
Why the HIV Reservoir Never Runs Dry: Clonal Expansion and the Characteristics of HIV-Infected Cells Challenge Strategies to Cure and Control HIV Infection



Abstract
:1. Introduction
1.1. HIV Replication and Establishing the Proviral State
- Sites of integration are widely distributed in the genome. Integrations generally take place at the 5′AAT, 5′AAA, and 5′ TAA sequences. There are, in general, no other sequence specificities and, in general, no specific “hot spots” for integration;
- Integration preferences exist, generally on the basis of the LEDGF/p75 chaperone preference for regions of active transcription. Proviruses are more commonly found in introns over exons because of the greater relative size of introns in the human genome. Importantly, promoter regions are largely excluded from LEDGF/p75-directed integration. As such, the HIV promoter is rarely present within a host gene promoter, which makes the strong transcriptional effects of the HIV long terminal repeat (LTR) on the host genes, such as those which drive oncogene expression in neoplasms, less likely [51].
- The integration event is independent with respect to the orientation of the transcription of the host gene. Proviruses may be introduced with viral and host transcriptions in the same orientation, or in an opposite orientation.
- Integration is independent of the size of the provirus and intact and deleted HIV DNA forms are readily integrated.

1.2. Response to Antiretroviral Therapy In Vivo
2. Potential Mechanisms of HIV Persistence
2.1. Ongoing Cycles of HIV Replication
2.2. Persistence and Clonal Expansion of HIV-Infected Cells
2.3. Implications of Persistence Mechanisms
3. Laboratory Approaches to Studying Persistence
3.1. Nucleic Acid Analyses and Bioassayss
3.2. Genetic Characterization of HIV Variants
3.3. Analysis of Integration Sites
4. Clinical Approaches to Study Persistence
4.1. Natural History Studies
4.2. Interventional Studies to Assess Ongoing Replication
5. Proviral Landscape during ART
6. Persistence in the Context of Infected Individuals
6.1. Target Cells and Anatomic Sites
6.2. Specific Anatomic Compartments with HIV-Infected Cells
7. Implications of Persistence for Cure Strategies
- What is the role of clonal expansion in maintaining a stable level of HIV-infected cells during prolonged ART?
- Do interventions designed to reduce the HIV reservoir affect the clonal expansion of HIV-infected cells? Does latency reversal lead to the clonal expansion of infected cells? Does block-and-lock result in a loss of clones?
- How does the HIV-infected cell population respond to cytotoxic chemotherapy? Understanding whether HIV-infected cells are preferentially lost or whether clonal expansion results in repopulation post-chemotherapy will provide new insights regarding eradication requirements;
- Are clones widely distributed in anatomic compartments? Does clonal expansion occur in all tissues?
- Are there unique strategies to target clonal expansion? For which cure approaches would this be necessary?
8. Conclusions
- The principal mechanism of HIV persistence is long-lived immune cells that undergo clonal expansion. This process is common and includes the expansion of HIV-infected cells;
- ARV intensification and phylogenetic studies suggest that the infection of new cells is rare during ART;
- Both replication-competent and defective proviruses undergo clonal expansion and contribute to morbidity in PLWH;
- There are four potential mechanisms of the clonal expansion of HIV-infected CD4 T cells. Integration may play a role in some instances of clonal expansion and its inhibition alters HIV decay dynamics;
- Clones come to dominate the proviral landscape on ART, but individual clones wax and wane over time.
