M. Infectious bursal disease virus in chickens: Prevalence, impact, and management strategies. Vet. Med. 2019, 10, 85–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spackman, E. A brief introduction to the avian influenza virus. Methods Mol. Biol. 2008, 436, 1–6. [Google Scholar] [PubMed]
- Ledur, M.C.; Fairfull, R.W.; McMillan, I.; Asseltine, L. Genetic effects of aging on egg production traits in the first laying cycle of White Leghorn strains and strain crosses. Poult. Sci. 2000, 79, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Fleming, D.S.; Koltes, J.E.; Fritz-Waters, E.R.; Rothschild, M.F.; Schmidt, C.J.; Ashwell, C.M.; Persia, M.E.; Reecy, J.M.; Lamont, S.J. Single nucleotide variant discovery of highly inbred Leghorn and Fayoumi chicken breeds using pooled whole genome resequencing data reveals insights into phenotype differences. BMC Genom. 2016, 17, 812. [Google Scholar] [CrossRef] [PubMed]
- Abernathy, J.; Li, X.; Jia, X.; Chou, W.; Lamont, S.J.; Crooijmans, R.; Zhou, H. Copy number variation in Fayoumi and Leghorn chickens analyzed using array comparative genomic hybridization. Anim. Genet 2014, 45, 400–411. [Google Scholar] [CrossRef]
- Li, J.; Li, R.; Wang, Y.; Hu, X.; Zhao, Y.; Li, L.; Feng, C.; Gu, X.; Liang, F.; Lamont, S.J.; et al. Genome-wide DNA methylome variation in two genetically distinct chicken lines using MethylC-seq. BMC Genom. 2015, 16, 851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Lupiani, B.; Reddy, S.M.; Lamont, S.J.; Zhou, H. RNA-seq analysis revealed novel genes and signaling pathway associated with disease resistance to avian influenza virus infection in chickens. Poult. Sci. 2014, 93, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Hilleman, M.R. Strategies and mechanisms for host and pathogen survival in acute and persistent viral infections. Proc. Natl. Acad. Sci. USA 2004, 101, 14560–14566. [Google Scholar] [CrossRef] [Green Version]
- Harwig, A.; Landick, R.; Berkhout, B. The Battle of RNA Synthesis: Virus versus Host. Viruses 2017, 9, 309. [Google Scholar] [CrossRef] [Green Version]
- Drakesmith, H.; Prentice, A. Viral infection and iron metabolism. Nat. Rev. Microbiol. 2008, 6, 541–552. [Google Scholar] [CrossRef]
- Zhao, Z.; Xia, J.; Tastan, O.; Singh, I.; Kshirsagar, M.; Carbonell, J.; Klein-Seetharaman, J. Virus interactions with human signal transduction pathways. Int. J. Comput. Biol. Drug. Des. 2011, 4, 83–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unver, T.; Budak, H. Virus-induced gene silencing, a post transcriptional gene silencing method. Int. J. Plant Genom. 2009, 2009, 198680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abernathy, E.; Glaunsinger, B. Emerging roles for RNA degradation in viral replication and antiviral defense. Virology 2015, 479–480, 600–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Li, X.; Huo, Y.; Yu, Y.; Zhang, Q.; Chen, G.; Zhang, Y.; Fraser, N.W.; Wu, D.; Zhou, J. Cellular responses to HSV-1 infection are linked to specific types of alterations in the host transcriptome. Sci. Rep. 2016, 6, 28075. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, A.J.; Erhard, F.; L’Hernault, A.; Bonfert, T.; Schilhabel, M.; Crump, C.; Rosenstiel, P.; Efstathiou, S.; Zimmer, R.; Friedel, C.C.; et al. Widespread disruption of host transcription termination in HSV-1 infection. Nat. Commun. 2015, 6, 7126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sessions, O.M.; Tan, Y.; Goh, K.C.; Liu, Y.; Tan, P.; Rozen, S.; Ooi, E.E. Host cell transcriptome profile during wild-type and attenuated dengue virus infection. PLoS Negl. Trop. Dis. 2013, 7, e2107. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Huo, Y.; Yang, L.; Chen, G.; Luo, M.; Yang, J.; Zhou, J. ZIKV infection effects changes in gene splicing, isoform composition and lncRNA expression in human neural progenitor cells. Virol. J. 2017, 14, 217. [Google Scholar] [CrossRef] [Green Version]
