
Adenovirus Biology, Recombinant Adenovirus, and Adenovirus Usage in Gene Therapy

Abstract
:1. Introduction
2. Biology of Wild-Type Adenoviruses and the Principle behind Recombinant Adenoviruses
2.1. Biology of Adenoviruses
2.2. Principle of Recombinant Adenoviruses
2.3. CRA Construction
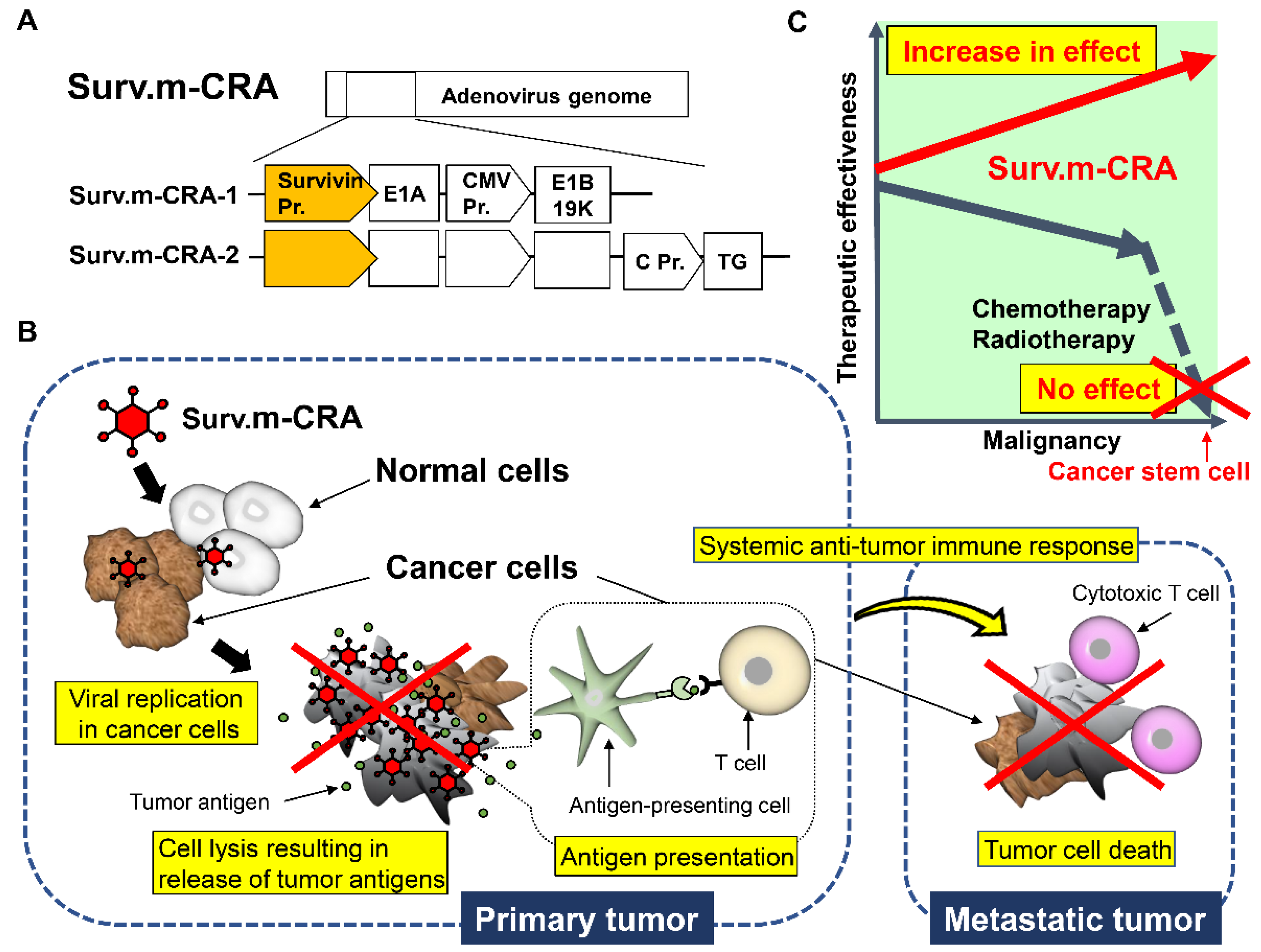
2.4. CRAs That Can Specifically Target Tumors with Multiple Factors (m-CRA)
3. Application of Recombinant Adenoviruses
3.1. Replication-Defective Adenovirus-Based Cancer Gene Therapy and Vaccines
3.2. History and Current Status of CRA-Based Oncolytic Virotherapy and Immunotherapy
3.3. Survivin Responsive m-CRA
3.4. Application of Recombinant Adenoviruses to Pluripotent Stem Cell-Based Regenerative Medicine
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- FDA. Approved Cellular and Gene Therapy Products. 2020. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products (accessed on 12 December 2021).