- Successful HIV cure strategies will need to overcome the effects of clonal expansion and target the anatomic sites in which latently infected cells persist.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Disclosure Statement
References
- Hammer, S.M.; Ribaudo, H.; Bassett, R.; Mellors, J.W.; Demeter, L.M.; Coombs, R.W.; Currier, J.; Morse, G.D.; Geber, J.G.; Martinez, A.I.; et al. A randomized, placebo-controlled trial of abacavir intensification in HIV-1-infected adults with virologic suppression on a protease inhibitor-containing regimen. HIV Clin. Trials. 2010, 11, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Kearney, M.F.; Spindler, J.; Shao, W.; Yu, S.; Anderson, E.M.; O’Shea, A.; Rehm, C.; Poethke, C.; Kovacs, N.; Mellors, J.W.; et al. Lack of Detectable HIV-1 Molecular Evolution during Suppressive Antiretroviral Therapy. PLoS Pathog. 2014, 10, e1004010. [Google Scholar] [CrossRef] [Green Version]
- Bozzi, G.; Simonetti, F.R.; Watters, S.A.; Anderson, E.M.; Gouzoulis, M.; Kearney, M.F.; Rote, P.; Lange, C.; Shao, W.; Gorelick, R.; et al. No evidence of ongoing HIV replication or compartmentalization in tissues during combination antiretroviral therapy: Implications for HIV eradication. Sci. Adv. 2019, 5, eaav2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, T.-W.; Moir, S.; Fauci, A.S. HIV reservoirs as obstacles and opportunities for an HIV cure. Nat. Immunol. 2015, 16, 584–589. [Google Scholar] [CrossRef]
- Croxford, S.; Kitching, A.; Desai, S.; Kall, M.; Edelstein, M.; Skingsley, A.; Burns, F.; Copas, A.; Brown, E.A.; Sullivan, A.K.; et al. Mortality and causes of death in people diagnosed with HIV in the era of highly active antiretroviral therapy compared with the general population: An analysis of a national observational cohort. Lancet Public Health 2017, 2, e35–e46. [Google Scholar] [CrossRef] [Green Version]
- Michaels, S.H.; Clark, R.; Kissinger, P. Declining Morbidity and Mortality among Patients with Advanced Human Immunodeficiency Virus Infection. N. Engl. J. Med. 1998, 339, 405–406. [Google Scholar] [CrossRef] [PubMed]
- Imamichi, H.; Dewar, R.L.; Adelsberger, J.W.; Rehm, C.A.; O’Doherty, U.; Paxinos, E.E.; Fauci, A.S.; Lane, H.C. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2016, 113, 8783–8788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Currier, J.S.; Baden, L.R. Getting Smarter—The Toxicity of Undertreated HIV Infection. N. Engl. J. Med. 2006, 355, 2359–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, R.A.; Jones, R.B.; Pertea, M.; Bruner, K.M.; Martin, A.R.; Thomas, A.; Capoferri, A.A.; Beg, S.A.; Huang, S.-H.; Karandish, S.; et al. Defective HIV-1 Proviruses Are Expressed and Can Be Recognized by Cytotoxic T Lymphocytes, which Shape the Proviral Landscape. Cell Host Microbe 2017, 21, 494–506.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodari, A.; Darcis, G.; Van Lint, C.M. The Current Status of Latency Reversing Agents for HIV-1 Remission. Annu. Rev. Virol. 2021, 8, 491–514. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; McMahon, J.; Chang, J.J.; Audsley, J.; Rhodes, A.; Tennakoon, S.; Dantanarayana, A.; Spelman, T.; Schmidt, T.; Kent, S.J.; et al. The effect of antiretroviral intensification with dolutegravir on residual virus replication in HIV-infected individuals: A randomised, placebo-controlled, double-blind trial. Lancet HIV 2018, 5, e221–e230. [Google Scholar] [CrossRef]