- Fabozzi, G.; Oler, A.J.; Liu, P.; Chen, Y.; Mindaye, S.; Dolan, M.A.; Kenney, H.; Gucek, M.; Zhu, J.; Rabin, R.L.; et al. Strand-Specific Dual RNA Sequencing of Bronchial Epithelial Cells Infected with Influenza A/H3N2 Viruses Reveals Splicing of Gene Segment 6 and Novel Host-Virus Interactions. J. Virol. 2018, 92, e00518. [Google Scholar] [CrossRef] [Green Version]
- Niu, X.; Wang, Y.; Li, M.; Zhang, X.; Wu, Y. Transcriptome analysis of avian reovirus-mediated changes in gene expression of normal chicken fibroblast DF-1 cells. BMC Genom. 2017, 18, 911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Qiu, X.; Song, C.; Sun, Y.; Meng, C.; Liao, Y.; Tan, L.; Ding, Z.; Liu, X.; Ding, C. Deep Sequencing-Based Transcriptome Profiling Reveals Avian Interferon-Stimulated Genes and Provides Comprehensive Insight into Newcastle Disease Virus-Induced Host Responses. Viruses 2018, 10, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, X.; Wang, Y.; Tian, K.; Ye, F.; Yin, H.; Zhao, X.; Xu, H.; Huang, Y.; Liu, H.; Hsieh, J.C.; et al. Integrated host and viral transcriptome analyses reveal pathology and inflammatory response mechanisms to ALV-J injection in SPF chickens. Sci. Rep. 2017, 7, 46156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deist, M.S.; Gallardo, R.A.; Bunn, D.A.; Dekkers, J.C.M.; Zhou, H.; Lamont, S.J. Resistant and susceptible chicken lines show distinctive responses to Newcastle disease virus infection in the lung transcriptome. BMC Genom. 2017, 18, 989. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kaiser, M.G.; Deist, M.S.; Gallardo, R.A.; Bunn, D.A.; Kelly, T.R.; Dekkers, J.C.M.; Zhou, H.; Lamont, S.J. Transcriptome Analysis in Spleen Reveals Differential Regulation of Response to Newcastle Disease Virus in Two Chicken Lines. Sci. Rep. 2018, 8, 1278. [Google Scholar] [CrossRef] [Green Version]
- Deist, M.S.; Gallardo, R.A.; Bunn, D.A.; Kelly, T.R.; Dekkers, J.C.M.; Zhou, H.; Lamont, S.J. Novel Mechanisms Revealed in the Trachea Transcriptome of Resistant and Susceptible Chicken Lines following Infection with Newcastle Disease Virus. Clin. Vaccine Immunol. 2017, 24, e00027. [Google Scholar] [CrossRef]
- Saelao, P.; Wang, Y.; Gallardo, R.A.; Lamont, S.J.; Dekkers, J.M.; Kelly, T.; Zhou, H. Novel insights into the host immune response of chicken Harderian gland tissue during Newcastle disease virus infection and heat treatment. BMC Vet. Res. 2018, 14, 280. [Google Scholar] [CrossRef]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, Y.; Wang, E.T.; Silterra, J.; Schwartz, S.; Wong, B.; Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P.; Airoldi, E.M.; Burge, C.B. Quantitative visualization of alternative exon expression from RNA-seq data. Bioinformatics 2015, 31, 2400–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, Y.; Wang, E.T.; Airoldi, E.M.; Burge, C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 2010, 7, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Foissac, S.; Sammeth, M. ASTALAVISTA: Dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucl. Acids Res. 2007, 35, W297–W299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cvitkovic, I.; Jurica, M.S. Spliceosome database: A tool for tracking components of the spliceosome. Nucl. Acids Res. 2013, 41, D132–D141. [Google Scholar] [CrossRef] [Green Version]
