
A Novel Terrestrial Rabies Virus Lineage Occurring in South America: Origin, Diversification, and Evidence of Contact between Wild and Domestic Cycles

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Acquisition and Sequencing
2.2. Bayesian Phylogenetic Analysis of Dog-Related Variants
2.3. Phylogeographic Analysis of AgV2
2.4. Temporal Analysis
2.5. Network Analysis
2.6. Ancestral States Reconstruction
2.7. Recombination Analysis
2.8. Selection Analysis
2.9. Amino Acid Ancestral Reconstruction
3. Results
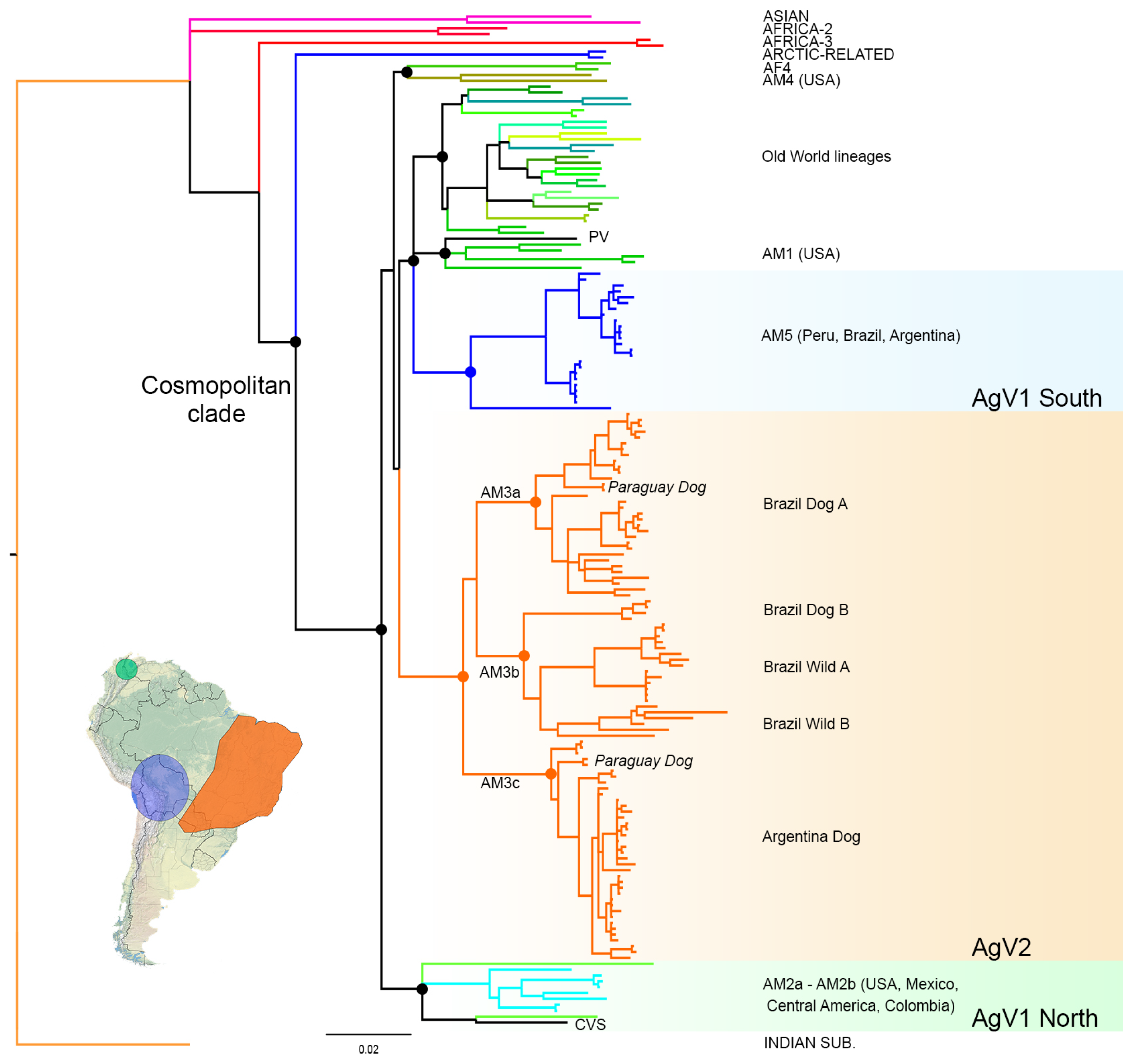
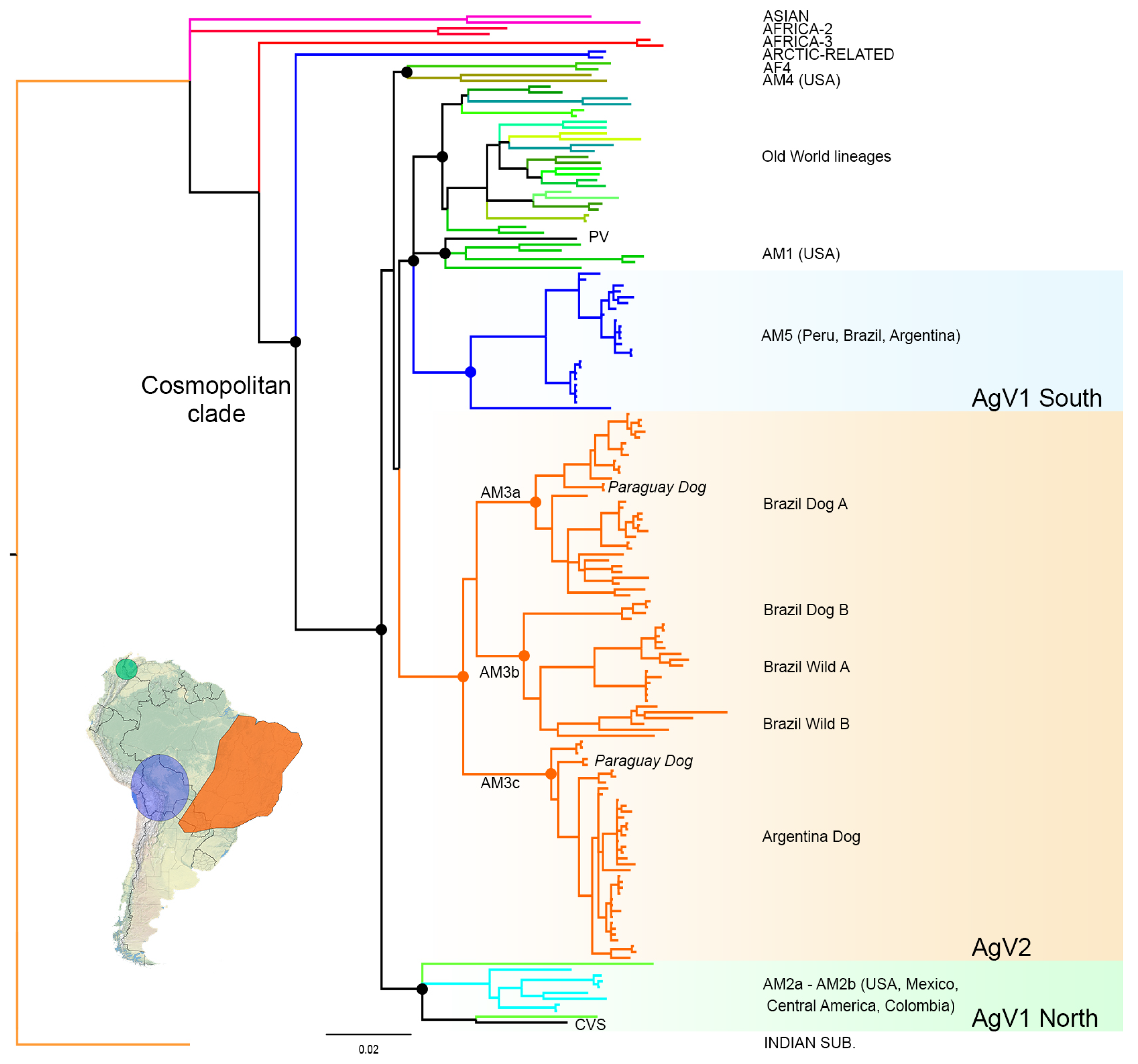
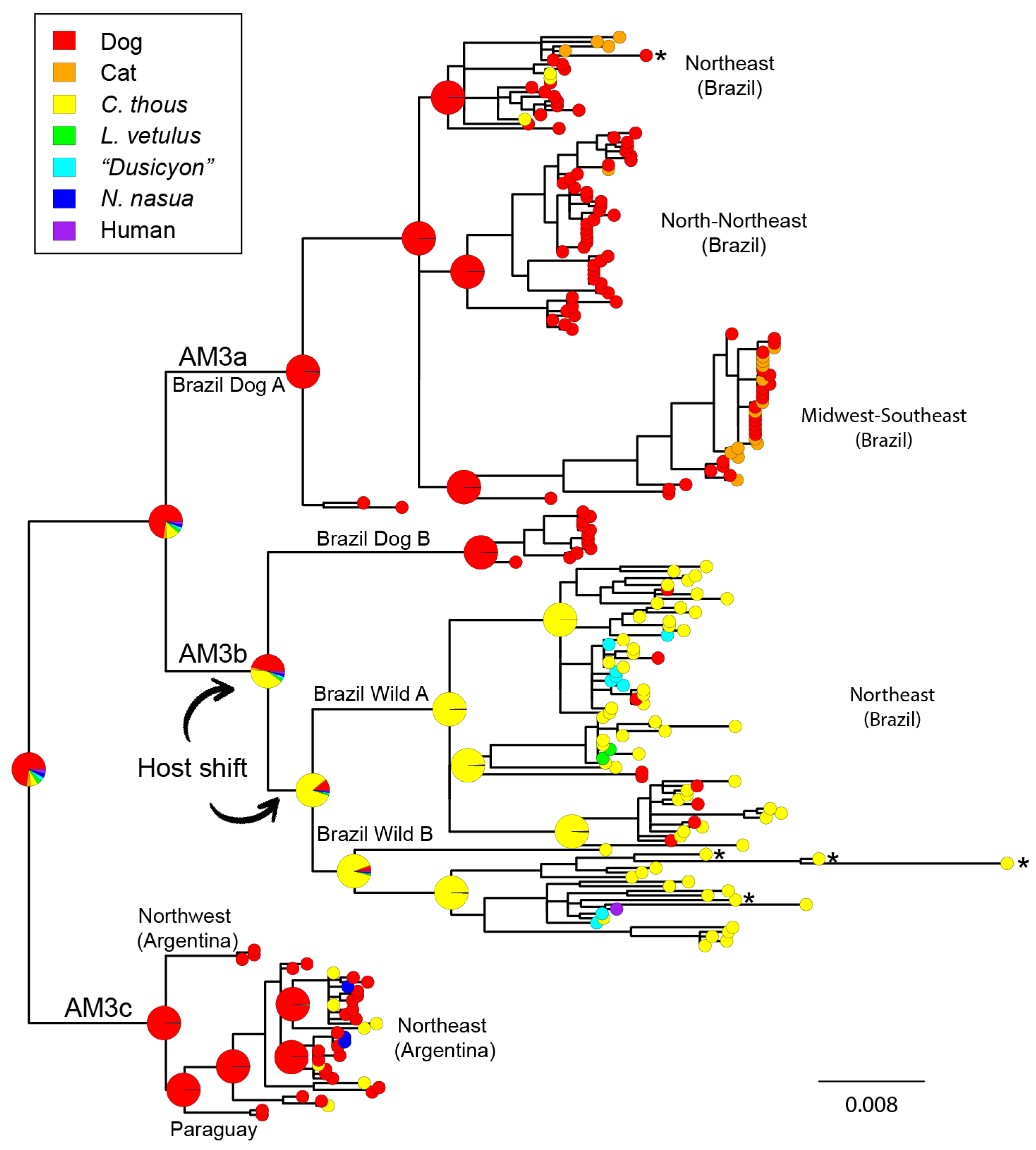
3.1. Phylogenetic Analysis and Origins of the AgV2 Lineage
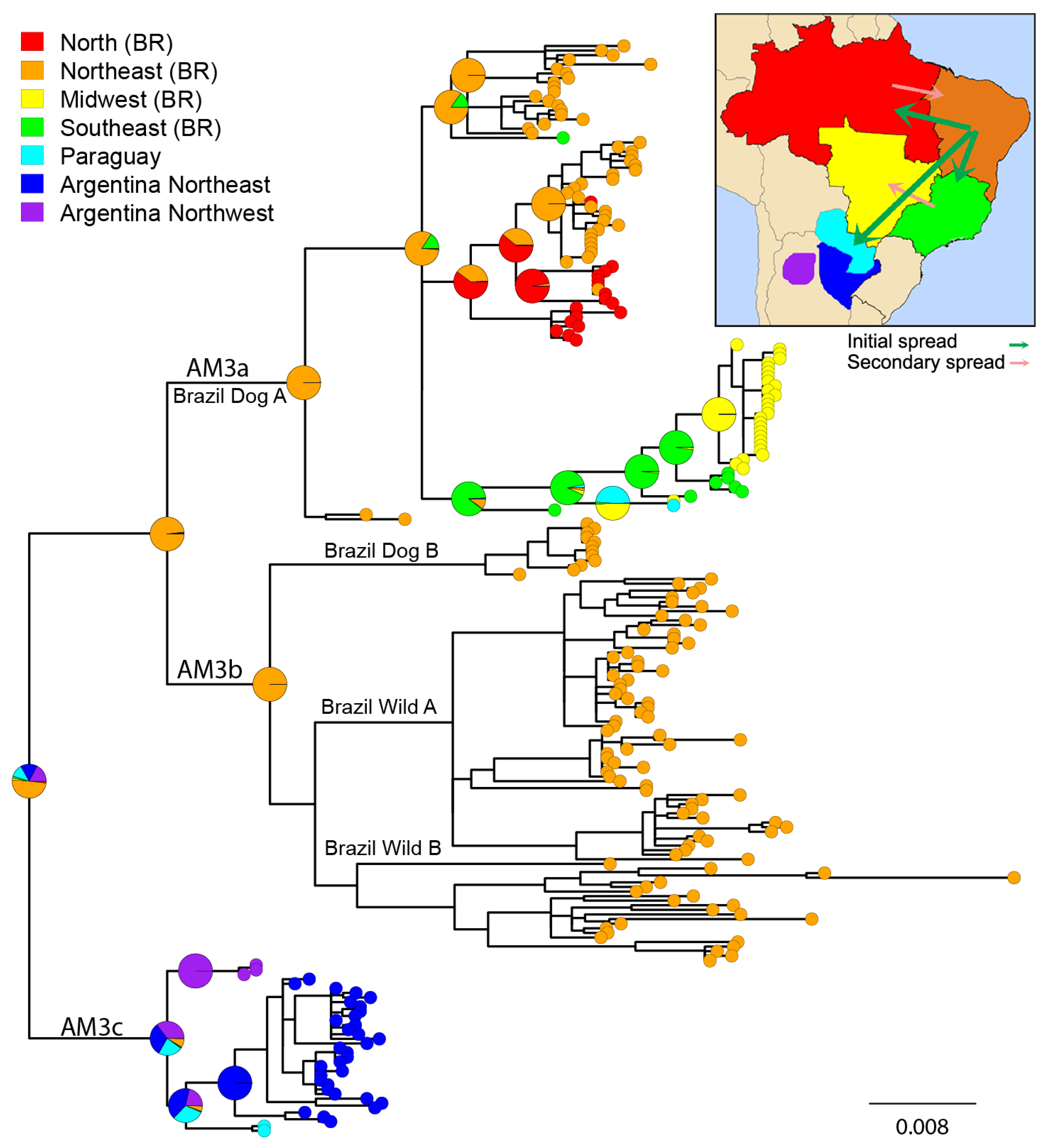
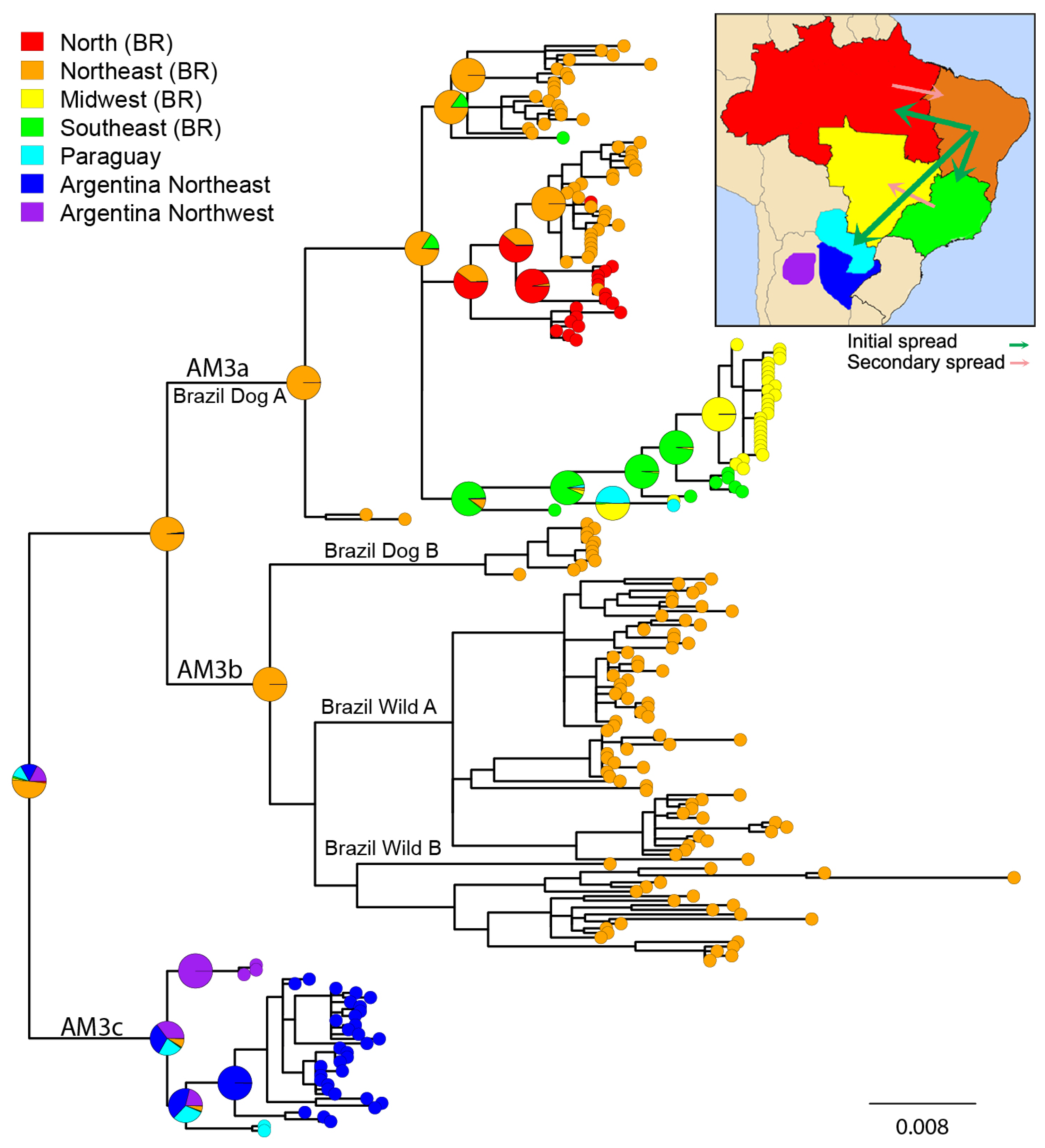
3.2. Phylogeographic Analysis
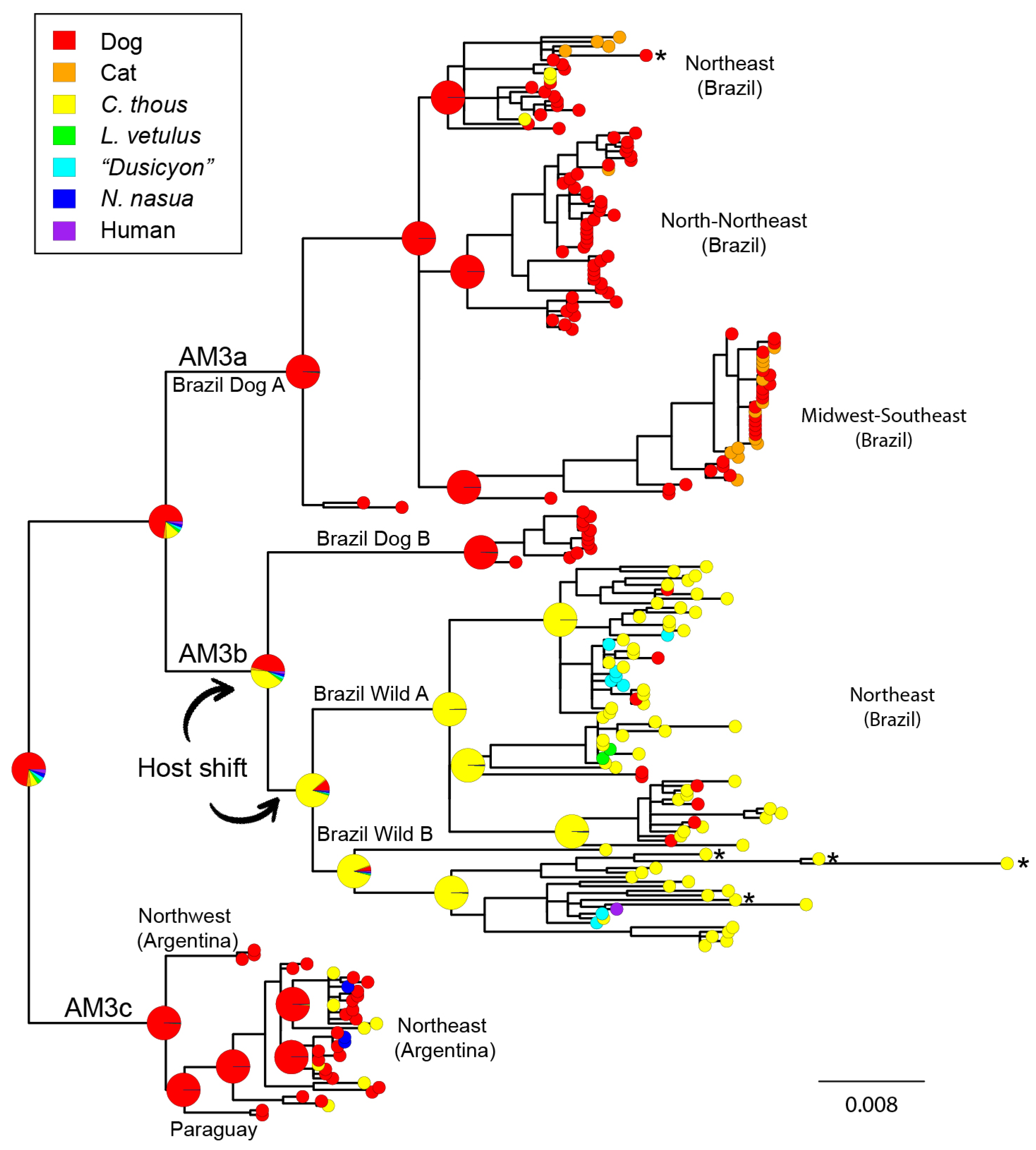
3.3. Cross-Species Transmission Analysis
3.4. Recombination Analysis
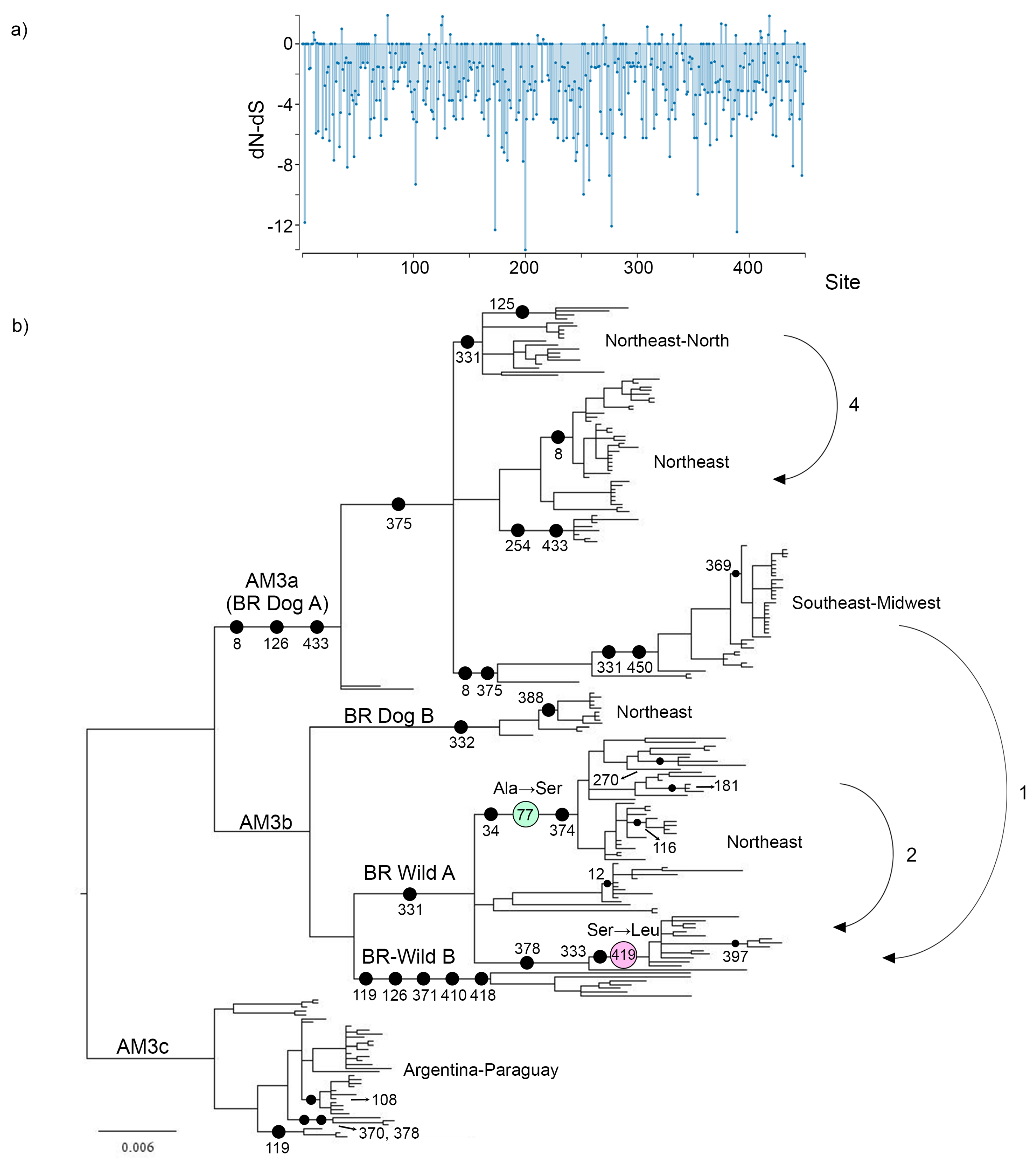
3.5. Selection Analysis
3.6. Amino Acid Replacements
4. Discussion
4.1. Origins and Diversification of AgV2
4.2. Cross-Species Transmission
4.3. Host Shifting and Adaptation
4.4. Interaction between Domestic and Wild Cycles
4.5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Badrane, H. Host Switching in. Society 2001, 75, 8096–8104. [Google Scholar] [CrossRef]
- Holmes, E.C.; Woelk, C.H.; Kassis, R.; Bourhy, H. Genetic constraints and the adaptive evolution of rabies virus in nature. Virology 2002, 292, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Rupprecht, C.E.; Turmelle, A.; Kuzmin, I.V. A perspective on lyssavirus emergence and perpetuation. Curr. Opin. Virol. 2011, 1, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Suchard, M.A.; Rambaut, A.; Streicker, D.G.; Lemey, P. Simultaneously reconstructing viral crossspecies transmission history and identifying the underlying constraints. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368. [Google Scholar] [CrossRef] [Green Version]
- Bourhy, H.; Reynes, J.M.; Dunham, E.J.; Dacheux, L.; Larrous, F.; Huong, V.T.Q.; Xu, G.; Yan, J.; Miranda, M.E.G.; Holmes, E.C. The origin and phylogeography of dog rabies virus. J. Gen. Virol. 2008, 89, 2673–2681. [Google Scholar] [CrossRef] [PubMed]
- Troupin, C.; Dacheux, L.; Tanguy, M.; Sabeta, C.; Blanc, H.; Bouchier, C.; Vignuzzi, M.; Duchene, S.; Holmes, E.C.; Bourhy, H. Large-Scale Phylogenomic Analysis Reveals the Complex Evolutionary History of Rabies Virus in Multiple Carnivore Hosts. PLoS Pathog. 2016, 12, e1006041. [Google Scholar] [CrossRef]
- Cisterna, D.; Bonaventura, R.; Caillou, S.; Pozo, O.; Andreau, M.L.; Dalla Fontana, L.; Novaro, L. Antigenic and molecular characterization of rabies virus in Argentina. Virus Res. 2005, 109, 139–147. [Google Scholar] [CrossRef]