- Davison, A.J.; Benkő, M.; Harrach, B. Genetic content and evolution of adenoviruses. J. Gen. Virol. 2003, 84, 2895–2908. [Google Scholar] [CrossRef]
- Saha, B.; Parks, R.J. Recent Advances in Novel Antiviral Therapies against Human Adenovirus. Microorganisms 2020, 8, 1284. [Google Scholar] [CrossRef] [PubMed]
- Bányai, K.; Martella, V.; Meleg, E.; Kisfali, P.; Péterfi, Z.; Benkö, M.; Melegh, B.; Szucs, G. Searching for HAdV-52, the putative gastroenteritis-associated human adenovirus serotype in Southern Hungary. New Microbiol. 2009, 32, 185–188. [Google Scholar]
- Yang, C.; Zhu, C.; Qian, Y.; Deng, J.; Zhang, B.; Zhu, R.; Wang, F.; Sun, Y.; Chen, D.; Guo, Q.; et al. Application of Human Adenovirus Genotyping by Phylogenetic Analysis in an Outbreak to Identify Nosocomial Infection. Virol. Sin. 2020, 36, 393–401. [Google Scholar] [CrossRef]
- Yang, Z.R.; Wang, H.F.; Zhao, J.; Peng, Y.Y.; Wang, J.; Guinn, B.; Huang, L.Q. Recent developments in the use of adenoviruses and immunotoxins in cancer gene therapy. Cancer Gene Ther. 2007, 14, 599–615. [Google Scholar] [CrossRef] [Green Version]
- Saha, B.; Wong, C.M.; Parks, R.J. The Adenovirus Genome Contributes to the Structural Stability of the Virion. Viruses 2014, 6, 3563–3583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, W.P.; Huebner, R.J.; Gilmore, L.K.; Parrott, R.H.; Ward, T.G. Isolation of a Cytopathogenic Agent from Human Adenoids Undergoing Spontaneous Degeneration in Tissue Culture. Exp. Biol. Med. 1953, 84, 570–573. [Google Scholar] [CrossRef]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a Common Receptor for Coxsackie B Viruses and Adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef]
- Tomko, R.P.; Xu, R.; Philipson, L. HCAR and MCAR: The human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc. Natl. Acad. Sci. USA 1997, 94, 3352–3356. [Google Scholar] [CrossRef] [Green Version]
- Gaden, F.; Franqueville, L.; Magnusson, M.; Hong, S.S.; Merten, M.D.; Lindholm, L.; Boulanger, P. Gene Transduction and Cell Entry Pathway of Fiber-Modified Adenovirus Type 5 Vectors Carrying Novel Endocytic Peptide Ligands Selected on Human Tracheal Glandular Cells. J. Virol. 2004, 78, 7227–7247. [Google Scholar] [CrossRef] [Green Version]
- Wiethoff, C.M.; Wodrich, H.; Gerace, L.; Nemerow, G.R. Adenovirus Protein VI Mediates Membrane Disruption following Capsid Disassembly. J. Virol. 2005, 79, 1992–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremner, K.H.; Scherer, J.; Yi, J.; Vershinin, M.; Gross, S.P.; Vallee, R.B. Adenovirus Transport via Direct Interaction of Cytoplasmic Dynein with the Viral Capsid Hexon Subunit. Cell Host Microbe 2009, 6, 523–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiethoff, C.M.; Nemerow, G.R. Adenovirus membrane penetration: Tickling the tail of a sleeping dragon. Virology 2015, 479-480, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charman, M.; Herrmann, C.; Weitzman, M.D. Viral and cellular interactions during adenovirus DNA replication. FEBS Lett. 2019, 593, 3531–3550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikoma, T.; Takahashi, T.; Nagano, S.; Li, Y.-M.; Ohno, Y.; Ando, K.; Fujiwara, T.; Fujiwara, H.; Kosai, K.-I. A Definitive Role of RhoC in Metastasis of Orthotopic Lung Cancer in Mice. Clin. Cancer Res. 2004, 10, 1192–1200. [Google Scholar] [CrossRef] [Green Version]
- Kosai, K.I.; Finegold, M.J.; Thi-Huynh, B.T.; Tewson, M.; Ou, C.N.; Bowles, N.; Woo, S.L.; Schwall, R.H.; Darlington, G.J. Ret-rovirus-mediated in vivo gene transfer in the replicating liver using recombinant hepatocyte growth factor without liver injury or partial hepatectomy. Hum. Gene Ther. 1998, 9, 1293–1301. [Google Scholar] [CrossRef]
- Zhong, L.; Granelli-Piperno, A.; Choi, Y.; Steinman, R.M. Recombinant adenovirus is an efficient and non-perturbing genetic vector for human dendritic cells. Eur. J. Immunol. 1999, 29, 964–972. [Google Scholar] [CrossRef]