- Dillies, M.A.; Rau, A.; Aubert, J.; Hennequet-Antier, C.; Jeanmougin, M.; Servant, N.; Keime, C.; Marot, G.; Castel, D.; Estelle, J.; et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform. 2013, 14, 671–683. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.S.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the alternative splicing landscape in plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.Y.; Lin, W.D.; Tu, S.L. Genome-Wide Analysis of Heat-Sensitive Alternative Splicing in Physcomitrella patens. Plant Physiol. 2014, 165, 826–840. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Jia, Q.; Song, Y.; Fu, H.; Wei, G.; Ni, T. Alternative Polyadenylation: Methods, Findings, and Impacts. Genom. Proteom. Bioinf. 2017, 15, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Di Giammartino, D.C.; Nishida, K.; Manley, J.L. Mechanisms and consequences of alternative polyadenylation. Mol. Cell 2011, 43, 853–866. [Google Scholar] [CrossRef] [Green Version]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet 2013, 14, 496–506. [Google Scholar] [CrossRef]
- Xia, Z.; Donehower, L.A.; Cooper, T.A.; Neilson, J.R.; Wheeler, D.A.; Wagner, E.J.; Li, W. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3’-UTR landscape across seven tumour types. Nat. Commun. 2014, 5, 5274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Wu, W.; Li, H.; Cheng, Y.; Wei, N.; Zong, J.; Feng, X.; Xie, Z.; Chen, D.; Manley, J.L.; et al. Transcriptome analysis of alternative splicing events regulated by SRSF10 reveals position-dependent splicing modulation. Nucl. Acids Res. 2014, 42, 4019–4030. [Google Scholar] [CrossRef]
- Katyal, S.; Gao, Z.; Liu, R.Z.; Godbout, R. Evolutionary conservation of alternative splicing in chicken. Cytogenet. Genome Res. 2007, 117, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Herbert, K.M.; Nag, A. A Tale of Two RNAs during Viral Infection: How Viruses Antagonize mRNAs and Small Non-Coding RNAs in The Host Cell. Viruses 2016, 8, 154. [Google Scholar] [CrossRef]
- Furusawa, Y.; Yamada, S.; Kawaoka, Y. Host Factor Nucleoporin 93 Is Involved in the Nuclear Export of Influenza Virus RNA. Front. Microbiol. 2018, 9, 1675. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, G.; Bognanni, C.; Mühlemann, O. Virus Escape and Manipulation of Cellular Nonsense-Mediated mRNA Decay. Viruses 2017, 9, 24. [Google Scholar] [CrossRef]
- Lee, K.M.; Chen, C.J.; Shih, S.R. Regulation Mechanisms of Viral IRES-Driven Translation. Trends Microbiol. 2017, 25, 546–561. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Kuo, R.L.; Lin, J.Y.; Huang, P.N.; Huang, Y.; Liu, H.; Arnold, J.J.; Chen, S.J.; Wang, R.Y.; Cameron, C.E.; et al. Cytoplasmic viral RNA-dependent RNA polymerase disrupts the intracellular splicing machinery by entering the nucleus and interfering with Prp8. PLoS Pathog. 2014, 10, e1004199. [Google Scholar] [CrossRef]
- Pabis, M.; Corsini, L.; Vincendeau, M.; Tripsianes, K.; Gibson, T.J.; Brack-Werner, R.; Sattler, M. Modulation of HIV-1 gene expression by binding of a ULM motif in the Rev protein to UHM-containing splicing factors. Nucl. Acids Res. 2019, 47, 4859–4871. [Google Scholar] [CrossRef]