- Páez, A.; Velasco-Villa, A.; Rey, G.; Rupprecht, C.E. Molecular epidemiology of rabies in Colombia 1994-2005 based on partial nucleoprotein gene sequences. Virus Res. 2007, 130, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Amarilla, A.C.; Pompei, J.C.; Araujo, D.B.; Vázquez, F.A.; Galeano, R.R.; Delgado, L.M.; Bogado, G.; Colman, M.; Sanabria, L.; Iamamoto, K.; et al. Re-emergence of rabies virus maintained by canid populations in Paraguay. Zoonoses Public Health 2018, 65, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Galhardo, J.A.; De Azevedo, C.S.; Remonti, B.R.; Neres Gonçalves, V.M.; Azevedo Marques, N.T.; Borges, L.O.; Ahad Das Neves, D. Canine rabies in the Brazil-Bolivia border region from 2006 to 2014. Ann. Glob. Health 2019, 85, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Carnieli, P.; Fahl, W.d.O.; Castilho, J.G.; Oliveira, R.d.N.; Macedo, C.I.; Durymanova, E.; Jorge, R.S.; Morato, R.G.; Spíndola, R.O.; Machado, L.M.; et al. Characterization of Rabies virus isolated from canids and identification of the main wild canid host in Northeastern Brazil. Virus Res. 2008, 131, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Carnieli, P.; Castilho, J.G.; Fahl, W.d.O.; Véras, N.M.C.; Carrieri, M.L.; Kotait, I. Molecular characterization of Rabies Virus isolates from dogs and crab-eating foxes in Northeastern Brazil. Virus Res. 2009, 141, 81–89. [Google Scholar] [CrossRef]
- Ito, M.; Itou, T.; Shoji, Y.; Sakai, T.; Ito, F.H.; Arai, Y.T.; Takasaki, T.; Kurane, I. Discrimination between dog-related and vampire bat-related rabies viruses in Brazil by strain-specific reverse transcriptase-polymerase chain reaction and restriction fragment length polymorphism analysis. J. Clin. Virol. 2003, 26, 317–330. [Google Scholar] [CrossRef]
- Shoji, Y.; Kobayashi, Y.; Sato, G.; Gomes, A.A.B.; Itou, T.; Ito, F.H.; Sakai, T. Genetic and phylogenetic characterization of rabies virus isolates from wildlife and livestock in Paraiba, Brazil. Acta Virol. 2006, 50, 33–37. [Google Scholar]
- Kobayashi, Y.; Inoue, N.; Sato, G.; Itou, T.; Santos, H.P.; Brito, C.J.; Gomes, A.A.; Santos, M.F.; Silva, M.V.; Mota, C.S.; et al. Phylogenetic characterization of rabies virus isolates from carnivora in Brazil. J. Vet. Med. Sci. 2007, 69, 691–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Souza, D.N.; Carnieli, P.; Macedo, C.I.; de Novaes Oliveira, R.; de Carvalho Ruthner Batista, H.B.; Rodrigues, A.C.; Pereira, P.M.C.; Achkar, S.M.; Vieira, L.F.P.; Kawai, J.G.C. Phylogenetic analysis of rabies virus isolated from canids in North and Northeast Brazil. Arch. Virol. 2016, 162, 71–77. [Google Scholar] [CrossRef]