- Jones, R.B.; Walker, B.D. HIV-specific CD8+ T cells and HIV eradication. J. Clin. Investig. 2016, 126, 455–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vansant, G.; Bruggemans, A.; Janssens, J.; Debyser, Z. Block-And-Lock Strategies to Cure HIV Infection. Viruses 2020, 12, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, Y.-H.J.; Ho, Y.-C. Shock-and-kill versus block-and-lock: Targeting the fluctuating and heterogeneous HIV-1 gene expression. Proc. Natl. Acad. Sci. USA 2021, 118, e2103692118. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Lian, X.; Gao, C.; Sun, X.; Einkauf, K.B.; Chevalier, J.M.; Chen, S.M.Y.; Hua, S.; Rhee, B.; Chang, K.; et al. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature 2020, 585, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Blazkova, J.; Gao, F.; Marichannegowda, M.H.; Justement, J.S.; Shi, V.; Whitehead, E.J.; Schneck, R.F.; Huiting, E.D.; Gittens, K.; Cottrell, M.; et al. Distinct mechanisms of long-term virologic control in two HIV-infected individuals after treatment interruption of anti-retroviral therapy. Nat. Med. 2021, 27, 1893–1898. [Google Scholar] [CrossRef]
- Parikh, U.M.; McCormick, K.; van Zyl, G.; Mellors, J.W. Future technologies for monitoring HIV drug resistance and cure. Curr. Opin. HIV AIDS 2017, 12, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mohsen, M.; Richman, D.; Siliciano, R.F.; Nussenzweig, M.C.; Howell, B.; Martinez-Picado, J.; Chomont, N.; Bar, K.J.; Yu, X.G.; Lichterfeld, M.; et al. Recommendations for measuring HIV reservoir size in cure-directed clinical trials. Nat. Med. 2020, 26, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Siliciano, R.F. Assays to Measure Latency, Reservoirs, and Reactivation. HIV-1 Latency 2017, 417, 23–41. [Google Scholar] [CrossRef]
- Rosenbloom, D.I.S.; Elliott, O.; Hill, A.L.; Henrich, T.J.; Siliciano, J.M.; Siliciano, R.F. Designing and Interpreting Limiting Dilution Assays: General Principles and Applications to the Latent Reservoir for Human Immunodeficiency Virus-1. Open Forum Infect. Dis. 2015, 2, ofv123. [Google Scholar] [CrossRef] [Green Version]
- Falcinelli, S.D.; Ceriani, C.; Margolis, D.M.; Archin, N.M. New Frontiers in Measuring and Characterizing the HIV Reservoir. Front. Microbiol. 2019, 10, 2878. [Google Scholar] [CrossRef] [Green Version]
- Lusic, M.; Siliciano, R.F. Nuclear landscape of HIV-1 infection and integration. Nat. Rev. Genet. 2016, 15, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.N.; Singh, P.K. Cellular and molecular mechanisms of HIV-1 integration targeting. Cell. Mol. Life Sci. 2018, 75, 2491–2507. [Google Scholar] [CrossRef] [PubMed]
- Symons, J.; Cameron, P.U.; Lewin, S.R. HIV integration sites and implications for maintenance of the reservoir. Curr. Opin. HIV AIDS 2018, 13, 152–159. [Google Scholar] [CrossRef]
- Deruaz, M.; Tager, A.M. Humanized mouse models of latent HIV infection. Curr. Opin. Virol. 2017, 25, 97–104. [Google Scholar] [CrossRef]
- Masse-Ranson, G.; Mouquet, H.; Di Santo, J.P. Humanized mouse models to study pathophysiology and treatment of HIV infection. Curr. Opin. HIV AIDS 2018, 13, 143–151. [Google Scholar] [CrossRef]
- Whitney, J.B.; Jones, R.B. In Vitro and In Vivo Models of HIV Latency. HIV Vaccines Cure 2018, 1075, 241–263. [Google Scholar] [CrossRef]
- Del Prete, G.Q.; Lifson, J.D.; Keele, B.F. Nonhuman primate models for the evaluation of HIV-1 preventive vaccine strategies: Model parameter considerations and consequences. Curr. Opin. HIV AIDS 2016, 11, 546–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Overbaugh, J. CD4–HIV-1 Envelope Interactions: Critical Insights for the Simian/HIV/Macaque Model. AIDS Res. Hum. Retrovir. 2018, 34, 778–779. [Google Scholar] [CrossRef] [PubMed]