- De Maio, F.A.; Risso, G.; Iglesias, N.G.; Shah, P.; Pozzi, B.; Gebhard, L.G.; Mammi, P.; Mancini, E.; Yanovsky, M.J.; Andino, R.; et al. The Dengue Virus NS5 Protein Intrudes in the Cellular Spliceosome and Modulates Splicing. PLoS Pathog. 2016, 12, e1005841. [Google Scholar] [CrossRef]
- Chang, M.X.; Zhang, J. Alternative Pre-mRNA Splicing in Mammals and Teleost Fish: A Effective Strategy for the Regulation of Immune Responses Against Pathogen Infection. Int. J Mol. Sci. 2017, 18, 1530. [Google Scholar] [CrossRef] [PubMed]
- De Arras, L.; Laws, R.; Leach, S.M.; Pontis, K.; Freedman, J.H.; Schwartz, D.A.; Alper, S. Comparative genomics RNAi screen identifies Eftud2 as a novel regulator of innate immunity. Genetics 2014, 197, 485–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, B.P.; Danhorn, T.; De Arras, L.; Flatley, B.R.; Marcus, R.A.; Farias-Hesson, E.; Leach, S.M.; Alper, S. Regulation of toll-like receptor signaling by the SF3a mRNA splicing complex. PLoS Genet. 2015, 11, e1004932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gack, M.U.; Kirchhofer, A.; Shin, Y.C.; Inn, K.S.; Liang, C.; Cui, S.; Myong, S.; Ha, T.; Hopfner, K.P.; Jung, J.U. Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc. Natl. Acad. Sci. USA 2008, 105, 16743–16748. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.H.; Fung, S.Y.; Gao, W.W.; Deng, J.J.; Cheng, Y.; Chaudhary, V.; Yuen, K.S.; Ho, T.H.; Chan, C.P.; Zhang, Y.; et al. A novel transcript isoform of STING that sequesters cGAMP and dominantly inhibits innate nucleic acid sensing. Nucl. Acids Res. 2018, 46, 4054–4071. [Google Scholar] [CrossRef]
- Deng, W.; Shi, M.; Han, M.; Zhong, J.; Li, Z.; Li, W.; Hu, Y.; Yan, L.; Wang, J.; He, Y.; et al. Negative regulation of virus-triggered IFN-beta signaling pathway by alternative splicing of TBK1. J. Biol. Chem. 2008, 283, 35590–35597. [Google Scholar] [CrossRef] [Green Version]
- Lakhdari, O.; McAllister, C.S.; Wang, M.; Minev, I.; Prince, L.S.; Eckmann, L.; Kagnoff, M.F. TLR3 signaling is downregulated by a MAVS isoform in epithelial cells. Cell Immunol. 2016, 310, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Qi, N.; Shi, Y.; Zhang, R.; Zhu, W.; Yuan, B.; Li, X.; Wang, C.; Zhang, X.; Hou, F. Multiple truncated isoforms of MAVS prevent its spontaneous aggregation in antiviral innate immune signalling. Nat. Commun. 2017, 8, 15676. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, J.; Muñoz-Moreno, R.; Kim, M.; Sakthivel, R.; Mo, W.; Shao, D.; Anantharaman, A.; García-Sastre, A.; Conrad, N.K.; et al. Influenza Virus NS1 Protein-RNA Interactome Reveals Intron Targeting. J. Virol. 2018, 92, e01634. [Google Scholar] [CrossRef] [Green Version]
- Chhangawala, S.; Rudy, G.; Mason, C.E.; Rosenfeld, J.A. The impact of read length on quantification of differentially expressed genes and splice junction detection. Genome Biol. 2015, 16, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, K.C.; Garcia-Blanco, M.A. Role of Alternative Splicing in Regulating Host Response to Viral Infection. Cells 2021, 10, 1720. [Google Scholar] [CrossRef] [PubMed]
- Frankiw, L.; Mann, M.; Li, G.; Joglekar, A.; Baltimore, D. Alternative splicing coupled with transcript degradation modulates OAS1g antiviral activity. Rna 2020, 26, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Savan, R. Alternative Splicing in Innate Antiviral Immunity. J. Interferon Cytokine Res. 2018, 38, 317–318. [Google Scholar] [CrossRef]
- Xu, L.; Yu, D.; Peng, L.; Wu, Y.; Fan, Y.; Gu, T.; Yao, Y.L.; Zhong, J.; Chen, X.; Yao, Y.G. An Alternative Splicing of Tupaia STING Modulated Anti-RNA Virus Responses by Targeting MDA5-LGP2 and IRF3. J. Immunol. 2020, 204, 3191–3204. [Google Scholar] [CrossRef] [PubMed]