- Carnieli, P.; Brandão, P.E.; Carrieri, M.L.; Castilho, J.G.; Macedo, C.I.; Machado, L.M.; Rangel, N.; de Carvalho, R.C.; de Carvalho, V.A.; Montebello, L.; et al. Molecular epidemiology of rabies virus strains isolated from wild canids in Northeastern Brazil. Virus Res. 2006, 120, 113–120. [Google Scholar] [CrossRef]
- Favoretto, S.R.; De Mattos, C.C.; De Mattos, C.A.; Campos, A.C.; Sacramento, D.R.; Durigon, E.L. The emergence of wildlife species as a source of human rabies infection in Brazil. Epidemiol. Infect. 2013, 141, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Carnieli, P.; de Novaes Oliveira, R.; Macedo, C.I.; Castilho, J.G. Phylogeography of rabies virus isolated from dogs in Brazil between 1985 and 2006. Arch. Virol. 2011, 156, 1007–1012. [Google Scholar] [CrossRef]
- Carnieli, P.; Ruthner Batista, H.B.; de Novaes Oliveira, R.; Castilho, J.G.; Vieira, L.F.P. Phylogeographic dispersion and diversification of rabies virus lineages associated with dogs and crab-eating foxes (Cerdocyon thous) in Brazil. Arch. Virol. 2013, 158, 2307–2313. [Google Scholar] [CrossRef]
- Diaz, A.M.; Papo, S.; Rodriguez, A.; Smith, J.S. Antigenic analysis of rabies-virus isolates from Latin America and the Caribbean. Zentralblatt Fur Veterinarmedizin. Reihe B. J. Vet. Med. Ser. B 1994, 41, 153–160. [Google Scholar] [CrossRef]
- Mochizuki, N.; Kobayashi, Y.; Sato, G.; Itou, T.; Gomes, A.A.B.; Ito, F.H.; Sakai, T. Complete genome analysis of a rabies virus isolate from Brazilian wild fox. Arch. Virol. 2009, 154, 1475–1488. [Google Scholar] [CrossRef] [PubMed]
- Palacios, G.; Quan, P.L.; Jabado, O.J.; Conlan, S.; Hirschberg, D.L.; Liu, Y.; Zhai, J.; Renwick, N.; Hui, J.; Hegyi, H.; et al. Panmicrobial Oligonucleotide Array for Diagnosis of Infectious Diseases. Emerg. Infect. Dis. 2007, 13, 73–81. [Google Scholar] [CrossRef]
- Morlan, J.D.; Qu, K.; Sinicropi, D.V. Selective depletion of rRNA enables whole transcriptome profiling of archival fixed tissue. PLoS ONE 2012, 7, e42882. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boisvert, S.; Raymond, F.; Godzaridis, É.; Laviolette, F.; Corbeil, J. Ray Meta: Scalable de novo metagenome assembly and profiling. Genome Biol. 2012, 13, R122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Ladner, J.T.; Wiley, M.R.; Mate, S.; Dudas, G.; Prieto, K.; Lovett, S.; Nagle, E.R.; Beitzel, B.; Gilbert, M.L.; Fakoli, L.; et al. Evolution and Spread of Ebola Virus in Liberia, 2014–2015. Cell Host Microbe 2015, 18, 659–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Dalponte, J.C.; Courtenay, O. Hoary fox Pseudalopex vetulus (Lund, 1842). In Canids: Foxes, Wolves, Jackals and Dogs. Status Survey and Conservation Action Plan; IUCN: Gland, Switzerland; Cambridge, UK, 2004; pp. 72–76. [Google Scholar]