- Humphries, E.H.; Glover, C.; Reichmann, M.E. Rous sarcoma virus infection of synchronized cells establishes provirus integration during S-phase DNA synthesis prior to cellular division. Proc. Natl. Acad. Sci. USA 1981, 78, 2601–2605. [Google Scholar] [CrossRef] [Green Version]
- Gingeras, T.; Sciaky, D.; Gelinas, R.E.; Bing-Dong, J.; Yen, C.E.; Kelly, M.M.; Bullock, P.A.; Parsons, B.; O’Neill, K.E.; Roberts, R. Nucleotide sequences from the adenovirus-2 genome. J. Biol. Chem. 1982, 257, 13475–13491. [Google Scholar] [CrossRef]
- Abudoureyimu, M.; Lai, Y.; Tian, C.; Wang, T.; Wang, R.; Chu, X. Oncolytic Adenovirus—A nova for gene-targeted oncolytic viral therapy in HCC. Front. Oncol. 2019, 9, 1182. [Google Scholar] [CrossRef]
- Rao, L.; Debbas, M.; Sabbatini, P.; Hockenbery, D.; Korsmeyer, S.; White, E. The adenovirus E1A proteins induce apoptosis, which is inhibited by the E1B 19-kDa and Bcl-2 proteins. Proc. Natl. Acad. Sci. USA 1992, 89, 7742–7746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, W.C. Adenoviruses: Update on structure and function. J. Gen. Virol. 2009, 90, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kleinberger, T. En Guard! The Interactions between Adenoviruses and the DNA Damage Response. Viruses 2020, 12, 996. [Google Scholar] [CrossRef]
- Li, S.; Ou, M.; Wang, G.; Tang, L. Application of conditionally replicating adenoviruses in tumor early diagnosis technology, gene-radiation therapy and chemotherapy. Appl. Microbiol. Biotechnol. 2016, 100, 8325–8335. [Google Scholar] [CrossRef]
- Peter, M.; Kühnel, F. Oncolytic Adenovirus in Cancer Immunotherapy. Cancers 2020, 12, 3354. [Google Scholar] [CrossRef] [PubMed]
- Bett, A.J.; Prevec, L.; Graham, F.L. Packaging capacity and stability of human adenovirus type 5 vectors. J. Virol. 1993, 67, 5911–5921. [Google Scholar] [CrossRef] [Green Version]
- Bett, A.J.; Haddara, W.; Prevec, L.; Graham, F.L. An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc. Natl. Acad. Sci. USA 1994, 91, 8802–8806. [Google Scholar] [CrossRef] [Green Version]
- Khai, N.C.; Takahashi, T.; Ushikoshi, H.; Nagano, S.; Yuge, K.; Esaki, M.; Kawai, T.; Goto, K.; Murofushi, Y.; Fujiwara, T.; et al. In vivo hepatic HB-EGF gene transduction inhibits Fas-induced liver injury and induces liver regeneration in mice: A comparative study to HGF. J. Hepatol. 2006, 44, 1046–1054. [Google Scholar] [CrossRef]
- Matsuda, E.; Obama, Y.; Kosai, K.-I. Safe and low-dose but therapeutically effective adenovirus-mediated hepatocyte growth factor gene therapy for type 1 diabetes in mice. Life Sci. 2021, 268, 119014. [Google Scholar] [CrossRef] [PubMed]
- Ushikoshi, H.; Takahashi, T.; Chen, X.; Khai, N.C.; Esaki, M.; Goto, K.; Takemura, G.; Maruyama, R.; Minatoguchi, S.; Fujiwara, T.; et al. Local overexpression of HB-EGF exacerbates remodeling following myocardial infarction by activating noncardiomyocytes. Lab. Investig. 2005, 85, 862–873. [Google Scholar] [CrossRef] [Green Version]
- Yuge, K.; Takahashi, T.; Khai, N.C.; Goto, K.; Fujiwara, T.; Fujiwara, H.; Kosai, K.-I. Intramuscular injection of adenoviral hepatocyte growth factor at a distal site ameliorates dextran sodium sulfate-induced colitis in mice. Int. J. Mol. Med. 2014, 33, 1064–1074. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Khai, N.C.; Wang, Y.; Irie, R.; Takamatsu, H.; Matsufuji, H.; Kosai, K.-I. Heparin-binding epidermal growth factor-like growth factor and hepatocyte growth factor inhibit cholestatic liver injury in mice through different mechanisms. Int. J. Mol. Med. 2016, 38, 1673–1682. [Google Scholar] [CrossRef] [Green Version]