- Prochasson, L.; Jalinot, P.; Mocquet, V. The Complex Relationship between HTLV-1 and Nonsense-Mediated mRNA Decay (NMD). Pathogens 2020, 9, 287. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, K.A.; Leon, K.E.; Khalid, M.M.; Tomar, S.; Jimenez-Morales, D.; Dunlap, M.; Kaye, J.A.; Shah, P.S.; Finkbeiner, S.; Krogan, N.J.; et al. The Cellular NMD Pathway Restricts Zika Virus Infection and Is Targeted by the Viral Capsid Protein. mBio 2018, 9, e02126. [Google Scholar] [CrossRef] [Green Version]
- Raxwal, V.K.; Simpson, C.G.; Gloggnitzer, J.; Entinze, J.C.; Guo, W.; Zhang, R.; Brown, J.W.S.; Riha, K. Nonsense-Mediated RNA Decay Factor UPF1 Is Critical for Posttranscriptional and Translational Gene Regulation in Arabidopsis. Plant. Cell 2020, 32, 2725–2741. [Google Scholar] [CrossRef]
- Ge, Y.; Porse, B.T. The functional consequences of intron retention: Alternative splicing coupled to NMD as a regulator of gene expression. Bioessays 2014, 36, 236–243. [Google Scholar] [CrossRef]
- Kalyna, M.; Simpson, C.G.; Syed, N.H.; Lewandowska, D.; Marquez, Y.; Kusenda, B.; Marshall, J.; Fuller, J.; Cardle, L.; McNicol, J.; et al. Alternative splicing and nonsense-mediated decay modulate expression of important regulatory genes in Arabidopsis. Nucl. Acids Res. 2012, 40, 2454–2469. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total | B1 | B2 | B3 | H1 | H2 | H3 | L1 | L2 | L3 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Mapped Reads | ||||||||||
| Unique left (%) | 299,224,346 (91.2%) | 42,058,610 (90.3%) | 42,726,173 (90.1%) | 33,598,545 (91.9%) | 24,791,424 (90.8%) | 28,441,340 (91.8%) | 29,097,409 (91.0%) | 34,714,376 (91.4%) | 31,484,844 (92.6%) | 32,311,625 (91.3%) |
| Nonunique left (%) | 28,796,379 (8.8%) | 4,309,183 (9.7%) | 4,705,478 (9.9%) | 2,954,136 (8.1%) | 2,516,090 (9.2%) | 2,546,773 (8.2%) | 2,863,948 (9.0%) | 3,285,909 (8.6%) | 2,525,643 (7.4%) | 3,089,219 (8.7%) |
| Unique right (%) | 285,044,438 (91.3%) | 39,971,739 (90.3%) | 41,096,311 (90.1%) | 30,961,117 (91.9%) | 23,667,626 (90.8%) | 27,053,760 (91.8%) | 27,702,817 (91.1%) | 33,141,424 (91.4%) | 30,353,878 (92.6%) | 31,095,766 (91.3%) |
| Nonunique left (%) | 27,808,310 (8.7%) | 4,542,921 (9.7%) | 4,495,046 (9.9%) | 2,720,323 (8.1%) | 2,397,429 (9.2%) | 2,407,222 (8.2%) | 2,716,835 (8.9%) | 3,130,883 (8.6%) | 2,429,221 (7.4%) | 2,968,430 (8.7%) |
| Overall alignment | 79.00% | 86.80% | 87.20% | 85.80% | 66.50% | 66.70% | 69.70% | 82.70% | 82.80% | 82.60% |
| Total aligned pairs | 293,168,743 | 41,610,655 | 42,736,163 | 31,659,213 | 24,439,859 | 27,508,308 | 28,424,075 | 33,979,545 | 30,741,417 | 32,069,508 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, W.; Sun, Y.; Qiu, X.; Meng, C.; Song, C.; Tan, L.; Liao, Y.; Liu, X.; Ding, C. Genome-Wide Analysis of Alternative Splicing during Host-Virus Interactions in Chicken. Viruses 2021, 13, 2409. https://doi.org/10.3390/v13122409
Liu W, Sun Y, Qiu X, Meng C, Song C, Tan L, Liao Y, Liu X, Ding C. Genome-Wide Analysis of Alternative Splicing during Host-Virus Interactions in Chicken. Viruses. 2021; 13(12):2409. https://doi.org/10.3390/v13122409
Chicago/Turabian StyleLiu, Weiwei, Yingjie Sun, Xusheng Qiu, Chunchun Meng, Cuiping Song, Lei Tan, Ying Liao, Xiufan Liu, and Chan Ding. 2021. "Genome-Wide Analysis of Alternative Splicing during Host-Virus Interactions in Chicken" Viruses 13, no. 12: 2409. https://doi.org/10.3390/v13122409