- Cilento, M.E.; Kirby, K.A.; Sarafianos, S.G. Avoiding Drug Resistance in HIV Reverse Transcriptase. Chem. Rev. 2021, 121, 3271–3296. [Google Scholar] [CrossRef]
- Giacomelli, A.; Pezzati, L.; Rusconi, S. The crosstalk between antiretrovirals pharmacology and HIV drug resistance. Expert Rev. Clin. Pharmacol. 2020, 13, 739–760. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.-Y.; Grant, P.M.; Tzou, P.L.; Barrow, G.; Harrigan, P.R.; Ioannidis, J.P.A.; Shafer, R.W. A systematic review of the genetic mechanisms of dolutegravir resistance. J. Antimicrob. Chemother. 2019, 74, 3135–3149. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Gonzales, M.J.; Kantor, R.; Betts, B.J.; Ravela, J.; Shafer, R.W. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003, 31, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Mansky, L.M.; Temin, H.M. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 1995, 69, 5087–5094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodes, T.D.; Nikolaitchik, O.; Chen, J.; Powell, D.; Hu, W.-S. Genetic Recombination of Human Immunodeficiency Virus Type 1 in One Round of Viral Replication: Effects of Genetic Distance, Target Cells, Accessory Genes, and Lack of High Negative Interference in Crossover Events. J. Virol. 2005, 79, 1666–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawson, O.J.M.; Nikolaitchik, A.O.; Keele, B.F.; Pathak, V.; Hu, W.-S. Recombination is required for efficient HIV-1 replication and the maintenance of viral genome integrity. Nucleic Acids Res. 2018, 46, 10535–10545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.-S.; Hughes, S.H. HIV-1 Reverse Transcription. Cold Spring Harb. Perspect. Med. 2012, 2, a006882. [Google Scholar] [CrossRef] [Green Version]
- Schlub, T.; Grimm, A.J.; Smyth, R.P.; Cromer, D.; Chopra, A.; Mallal, S.; Venturi, V.; Waugh, C.; Mak, J.; Davenport, M.P. Fifteen to Twenty Percent of HIV Substitution Mutations Are Associated with Recombination. J. Virol. 2014, 88, 3837–3849. [Google Scholar] [CrossRef] [Green Version]
- Smyth, R.; Schlub, T.; Grimm, A.J.; Waugh, C.; Ellenberg, P.; Chopra, A.; Mallal, S.; Cromer, D.; Mak, J.; Davenport, M.P.; et al. Identifying Recombination Hot Spots in the HIV-1 Genome. J. Virol. 2013, 88, 2891–2902. [Google Scholar] [CrossRef] [Green Version]
- Rawson, J.M.; Mansky, L.M. Retroviral Vectors for Analysis of Viral Mutagenesis and Recombination. Viruses 2014, 6, 3612–3642. [Google Scholar] [CrossRef] [Green Version]
- Batorsky, R.; Kearney, M.F.; Palmer, S.E.; Maldarelli, F.; Rouzine, I.M.; Coffin, J.M. Estimate of effective recombination rate and average selection coefficient for HIV in chronic infection. Proc. Natl. Acad. Sci. USA 2011, 108, 5661–5666. [Google Scholar] [CrossRef] [Green Version]
- Coffin, J.M. HIV population dynamics in vivo: Implications for genetic variation, pathogenesis, and therapy. Science 1995, 267, 483–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdick, R.C.; Li, C.; Munshi, M.; Rawson, J.M.O.; Nagashima, K.; Hu, W.-S.; Pathak, V.K. HIV-1 uncoats in the nucleus near sites of integration. Proc. Natl. Acad. Sci. USA 2020, 117, 5486–5493. [Google Scholar] [CrossRef] [PubMed]