- Bressy, C.; Hastie, E.; Grdzelishvili, V.Z. Combining Oncolytic Virotherapy with p53 Tumor Suppressor Gene Therapy. Mol. Ther. Oncolytics 2017, 5, 20–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.H.; Wang, Y.; Kosai, K.; Finegold, M.J.; Rich, S.S.; Woo, S.L. Combination gene therapy for liver metastasis of colon carcinoma in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 2577–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.H.; Kosai, K.; Xu, B.; Pham-Nguyen, K.; Contant, C.; Finegold, M.J.; Woo, S.L. Combination suicide and cytokine gene therapy for hepatic metastases of colon carcinoma: Sustained antitumor immunity prolongs animal survival. Cancer Res. 1996, 56, 3758–3762. [Google Scholar] [PubMed]
- Kwong, Y.-L.; Chen, S.-H.; Kosai, K.; Finegold, M.; Woo, S.L. Combination therapy with suicide and cytokine genes for hepatic metastases of lung cancer. Chest 1997, 112, 1332–1337. [Google Scholar] [CrossRef]
- Block, A.; Chen, S.H.; Kosai, K.; Finegold, M.; Woo, S.L. Adenoviral-mediated herpes simplex virus thymidine kinase gene transfer: Regression of hepatic metastasis of pancreatic tumors. Pancreas 1997, 15, 25–34. [Google Scholar] [CrossRef]
- Fukunaga, M.; Takamori, S.; Hayashi, A.; Shirouzu, K.; Kosai, K.-I. Adenoviral herpes simplex virus thymidine kinase gene therapy in an orthotopic lung cancer model. Ann. Thorac. Surg. 2002, 73, 1740–1746. [Google Scholar] [CrossRef]
- Kwong, Y.L.; Chen, S.H.; Kosai, K.; Finegold, M.J.; Woo, S.L. Adenoviral-mediated suicide gene therapy for hepatic metastases of breast cancer. Cancer Gene Ther. 1996, 3, 339–344. [Google Scholar] [PubMed]
- Terazaki, Y.; Yano, S.; Nagano, S.; Fukunaga, M.; Guo, Z.S.; Yuge, K.; Komiya, S.; Shirouzu, K.; Kosai, K. An optimal therapeutic expression level is crucial for suicide gene therapy for hepatic metastatic cancer in mice. Hepatology 2003, 37, 155–163. [Google Scholar] [CrossRef]
- Huang, H.-P.; Chen, S.H.; Kosai, K.; Finegold, M.J.; Woo, S.L. Gene therapy for hepatocellular carcinoma: Long-term remission of primary and metastatic tumors in mice by interleukin-2 gene therapy in vivo. Gene Ther. 1996, 3, 980–987. [Google Scholar]
- Nagano, S.; Yuge, K.; Fukunaga, M.; Terazaki, Y.; Fujiwara, H.; Komiya, S.; Kosai, K. Gene therapy eradicating distant dissemi-nated micro-metastases by optimal cytokine expression in the primary lesion only: Novel concepts for successful cytokine gene therapy. Int. J. Oncol. 2004, 24, 549–558. [Google Scholar] [PubMed]
- O’Malley, B.W., Jr.; Sewell, D.A.; Li, D.; Kosai, K.-I.; Chen, S.-H.; Woo, S.L.; Duan, L. The Role of Interleukin-2 in Combination Adenovirus Gene Therapy for Head and Neck Cancer. Mol. Endocrinol. 1997, 11, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Caruso, M.; Pham-Nguyen, K.; Kwong, Y.L.; Xu, B.; Kosai, K.I.; Finegold, M.; Woo, S.L.; Chen, S.H. Adenovirus-mediated interleukin-12 gene therapy for metastatic colon carcinoma. Proc. Natl. Acad. Sci. USA 1996, 93, 11302–11306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.-Y.; Huang, Q.; Kung, H.-F. Cytokine and immuno-gene therapy for solid tumors. Cell. Mol. Immunol. 2005, 2, 81–91. [Google Scholar]
- Raper, S.E.; Chirmule, N.; Lee, F.; Wivel, N.A.; Bagg, A.; Gao, G.-P.; Wilson, J.; Batshaw, M.L. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003, 80, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Kreppel, F.; Hagedorn, C. Capsid and Genome Modification Strategies to Reduce the Immunogenicity of Adenoviral Vectors. Int. J. Mol. Sci. 2021, 22, 2417. [Google Scholar] [CrossRef]
- Parks, R.J.; Graham, F.L. A helper-dependent system for adenovirus vector production helps define a lower limit for efficient DNA packaging. J. Virol. 1997, 71, 3293–3298. [Google Scholar] [CrossRef] [Green Version]
- Schmid, M.; Ernst, P.; Honegger, A.; Suomalainen, M.; Zimmermann, M.; Braun, L.; Stauffer, S.; Thom, C.; Dreier, B.; Eibauer, M.; et al. Adenoviral vector with shield and adapter increases tumor specificity and escapes liver and immune control. Nat. Commun. 2018, 9, 450. [Google Scholar] [CrossRef]