- Dalponte, J.C. Lycalopex vetulus (Carnivora: Canidae). Mamm. Species 2009, 847, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Olifiers, N.; Delciellos, A.C. New record of lycalopex vetulus (Carnivora, Canidae) in Northeastern Brazil. Oecologia Aust. 2013, 17, 533–537. [Google Scholar] [CrossRef] [Green Version]
- Lemos, F.G.; Azevedo, F.C.D.; Costa, H.C.; Joares, A. Human threats to hoary and crab-eating foxes in central Brazil. Canid News 2011, 14, 1–6. [Google Scholar]
- Sievers, F.; Higgins, D.G. Clustal omega. Curr. Protoc. Bioinform. 2014, 48, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Nylander, J. Mr Modeltest 2.2. Program Distributed by the Author; Evolutionary Biology Centre, Uppsala University: Uppsala, Sweden, 2004. [Google Scholar]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A. FigTree—Tree Figure Drawing Tool. 2018. [Google Scholar]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, 1–7. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. 2021. [Google Scholar]
- Revell, L.J. Phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Rstudio, T. RStudio: Integrated Development for R. 2020. [Google Scholar]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6. [Google Scholar] [CrossRef]
- Heritage, S. MBASR: Workflow-simplified ancestral state reconstruction of discrete traits with MrBayes in the R environment. bioRxiv 2021. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Kosakovsky Pond, S.L. Less is more: An adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian AppRoximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wertheim, J.O.; Murrell, B.; Smith, M.D.; Pond, S.L.; Scheffler, K. RELAX: Detecting relaxed selection in a phylogenetic framework. Mol. Biol. Evol. 2015, 32, 820–832. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Weaver, S.; Smith, M.D.; Wertheim, J.O.; Murrell, S.; Aylward, A.; Eren, K.; Pollner, T.; Martin, D.P.; Smith, D.M.; et al. Gene-wide identification of episodic selection. Mol. Biol. Evol. 2015, 32, 1365–1371. [Google Scholar] [CrossRef] [Green Version]
- Hampson, K.; Coudeville, L.; Lembo, T.; Sambo, M.; Kieffer, A.; Attlan, M.; Barrat, J.; Blanton, J.D.; Briggs, D.J.; Cleaveland, S.; et al. Estimating the Global Burden of Endemic Canine Rabies. PLoS Neglected Trop. Dis. 2015, 9, e0003709. [Google Scholar] [CrossRef] [Green Version]
- Velasco-Villa, A.; Escobar, L.E.; Sanchez, A.; Shi, M.; Streicker, D.G.; Gallardo-Romero, N.F.; Vargas-Pino, F.; Gutierrez-Cedillo, V.; Damon, I.; Emerson, G. Successful strategies implemented towards the elimination of canine rabies in the Western Hemisphere. Antiviral Res. 2017, 143, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Seidel, H.D. Rabies: A new look at an old disease. Prog. Med. Virol. 1993, 40, 82–106. [Google Scholar]