- Burdick, R.C.; Pathak, V.K. Efficient HIV-1 in vitro reverse transcription: Optimal capsid stability is required. Signal Transduct. Target. Ther. 2021, 6, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Burdick, R.C.; Nagashima, K.; Hu, W.-S.; Pathak, V.K. HIV-1 cores retain their integrity until minutes before uncoating in the nucleus. Proc. Natl. Acad. Sci. USA 2021, 118, e2019467118. [Google Scholar] [CrossRef]
- Koh, Y.; Wu, X.; Ferris, A.L.; Matreyek, K.; Smith, S.J.; Lee, K.; KewalRamani, V.N.; Hughes, S.H.; Engelman, A. Differential Effects of Human Immunodeficiency Virus Type 1 Capsid and Cellular Factors Nucleoporin 153 and LEDGF/p75 on the Efficiency and Specificity of Viral DNA Integration. J. Virol. 2012, 87, 648–658. [Google Scholar] [CrossRef] [Green Version]
- Achuthan, V.; Perreira, J.M.; Sowd, G.A.; Puray-Chavez, M.; McDougall, W.M.; Paulucci-Holthauzen, A.; Wu, X.; Fadel, H.J.; Poeschla, E.M.; Multani, A.S.; et al. Capsid-CPSF6 Interaction Licenses Nuclear HIV-1 Trafficking to Sites of Viral DNA Integration. Cell Host Microbe 2018, 24, 392–404.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos da Silva, E.; Shanmugapriya, S.; Malikov, V.; Gu, F.; Delaney, M.K.; Naghavi, M.H. HIV -1 Capsids Mimic a Microtubule Regulator to Coordinate Early Stages of Infection. EMBO J. 2020, 39, 1043–1049. [Google Scholar] [CrossRef]
- Lesbats, P.; Engelman, A.N.; Cherepanov, P. Retroviral DNA Integration. Chem. Rev. 2016, 116, 12730–12757. [Google Scholar] [CrossRef] [Green Version]
- Coffin, J.M.; Bale, M.J.; Wells, D.; Guo, S.; Luke, B.; Zerbato, J.M.; Sobolewski, M.D.; Sia, T.; Shao, W.; Wu, X.; et al. Integration in oncogenes plays only a minor role in determining the in vivo distribution of HIV integration sites before or during suppressive antiretroviral therapy. PLoS Pathog. 2021, 17, e1009141. [Google Scholar] [CrossRef]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef] [Green Version]
- Geis, F.K.; Goff, S.P. Unintegrated HIV-1 DNAs are loaded with core and linker histones and transcriptionally silenced. Proc. Natl. Acad. Sci. USA 2019, 116, 23735–23742. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Umunnakwe, C.; Sun, D.Q.; Nikolaitchik, O.A.; Pathak, V.K.; Berkhout, B.; Das, A.T.; Hu, W.-S. Impact of Nuclear Export Pathway on Cytoplasmic HIV-1 RNA Transport Mechanism and Distribution. mBio 2020, 11, e01578-20. [Google Scholar] [CrossRef]
- Lassen, K.G.; Ramyar, K.X.; Bailey, J.R.; Zhou, Y.; Siliciano, R.F. Nuclear Retention of Multiply Spliced HIV-1 RNA in Resting CD4+ T Cells. PLoS Pathog. 2006, 2, e68. [Google Scholar] [CrossRef]
- Byun, S.; Han, S.; Zheng, Y.; Planelles, V.; Lee, Y. The landscape of alternative splicing in HIV-1 infected CD4 T-cells. BMC Med. Genom. 2020, 13, 38. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, W.I.; Kräusslich, H.-G. HIV-1 Assembly, Budding, and Maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef] [PubMed]
- Hataye, J.M.; Casazza, J.P.; Best, K.; Liang, C.J.; Immonen, T.T.; Ambrozak, D.R.; Darko, S.; Henry, A.R.; Laboune, F.; Maldarelli, F.; et al. Principles Governing Establishment versus Collapse of HIV-1 Cellular Spread. Cell Host Microbe 2019, 26, 748–763.e20. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef]
- Chun, T.-W.; Finzi, D.; Margolick, J.; Chadwick, K.; Schwartz, D.; Siliciano, R.F. In vivo fate of HIV-1-infected T cells: Quantitative analysis of the transition to stable latency. Nat. Med. 1995, 1, 1284–1290. [Google Scholar] [CrossRef]