- Chen, C.Y.; May, S.M.; Barry, M.A. Targeting Adenoviruses with Factor X–Single-Chain Antibody Fusion Proteins. Hum. Gene Ther. 2010, 21, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Ling, Y.; Zhong, J.; Luo, J. Safety and effectiveness of SARS-CoV-2 vaccines: A systematic review and meta-analysis. J. Med. Virol. 2021, 93, 6486–6495. [Google Scholar] [CrossRef] [PubMed]
- Goradel, N.H.; Mohajel, N.; Malekshahi, Z.V.; Jahangiri, S.; Najafi, M.; Farhood, B.; Mortezaee, K.; Negahdari, B.; Arashkia, A. Oncolytic adenovirus: A tool for cancer therapy in combination with other therapeutic approaches. J. Cell. Physiol. 2019, 234, 8636–8646. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Alfred Yung, W.K.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Fueyo, J.; Gomez-Manzano, C.; Alemany, R.; Lee, P.S.; McDonnell, T.J.; Mitliagka, P.; Shi, Y.-X.; Levin, V.A.; Yung, W.K.A.; Kyritsis, A.P. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 2000, 19, 2–12. [Google Scholar] [CrossRef] [Green Version]
- Heise, C.; Hermiston, T.; Johnson, L.; Brooks, G.A.; Sampson-Johannes, A.; Williams, A.; Hawkins, L.; Kirn, D.H. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat. Med. 2000, 6, 1134–1139. [Google Scholar] [CrossRef]
- Garcia-Moure, M.; Martinez-Vélez, N.; Patiño-García, A.; Alonso, M.M. Oncolytic adenoviruses as a therapeutic approach for osteosarcoma: A new hope. J. Bone Oncol. 2016, 9, 41–47. [Google Scholar] [CrossRef]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An Adenovirus Mutant That Replicates Selectively in p53- Deficient Human Tumor Cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef]
- Zhang, K.-J.; Zhang, J.; Wu, Y.-M.; Qian, J.; Liu, X.-J.; Yan, L.-C.; Zhou, X.-M.; Xiao, R.-J.; Wang, Y.-G.; Cao, X.; et al. Complete eradication of hepatomas using an oncolytic adenovirus containing AFP promoter controlling E1A and an E1B deletion to drive IL-24 expression. Cancer Gene Ther. 2012, 19, 619–629. [Google Scholar] [CrossRef]
- Irving, J.; Wang, Z.; Powell, S.; O’Sullivan, C.; Mok, M.; Murphy, B.; Cardoza, L.; Lebkowski, J.S.; Majumdar, A.S. Conditionally replicative adenovirus driven by the human telomerase promoter provides broad-spectrum antitumor activity without liver toxicity. Cancer Gene Ther. 2004, 11, 174–185. [Google Scholar] [CrossRef] [Green Version]
- Wirth, T.; Zender, L.; Schulte, B.; Mundt, B.; Plentz, R.; Rudolph, K.L.; Manns, M.; Kubicka, S.; Kühnel, F. A telomerase-dependent conditionally replicating adenovirus for selective treatment of cancer. Cancer Res. 2003, 63, 3181–3188. [Google Scholar]
- Harada, J.N.; Berk, A.J. p53-Independent and -Dependent Requirements for E1B-55K in Adenovirus Type 5 Replication. J. Virol. 1999, 73, 5333–5344. [Google Scholar] [CrossRef] [Green Version]
- Rothmann, T.; Hengstermann, A.; Whitaker, N.J.; Scheffner, M.; Hausen, H.Z. Replication of ONYX-015, a Potential Anticancer Adenovirus, Is Independent of p53 Status in Tumor Cells. J. Virol. 1998, 72, 9470–9478. [Google Scholar] [CrossRef] [Green Version]
- Geoerger, B.; Vassal, G.; Opolon, P.; Dirven, C.M.; Morizet, J.; Laudani, L.; Grill, J.; Giaccone, G.; Vandertop, W.P.; Gerritsen, W.R.; et al. Oncolytic Activity of p53-Expressing Conditionally Replicative Adenovirus AdΔ24-p53 against Human Malignant Glioma. Cancer Res. 2004, 64, 5753–5759. [Google Scholar] [CrossRef] [Green Version]
- Nettelbeck, D.M.; Rivera, A.A.; Balagué, C.; Alemany, R.; Curiel, D.T. Novel oncolytic adenoviruses targeted to melanoma: Specific viral replication and cytolysis by expression of E1A mutants from the tyrosinase enhancer/promoter. Cancer Res. 2002, 62, 4663–4670. [Google Scholar]