- Bingham, J. Canine rabies ecology in Southern Africa. Emerg. Infect. Dis. 2005, 11, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, I.V.; Shi, M.; Orciari, L.A.; Yager, P.A.; Velasco-Villa, A.; Kuzmina, N.A.; Streicker, D.G.; Bergman, D.L.; Rupprecht, C.E. Molecular inferences suggest multiple host shifts of rabies viruses from bats to mesocarnivores in Arizona during 2001–2009. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef]
- Streicker, D.G.; Altizer, S.M.; Velasco-Villa, A.; Rupprecht, C.E. Variable evolutionary routes to host establishment across repeated rabies virus host shifts among bats. Proc. Natl. Acad. Sci. USA 2012, 109, 19715–19720. [Google Scholar] [CrossRef] [Green Version]
- Ding, N.Z.; Xu, D.S.; Sun, Y.Y.; He, H.B.; He, C.Q. A permanent host shift of rabies virus from Chiroptera to Carnivora associated with recombination. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kuiken, T.; Holmes, E.C.; McCauley, J.; Rimmelzwaan, G.F.; Williams, C.S.; Grenfell, B.T. Host species barriers to influenza virus infections. Science 2006, 312, 394–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borucki, M.K.; Chen-Harris, H.; Lao, V.; Vanier, G.; Wadford, D.A.; Messenger, S.; Allen, J.E. Ultra-Deep Sequencing of Intra-host Rabies Virus Populations during Cross-species Transmission. PLoS Neglected Trop. Dis. 2013, 7. [Google Scholar] [CrossRef] [Green Version]
- Gatti, A.; Bianchi, R.; Rosa, C.R.X.; Mendes, S.L. Diet of the crab-eating fox, Cerdocyon thous (Carnivora, Canidae) in Paulo Cesar Vinha State Park, Espírito Santo State, Brazil / La diète du renard crabier Cerdocyon thous (Carnivora, Canidae) au parc d’état Paulo Cesar Vinha, Espírito Santo, Brazil. Mammalia 2006, 70, 153–155. [Google Scholar] [CrossRef]
- Smith, J. Molecular Epidemiology. In Rabies; Jackson, A., Wunner, W., Eds.; Academic Press: San Diego, CA, USA, 2002; pp. 79–111. [Google Scholar]
- Scheffler, K.; Martin, D.P.; Seoighe, C. Robust inference of positive selection from recombining coding sequences. Bioinformatics 2006, 22, 2493–2499. [Google Scholar] [CrossRef] [Green Version]
- Arenas, M.; Posada, D. The effect of recombination on the reconstruction of ancestral sequences. Genetics 2010, 184, 1133–1139. [Google Scholar] [CrossRef] [Green Version]
- Lefeuvre, P.; Martin, D.P.; Harkins, G.; Lemey, P.; Gray, A.J.; Meredith, S.; Lakay, F.; Monjane, A.; Lett, J.M.; Varsani, A.; et al. The spread of tomato yellow leaf curl virus from the middle east to the world. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef] [Green Version]
- Prasanna, H.C.; Sinha, D.P.; Verma, A.; Singh, M.; Singh, B.; Rai, M.; Martin, D.P. The population genomics of begomoviruses: Global scale population structure and gene flow. Virol. J. 2010, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Deviatkin, A.A.; Lukashev, A.N. Recombination in the rabies virus and other lyssaviruses. Infect. Genet. Evol. 2018, 60, 97–102. [Google Scholar] [CrossRef]