- Finzi, D.; Blankson, J.N.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.C.; Smith, A.K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a Reservoir for HIV-1 in Patients on Highly Active Antiretroviral Therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef]
- Roche, M.; Tumpach, C.; Symons, J.; Gartner, M.; Anderson, J.L.; Khoury, G.; Cashin, K.; Cameron, P.U.; Churchill, M.J.; Deeks, S.G.; et al. CXCR4-Using HIV Strains Predominate in Naive and Central Memory CD4+ T Cells in People Living with HIV on Antiretroviral Therapy: Implications for How Latency Is Established and Maintained. J. Virol. 2020, 94, 01736-19. [Google Scholar] [CrossRef]
- Anderson, E.M.; Maldarelli, F. The role of integration and clonal expansion in HIV infection: Live long and prosper. Retrovirology 2018, 15, 71. [Google Scholar] [CrossRef]
- Hill, A.L.; Rosenbloom, D.I.S.; Nowak, M.A.; Siliciano, R.F. Insight into treatment of HIV infection from viral dynamics models. Immunol. Rev. 2018, 285, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.; Wiegand, A.P.; Maldarelli, F.; Bazmi, H.; Mican, J.M.; Polis, M.; Dewar, R.L.; Planta, A.; Liu, S.; Metcalf, J.A.; et al. New Real-Time Reverse Transcriptase-Initiated PCR Assay with Single-Copy Sensitivity for Human Immunodeficiency Virus Type 1 RNA in Plasma. J. Clin. Microbiol. 2003, 41, 4531–4536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cillo, A.R.; Vagratian, D.; Bedison, M.A.; Anderson, E.M.; Kearney, M.; Fyne, E.; Koontz, D.; Coffin, J.M.; Piatak, M.; Mellors, J.W. Improved Single-Copy Assays for Quantification of Persistent HIV-1 Viremia in Patients on Suppressive Antiretroviral Therapy. J. Clin. Microbiol. 2014, 52, 3944–3951. [Google Scholar] [CrossRef] [Green Version]
- Somsouk, M.; Dunham, R.M.; Cohen, M.; Albright, R.; Abdel-Mohsen, M.; Liegler, T.; Lifson, J.; Piatak, M.; Gorelick, R.; Huang, Y.; et al. The Immunologic Effects of Mesalamine in Treated HIV-Infected Individuals with Incomplete CD4+ T Cell Recovery: A Randomized Crossover Trial. PLoS ONE 2014, 9, e116306. [Google Scholar] [CrossRef] [Green Version]
- Tosiano, M.A.; Jacobs, J.L.; Shutt, K.A.; Cyktor, J.C.; Mellors, J.W. A Simpler and More Sensitive Single-Copy HIV-1 RNA Assay for Quantification of Persistent HIV-1 Viremia in Individuals on Suppressive Antiretroviral Therapy. J. Clin. Microbiol. 2019, 57, e01714-18. [Google Scholar] [CrossRef] [Green Version]
- Riddler, S.A.; Dragavon, J.; Bosch, R.J.; Bastow, B.; Bedison, A.M.; Vagratian, D.; Vaida, F.; Eron, J.J.; Gandhi, R.T.; Mellors, J.W. Continued Slow Decay of the Residual Plasma Viremia Level in HIV-1–Infected Adults Receiving Long-term Antiretroviral Therapy. J. Infect. Dis. 2015, 213, 556–560. [Google Scholar] [CrossRef] [Green Version]
- Andrade, A.; Rosenkranz, S.L.; Cillo, A.R.; Lu, D.; Daar, E.S.; Jacobson, J.M.; Lederman, M.; Acosta, E.P.; Campbell, T.; Feinberg, J.; et al. Three Distinct Phases of HIV-1 RNA Decay in Treatment-Naive Patients Receiving Raltegravir-Based Antiretroviral Therapy: ACTG A5248. J. Infect. Dis. 2013, 208, 884–891. [Google Scholar] [CrossRef] [Green Version]
- Palmer, S.; Maldarelli, F.; Wiegand, A.; Bernstein, B.; Hanna, G.; Brun, S.C.; Kempf, D.J.; Mellors, J.W.; Coffin, J.M.; King, M.S. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2008, 105, 3879–3884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perelson, A.S.; Ribeiro, R.M. Modeling the within-host dynamics of HIV infection. BMC Biol. 2013, 11, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedaghat, A.R.; Dinoso, J.B.; Shen, L.; Wilke, C.O.; Siliciano, R.F. Decay dynamics of HIV-1 depend on the inhibited stages of the viral life cycle. Proc. Natl. Acad. Sci. USA 2008, 105, 4832–4837. [Google Scholar] [CrossRef] [Green Version]