- Nemunaitis, J.; Tong, A.W.; Nemunaitis, M.; Senzer, N.; Phadke, A.P.; Bedell, C.; Adams, N.; Zhang, Y.-A.; Maples, P.B.; Chen, S.; et al. A Phase I Study of Telomerase-specific Replication Competent Oncolytic Adenovirus (Telomelysin) for Various Solid Tumors. Mol. Ther. 2010, 18, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Nagano, S.; Oshika, H.; Fujiwara, H.; Komiya, S.; Kosai, K. An efficient construction of conditionally replicating adenoviruses that target tumor cells with multiple factors. Gene Ther. 2005, 12, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, Y.; Wang, Y.; Nagano, S.; Kamizono, J.; Ikeda, M.; Komiya, S.; Kosai, K.-I. Assessment of an altered E1B promoter on the specificity and potency of triple-regulated conditionally replicating adenoviruses: Implications for the generation of ideal m-CRAs. Cancer Gene Ther. 2011, 18, 724–733. [Google Scholar] [CrossRef] [Green Version]
- Kamizono, J.; Nagano, S.; Murofushi, Y.; Komiya, S.; Fujiwara, H.; Matsuishi, T.; Kosai, K.-I. Survivin-Responsive Conditionally Replicating Adenovirus Exhibits Cancer-Specific and Efficient Viral Replication. Cancer Res. 2005, 65, 5284–5291. [Google Scholar] [CrossRef] [Green Version]
- Tanoue, K.; Wang, Y.; Ikeda, M.; Mitsui, K.; Irie, R.; Setoguchi, T.; Komiya, S.; Natsugoe, S.; Kosai, K.-I. Survivin-responsive conditionally replicating adenovirus kills rhabdomyosarcoma stem cells more efficiently than their progeny. J. Transl. Med. 2014, 12, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Kofune, H.; Uozumi, K.; Yoshimitsu, M.; Arima, N.; Ishitsuka, K.; Ueno, S.-I.; Kosai, K.-I. A survivin-responsive, conditionally replicating adenovirus induces potent cytocidal effects in adult T-cell leukemia/lymphoma. BMC Cancer 2019, 19, 516. [Google Scholar] [CrossRef]
- Mitsui, K.; Ide, K.; Takayama, A.; Wada, T.; Irie, R.; Kosai, K.-I. Conditionally replicating adenovirus prevents pluripotent stem cell–derived teratoma by specifically eliminating undifferentiated cells. Mol. Ther. Methods Clin. Dev. 2015, 2, 15026. [Google Scholar] [CrossRef]
- Wold, W.S.M.; Toth, K. Adenovirus Vectors for Gene Therapy, Vaccination and Cancer Gene Therapy. Curr. Gene Ther. 2013, 13, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Flinterman, M.; Gäken, J.; Farzaneh, F.; Tavassoli, M. E1A-mediated suppression of EGFR expression and induction of apoptosis in head and neck squamous carcinoma cell lines. Oncogene 2003, 22, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Berk, A.J. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene 2005, 24, 7673–7685. [Google Scholar] [CrossRef] [Green Version]
- Cheng, P.-H.; Wechman, S.L.; McMasters, K.M.; Zhou, H.S. Oncolytic Replication of E1b-Deleted Adenoviruses. Viruses 2015, 7, 5767–5779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gürlevik, E.; Woller, N.; Schache, P.; Malek, N.P.; Wirth, T.C.; Zender, L.; Manns, M.P.; Kubicka, S.; Kühnel, F. p53-dependent antiviral RNA-interference facilitates tumor-selective viral replication. Nucleic Acids Res. 2009, 37, e84. [Google Scholar] [CrossRef] [Green Version]
- Ramachandra, M.; Rahman, A.; Zou, A.; Vaillancourt, M.; Howe, J.A.; Antelman, D.; Sugarman, B.J.; Demers, G.W.; Engler, H.; Johnson, D.E.; et al. Re-engineering adenovirus regulatory pathways to enhance oncolytic specificity and efficacy. Nat. Biotechnol. 2001, 19, 1035–1041. [Google Scholar] [CrossRef]
- Kawashima, T.; Kagawa, S.; Kobayashi, N.; Shirakiya, Y.; Umeoka, T.; Teraishi, F.; Taki, M.; Kyo, S.; Tanaka, N.; Fujiwara, T. Telomerase-specific replication-selective virotherapy for human cancer. Clin. Cancer Res. 2004, 10, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Lanson, N.A.; Friedlander, P.L.; Schwarzenberger, P.; Kolls, J.K.; Wang, G. Replication of an adenoviral vector controlled by the human telomerase reverse transcriptase promoter causes tumor-selective tumor lysis. Cancer Res. 2003, 63, 7936–7941. [Google Scholar]