- He, C.Q.; Meng, S.L.; Yan, H.Y.; Ding, N.Z.; He, H.B.; Yan, J.X.; Xu, G.L. Isolation and Identification of a Novel Rabies Virus Lineage in China with Natural Recombinant Nucleoprotein Gene. PLoS ONE 2012, 7, e49992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Liu, Y.; Liu, J.; Zhai, J.; Xie, Y. Evidence for inter- and intra-clade recombinations in rabies virus. Infect. Genet. Evol. 2011, 11, 1906–1912. [Google Scholar] [CrossRef] [PubMed]
- Chare, E.R.; Gould, E.A.; Holmes, E.C. Phylogenetic analysis reveals a low rate of homologous recombination in negative-sense RNA viruses. J. Gen. Virol. 2003, 84, 2691–2703. [Google Scholar] [CrossRef] [PubMed]
- Parrish, C.R.; Holmes, E.C.; Morens, D.M.; Park, E.C.; Burke, D.S.; Calisher, C.H.; Laughlin, C.A.; Saif, L.J.; Daszak, P. Cross-Species Virus Transmission and the Emergence of New Epidemic Diseases. Microbiol. Mol. Biol. Rev. 2008, 72, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Han, G.Z.; Worobey, M. Homologous recombination in negative sense RNA viruses. Viruses 2011, 3, 1358–1373. [Google Scholar] [CrossRef]
- Kew, O.; Morris-Glasgow, V.; Landaverde, M.; Burns, C.; Shaw, J.; Garib, Z.; André, J.; Blackman, E.; Freeman, C.J.; Jorba, J.; et al. Outbreak of poliomyelitis in hispaniola associated with circulating type 1 vaccine-derived poliovirus. Science 2002, 296. [Google Scholar] [CrossRef] [Green Version]
- Chi, P.B.; Chattopadhyay, S.; Lemey, P.; Sokurenko, E.V.; Minin, V.N. Synonymous and nonsynonymous distances help untangle convergent evolution and recombination. Stat. Appl. Genet. Mol. Biol. 2015, 14, 375–389. [Google Scholar] [CrossRef] [Green Version]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Species | Country | Province | Locality | Year | Variant | Genbank Accession Number |
|---|---|---|---|---|---|---|---|
| AA35 | Dog | Argentina | Tucumán | Concepción | 1995 | AgV2 | OL451878 |
| AA38 | Dog | Argentina | Tucumán | Aguilares | 1996 | AgV2 | OL451879 |
| AA39 | Dog | Argentina | Tucumán | Chicligasta | 1996 | AgV2 | OL451880 |
| 552 | C. thous | Argentina | Chaco | El Espinillo | 1997 | AgV2 | OL451910 |
| AA53 | Dog | Argentina | Formosa | Pozo del Tigre | 2000 | AgV2 | OL451881 |
| AA71 | Dog | Argentina | Formosa | Pirané | 2000 | AgV2 | OL451882 |
| AA112 | Dog | Argentina | Chaco | Ciervo Petizo | 2001 | AgV2 | OL451883 |
| AA120 | Dog | Argentina | Chaco | Pampa Almirón | 2001 | AgV2 | OL451884 |
| A119 | C. thous | Argentina | Chaco | Pampa Almirón | 2001 | AgV2 | OL451912 |
| A166 | Dog | Argentina | Formosa | Potrero Norte | 2002 | AgV2 | OL451909 |
| AA148 | Dog | Argentina | Chaco | Pirané | 2002 | AgV2 | OL451913 |