- Cardozo, E.F.; Andrade, A.; Mellors, J.W.; Kuritzkes, D.R.; Perelson, A.S.; Ribeiro, R.M. Treatment with integrase inhibitor suggests a new interpretation of HIV RNA decay curves that reveals a subset of cells with slow integration. PLoS Pathog. 2017, 13, e1006478. [Google Scholar] [CrossRef] [PubMed]
- Andrade, A.; Guedj, J.; Rosenkranz, S.L.; Lu, D.; Mellors, J.; Kuritzkes, D.R.; Perelson, A.S.; Ribeiro, R.M. Early HIV RNA decay during raltegravir-containing regimens exhibits two distinct subphases (1a and 1b). AIDS 2015, 29, 2419–2426. [Google Scholar] [CrossRef]
- Smith, R. Adherence to antiretroviral HIV drugs: How many doses can you miss before resistance emerges? Proc. R. Soc. B Boil. Sci. 2005, 273, 617–624. [Google Scholar] [CrossRef] [Green Version]
- Winters, M.A.; Lloyd, R.M., Jr.; Shafer, R.W.; Kozal, M.J.; Miller, M.D.; Holodniy, M. Development of elvitegravir resistance and linkage of integrase inhibitor mutations with protease and reverse transcriptase resistance mutations. PLoS ONE 2012, 7, e40514. [Google Scholar] [CrossRef] [Green Version]
- Wensing, A.M.; Calvez, V.; Ceccherini-Silberstein, F.; Charpentier, C.; Gunthard, H.F.; Paredes, R.; Shafer, R.W.; Richman, D.D. 2019 update of the drug resistance mutations in HIV-1. Top Antivir. Med. 2019, 27, 111–121. [Google Scholar]
- Pingen, M.; Wensing, A.M.; Fransen, K.; De Bel, A.; de Jong, D.; Hoepelman, A.I.; Magiorkinis, E.; Paraskevis, D.; Lunar, M.M.; Poljak, M.; et al. Persistence of frequently transmitted drug-resistant HIV-1 variants can be explained by high viral replication capacity. Retrovirology 2014, 11, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Puertas, M.C.; Ploumidis, G.; Ploumidis, M.; Fumero, E.; Clotet, B.; Walworth, C.M.; Petropoulos, C.J.; Martinez-Picado, J. Pan-resistant HIV-1 emergence in the era of integrase strand-transfer inhibitors: A case report. Lancet Microbe 2020, 1, e130–e135. [Google Scholar] [CrossRef]
- Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Department of Health and Human Services 2020. Available online: https://clinicalinfo.hiv.gov/sites/default/files/inline-files/AdultandAdolescentGL.pdfm (accessed on 4 November 2020).
- Hare, S.; Vos, A.; Clayton, R.F.; Thuring, J.W.; Cummings, M.D.; Cherepanov, P. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 20057–20062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anstett, K.; Brenner, B.; Mesplede, T.; Wainberg, M.A. HIV drug resistance against strand transfer integrase inhibitors. Retrovirology 2017, 14, 1–16. [Google Scholar] [CrossRef]
- Goethals, O.; Clayton, R.; Van Ginderen, M.; Vereycken, I.; Wagemans, E.; Geluykens, P.; Dockx, K.; Strijbos, R.; Smits, V.; Vos, A.; et al. Resistance Mutations in Human Immunodeficiency Virus Type 1 Integrase Selected with Elvitegravir Confer Reduced Susceptibility to a Wide Range of Integrase Inhibitors. J. Virol. 2008, 82, 10366–10374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattaneo, D.; Gervasoni, C. Pharmacokinetics and Pharmacodynamics of Cabotegravir, a Long-Acting HIV Integrase Strand Transfer Inhibitor. Eur. J. Drug Metab. Pharmacokinet. 2018, 44, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.L.; Halvas, E.K.; Tosiano, M.A.; Mellors, J.W. Persistent HIV-1 Viremia on Antiretroviral Therapy: Measurement and Mechanisms. Front. Microbiol. 2019, 10, 2383. [Google Scholar] [CrossRef]