- Kurihara, T.; Brough, D.E.; Kovesdi, I.; Kufe, D.W. Selectivity of a replication-competent adenovirus for human breast carcinoma cells expressing the MUC1 antigen. J. Clin. Investig. 2000, 106, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Hallenbeck, P.L.; Chang, Y.-N.; Hay, C.; Golightly, D.; Stewart, D.; Lin, J.; Phipps, S.; Chiang, Y.L. A Novel Tumor-Specific Replication-Restricted Adenoviral Vector for Gene Therapy of Hepatocellular Carcinoma. Hum. Gene Ther. 1999, 10, 1721–1733. [Google Scholar] [CrossRef]
- Rodriguez, R.; Schuur, E.R.; Lim, H.Y.; Henderson, G.A.; Simons, J.W.; Henderson, D.R. Prostate attenuated replication competent adenovirus (ARCA) CN706: A selective cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Res. 1997, 57, 2559–2563. [Google Scholar]
- Gomez-Manzano, C.; Balague, C.; Alemany, R.; Lemoine, M.G.; Mitliagka, P.; Jiang, H.; Khan, A.; Alonso, M.; Lang, F.F.; Conrad, C.A.; et al. A novel E1A–E1B mutant adenovirus induces glioma regression in vivo. Oncogene 2004, 23, 1821–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.; Shen, A.; Boyle, L.; Kunich, J.; Pandey, K.; Lemmon, M.; Hermiston, T.; Giedlin, M.; McCormick, F.; Fattaey, A. Selectively replicating adenoviruses targeting deregulated E2F activity are potent, systemic antitumor agents. Cancer Cell 2002, 1, 325–337. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.M.; Sakamoto, G.T.; Henderson, D.R. Identification of the transcriptional regulatory sequences of human kallikrein 2 and their use in the construction of calydon virus 764, an attenuated replication competent adenovirus for prostate cancer therapy. Cancer Res. 1999, 59, 1498–1504. [Google Scholar] [PubMed]
- Chartier, C.; Degryse, E.; Gantzer, M.; Dieterle, A.; Pavirani, A.; Mehtali, M. Efficient generation of recombinant adenovirus vectors by homologous recombination in Escherichia coli. J. Virol. 1996, 70, 4805–4810. [Google Scholar] [CrossRef] [Green Version]
- Miyake, S.; Makimura, M.; Kanegae, Y.; Harada, S.; Sato, Y.; Takamori, K.; Tokuda, C.; Saito, I. Efficient generation of recombinant adenoviruses using adenovirus DNA-terminal protein complex and a cosmid bearing the full-length virus genome. Proc. Natl. Acad. Sci. USA 1996, 93, 1320–1324. [Google Scholar] [CrossRef] [Green Version]
- Mizuguchi, H.; Kay, M.A. Efficient Construction of a Recombinant Adenovirus Vector by an Improved In Vitro Ligation Method. Hum. Gene Ther. 1998, 9, 2577–2583. [Google Scholar] [CrossRef] [Green Version]
- Makower, D.; Rozenblit, A.; Kaufman, H.; Edelman, M.; Lane, M.E.; Zwiebel, J.; Haynes, H.; Wadler, S. Phase II clinical trial of intralesional administration of the oncolytic adenovirus ONYX-015 in patients with hepatobiliary tumors with correlative p53 studies. Clin. Cancer Res. 2003, 9, 693–702. [Google Scholar]
- Chiocca, E.A.; Abbed, K.M.; Tatter, S.; Louis, D.N.; Hochberg, F.H.; Barker, F.; Kracher, J.; Grossman, S.A.; Fisher, J.D.; Carson, K.; et al. A Phase I Open-Label, Dose-Escalation, Multi-Institutional Trial of Injection with an E1B-Attenuated Adenovirus, ONYX-015, into the Peritumoral Region of Recurrent Malignant Gliomas, in the Adjuvant Setting. Mol. Ther. 2004, 10, 958–966. [Google Scholar] [CrossRef]
- Guo, Z.S.; Lotze, M.T.; Zhu, Z.; Storkus, W.J.; Song, X.-T. Bi- and Tri-Specific T Cell Engager-Armed Oncolytic Viruses: Next-Generation Cancer Immunotherapy. Biomedicines 2020, 8, 204. [Google Scholar] [CrossRef]
- Goradel, N.H.; Baker, A.T.; Arashkia, A.; Ebrahimi, N.; Ghorghanlu, S.; Negahdari, B. Oncolytic virotherapy: Challenges and solutions. Curr. Probl. Cancer 2021, 45, 100639. [Google Scholar] [CrossRef]