| AA474 | C. thous | Argentina | Chaco | Presidencia Roca | 2004 | AgV2 | OL451885 |
| A696 | C. thous | Argentina | Chaco | Resistencia | 2006 | AgV2 | OL451911 |
| AA774 | Dog | Argentina | Formosa | Gran Guardia | 2007 | AgV2 | OL451886 |
| AA881 | Dog | Argentina | Formosa | Palo Santo | 2007 | AgV2 | OL451914 |
| AA886 | C. thous | Argentina | Formosa | Colonia Da Prato | 2008 | AgV2 | OL451887 |
| AA888 | Dog | Argentina | Formosa | NA | 2008 | AgV2 | OL451888 |
| AA911 | Dog | Argentina | Chaco | Pampa del Indio | 2008 | AgV2 | OL451889 |
| A874 | Dog | Argentina | Chaco | Capitán Solari | 2008 | AgV2 | OL451906 |
| AA929 | N. nasua | Argentina | Chaco | Laguna Limpia | 2009 | AgV2 | OL451890 |
| AA957 | Dog | Argentina | Chaco | Colonia Elisa | 2009 | AgV2 | OL451891 |
| AA959 | Dog | Argentina | Chaco | Colonia Elisa | 2009 | AgV2 | OL451892 |
| AA966 | N. nasua | Argentina | Formosa | Palo Santo | 2009 | AgV2 | OL451893 |
| AB124 | Dog | Argentina | Formosa | Comandante Fontana | 2012 | AgV2 | OL451894 |
| AB160 | Dog | Argentina | Formosa | Ibarreta | 2013 | AgV2 | OL451895 |
| AB193 | C. thous | Argentina | Chaco | La Verde | 2013 | AgV2 | OL451896 |
| AB353 | Dog | Argentina | Formosa | Ibarreta | 2013 | AgV2 | OL451897 |
| AB356 | C. thous | Argentina | Formosa | El Colorado | 2013 | AgV2 | OL451898 |
| AB357 | Dog | Argentina | Formosa | Laishi | 2013 | AgV2 | OL451899 |
| AB361 | N. nasua | Argentina | Formosa | Mariano Boedo | 2013 | AgV2 | OL451900 |
| AB362 | Dog | Argentina | Formosa | Comandante Fontana | 2013 | AgV2 | OL451901 |
| AB118 | Dog | Argentina | Formosa | S/D | 2015 | AgV2 | OL451902 |
| AB122 | Dog | Argentina | Chaco | Puerto Tirol | 2015 | AgV2 | OL451903 |
| 739 | Dog | Argentina | Chaco | Pampa Almirón | 2016 | AgV2 | OL451904 |
| 682 | Dog | Argentina | Chaco | Laguna Blanca | 2017 | AgV2 | OL451905 |
| 257 | Dog | Brazil | Mato Grosso do Sul | Pantanais | 1989 | AgV2 | OL451915 |
| 352 | Dog | Paraguay | Itapúa | Encarnación | 1991 | AgV2 | OL451908 |
| 353 | Dog | Paraguay | Itapúa | Encarnación | 1991 | AgV2 | OL451916 |
| 369 | Dog | Paraguay | Itapúa | Encarnación | 1992 | AgV2 | OL451907 |
| AA42 | Dog | Argentina | Salta | Orán | 1999 | AgV1 | OL451918 |
| AA47 | Cat | Argentina | Salta | Orán | 1999 | AgV1 | OL451919 |
| AA50 | Cat | Argentina | Salta | Orán | 1999 | AgV1 | OL451920 |
| AA21 | Dog | Argentina | Salta | Hipólito Yrigoyen | 2000 | AgV1 | OL451917 |
| AA126 | Dog | Argentina | Salta | NA | 2002 | AgV1 | OL451921 |
| AA178 | Dog | Argentina | Salta | Aguaray | 2002 | AgV1 | OL451922 |
| AA180 | Monkey | Argentina | Salta | Tartagal | 2002 | AgV1 | OL451923 |
| AA183 | Dog | Argentina | Salta | Tartagal | 2002 | AgV1 | OL451924 |
| M863 | Dog | Argentina | Jujuy | Palpalá | 2003 | AgV1 | OL451929 |