- Harrington, K.; Puzanov, I.; Hecht, J.R.; Hodi, F.S.; Szabo, Z.; Murugappan, S.; Kaufman, H.L. Clinical development of talimogene laherparepvec (T-VEC): A modified herpes simplex virus type-1–derived oncolytic immunotherapy. Expert Rev. Anticancer. Ther. 2015, 15, 1389–1403. [Google Scholar] [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef] [Green Version]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Murofushi, Y.; Nagano, S.; Kamizono, J.; Takahashi, T.; Fujiwara, H.; Komiya, S.; Matsuishi, T.; Kosai, K. Cell cycle-specific changes in hTERT promoter activity in normal and cancerous cells in adenoviral gene therapy: A promising implication of te-lomerase-dependent targeted cancer gene therapy. Int. J. Oncol. 2006, 29, 681–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [Green Version]
- Samadani, A.A.; Keymoradzdeh, A.; Shams, S.; Soleymanpour, A.; Norollahi, S.E.; Vahidi, S.; Rashidy-Pour, A.; Ashraf, A.; Mirzajani, E.; Khanaki, K.; et al. Mechanisms of cancer stem cell therapy. Clin. Chim. Acta 2020, 510, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. The molecular basis and potential role of survivin in cancer diagnosis and therapy. Trends Mol. Med. 2001, 7, 542–547. [Google Scholar] [CrossRef]
- Ide, K.; Mitsui, K.; Irie, R.; Matsushita, Y.; Ijichi, N.; Toyodome, S.; Kosai, K. A novel construction of lentiviral vectors for eliminating tumorigenic cells from pluripotent stem cells. Stem Cells 2018, 36, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Mitsui, K.; Ide, K.; Takahashi, T.; Kosai, K.-I. Viral Vector-Based Innovative Approaches to Directly Abolishing Tumorigenic Pluripotent Stem Cells for Safer Regenerative Medicine. Mol. Ther. Methods Clin. Dev. 2017, 5, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsui, K.; Takahashi, T.; Ide, K.; Matsuda, E.; Kosai, K.-I. Optimization of adenoviral gene transfer in human pluripotent stem cells. Biochem. Biophys. Res. Commun. 2021, 541, 78–83. [Google Scholar] [CrossRef]
- Takahashi, T.; Kawai, T.; Ushikoshi, H.; Nagano, S.; Oshika, H.; Inoue, M.; Kunisada, T.; Takemura, G.; Fujiwara, H.; Kosai, K.-I. Identification and Isolation of Embryonic Stem Cell-Derived Target Cells by Adenoviral Conditional Targeting. Mol. Ther. 2006, 14, 673–683. [Google Scholar] [CrossRef] [PubMed]
- McCormack, M.P.; Rabbitts, T.H. Activation of the T-Cell OncogeneLMO2after Gene Therapy for X-Linked Severe Combined Immunodeficiency. N. Engl. J. Med. 2004, 350, 913–922. [Google Scholar] [CrossRef]
- Lee, A.S.; Tang, C.; Rao, M.S.; Weissman, I.L.; Wu, J.C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 2013, 19, 998–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Type of Recombinant Adenovirus | Modification of Adenovirus | Advantage | Disadvantage | Applicable Diseases and Therapies | References |
|---|---|---|---|---|---|
| Replication-defective adenoviral vector |
|
|
|
| [28,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52] |
| Conditionally replicating adenovirus (CRA; oncolytic adenovirus) |
|
|
|
| [21,25,53,54,55,56,57,58,59,60,61,62,63,64,65,66] |
| CRA that can specifically target tumors with multiple factors (m-CRA) |
|
|
| [67,68,69,70,71,72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watanabe, M.; Nishikawaji, Y.; Kawakami, H.; Kosai, K.-i. Adenovirus Biology, Recombinant Adenovirus, and Adenovirus Usage in Gene Therapy. Viruses 2021, 13, 2502. https://doi.org/10.3390/v13122502
Watanabe M, Nishikawaji Y, Kawakami H, Kosai K-i. Adenovirus Biology, Recombinant Adenovirus, and Adenovirus Usage in Gene Therapy. Viruses. 2021; 13(12):2502. https://doi.org/10.3390/v13122502
Chicago/Turabian StyleWatanabe, Maki, Yuya Nishikawaji, Hirotaka Kawakami, and Ken-ichiro Kosai. 2021. "Adenovirus Biology, Recombinant Adenovirus, and Adenovirus Usage in Gene Therapy" Viruses 13, no. 12: 2502. https://doi.org/10.3390/v13122502