| M870 | Dog | Argentina | Jujuy | El Carmen | 2003 | AgV1 | OL451930 |
| M1009 | Dog | Argentina | Jujuy | S.S. de Jujuy | 2003 | AgV1 | OL451932 |
| M632 | Dog | Argentina | Jujuy | S.S. de Jujuy | 2005 | AgV1 | OL451926 |
| M654 | Dog | Argentina | Jujuy | S.S. de Jujuy | 2005 | AgV1 | OL451928 |
| M639 | Dog | Argentina | Jujuy | S.S. de Jujuy | 2006 | AgV1 | OL451927 |
| M876 | Dog | Argentina | Jujuy | S.S. de Jujuy | 2006 | AgV1 | OL451931 |
| M297 | Dog | Argentina | Salta | Orán | 2015 | AgV1 | OL451925 |
| (a) Site-Based Methods | |||||
|---|---|---|---|---|---|
| Length (Codons) | Type of Selection | FEL | SLAC | FUBAR | MEME |
| 450 | Positive | 2 | 0 | 0 | 3 |
| Negative | 193 | 162 | 311 | - | |
| Codons | 77,339 | - | - | 158, 419, 443 | |
| (b) Branch-Based Methods | |||||
| Method | Test Group | Reference Group | Result | ||
| aBSREL | All AgV2 | - | No EDS (p ≤ 0.05) | ||
| aBSREL | AM3b MRCA + Brazilian Wild A + B MRCA | - | No EDS (p ≤ 0.05) | ||
| aBSREL | AM3b MRCA + Brazilian Wild A + B | - | No EDS (p = 0.0057 ≤ 0.05) | ||
| Relax | AM3b MRCA + Brazilian Wild A + B | All remaining branches | Selection Relaxation not significant (K = 0.94, p = 0.360, LR = 0.84) | ||
| BUSTED | AM3b MRCA + Brazilian Wild A + B | All remaining branches | Evidence of gene-wide EDS (LRT, p-value = 0.022 ≤ 0.05) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caraballo, D.A.; Lema, C.; Novaro, L.; Gury-Dohmen, F.; Russo, S.; Beltrán, F.J.; Palacios, G.; Cisterna, D.M. A Novel Terrestrial Rabies Virus Lineage Occurring in South America: Origin, Diversification, and Evidence of Contact between Wild and Domestic Cycles. Viruses 2021, 13, 2484. https://doi.org/10.3390/v13122484
Caraballo DA, Lema C, Novaro L, Gury-Dohmen F, Russo S, Beltrán FJ, Palacios G, Cisterna DM. A Novel Terrestrial Rabies Virus Lineage Occurring in South America: Origin, Diversification, and Evidence of Contact between Wild and Domestic Cycles. Viruses. 2021; 13(12):2484. https://doi.org/10.3390/v13122484
Chicago/Turabian StyleCaraballo, Diego A., Cristina Lema, Laura Novaro, Federico Gury-Dohmen, Susana Russo, Fernando J. Beltrán, Gustavo Palacios, and Daniel M. Cisterna. 2021. "A Novel Terrestrial Rabies Virus Lineage Occurring in South America: Origin, Diversification, and Evidence of Contact between Wild and Domestic Cycles" Viruses 13, no. 12: 2484. https://doi.org/10.3390/v13122484
APA StyleCaraballo, D. A., Lema, C., Novaro, L., Gury-Dohmen, F., Russo, S., Beltrán, F. J., Palacios, G., & Cisterna, D. M. (2021). A Novel Terrestrial Rabies Virus Lineage Occurring in South America: Origin, Diversification, and Evidence of Contact between Wild and Domestic Cycles. Viruses, 13(12), 2484. https://doi.org/10.3390/v13122484