- Horsburgh, B.A.; Palmer, S. Measuring HIV Persistence on Antiretroviral Therapy. Adv. Exp. Med. Biol. 2018, 1075, 265–284. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, K.; Cary, D.C. Experimental Systems for Measuring HIV Latency and Reactivation. Viruses 2020, 12, 1279. [Google Scholar] [CrossRef]
- Estes, J.D.; Kityo, C.; Ssali, F.; Swainson, L.; Makamdop, K.N.; Del Prete, G.Q.; Deeks, S.G.; Luciw, P.A.; Chipman, J.G.; Beilman, G.J.; et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat. Med. 2017, 23, 1271–1276. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, S.F.C.; Guest, P.C. Multiplex Analyses Using Real-Time Quantitative PCR. Methods Mol. Biol. 2016, 1546, 125–133. [Google Scholar] [CrossRef]
- Anderson, E.M.; Maldarelli, F. Quantification of HIV DNA Using Droplet Digital PCR Techniques. Curr. Protoc. Microbiol. 2018, 51, e62. [Google Scholar] [CrossRef] [PubMed]
- Margot, N.; Koontz, D.; McCallister, S.; Mellors, J.W.; Callebaut, C. Measurement of plasma HIV-1 RNA below the limit of quantification (<20 copies/mL) of commercial assays with the integrase HIV RNA single-copy assay. J. Clin. Virol. 2018, 108, 50–52. [Google Scholar] [CrossRef]
- Avettand-Fènoël, V.; Hocqueloux, L.; Ghosn, J.; Cheret, A.; Frange, P.; Melard, A.; Viard, J.-P.; Rouzioux, C. Total HIV-1 DNA, a Marker of Viral Reservoir Dynamics with Clinical Implications. Clin. Microbiol. Rev. 2016, 29, 859–880. [Google Scholar] [CrossRef] [Green Version]
- Hong, F.; Aga, E.; Cillo, A.R.; Yates, A.L.; Besson, G.; Fyne, E.; Koontz, D.L.; Jennings, C.; Zheng, L.; Mellors, J.W. Novel Assays for Measurement of Total Cell-Associated HIV-1 DNA and RNA. J. Clin. Microbiol. 2016, 54, 902–911. [Google Scholar] [CrossRef] [Green Version]
- Sannier, G.; Dubé, M.; Kaufmann, D.E. Single-Cell Technologies Applied to HIV-1 Research: Reaching Maturity. Front. Microbiol. 2020, 11, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grau-Expósito, J.; Serra-Peinado, C.; Miguel, L.; Navarro, J.; Curran, A.; Burgos, J.; Ocaña, I.; Ribera, E.; Torrella, A.; Planas, B.; et al. A Novel Single-Cell FISH-Flow Assay Identifies Effector Memory CD4 + T cells as a Major Niche for HIV-1 Transcription in HIV-Infected Patients. mBio 2017, 8, e00876-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegand, A.; Spindler, J.; Hong, F.F.; Shao, W.; Cyktor, J.C.; Cillo, A.R.; Halvas, E.K.; Coffin, J.M.; Mellors, J.W.; Kearney, M.F. Single-cell analysis of HIV-1 transcriptional activity reveals expression of proviruses in expanded clones during ART. Proc. Natl. Acad. Sci. USA 2017, 114, E3659–E3668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Yeh, Y.-H.J.; Varabyou, A.; Collora, J.A.; Sherrill-Mix, S.; Talbot, C.C.; Mehta, S.; Albrecht, K.; Hao, H.; Zhang, H.; et al. Single-cell transcriptional landscapes reveal HIV-1–driven aberrant host gene transcription as a potential therapeutic target. Sci. Transl. Med. 2020, 12, eaaz0802. [Google Scholar] [CrossRef] [PubMed]
- Deleage, C.; Chan, C.N.; Busman-Sahay, K.; Estes, J.D. Next-generation in situ hybridization approaches to define and quantify HIV and SIV reservoirs in tissue microenvironments. Retrovirology 2018, 15, 1–10. [Google Scholar] [CrossRef]
- Vasquez, J.J.; Hussien, R.; Aguilar-Rodriguez, B.; Junger, H.; Dobi, D.; Henrich, T.J.; Thanh, C.; Gibson, E.; Hogan, L.; McCune, J.; et al. Elucidating the Burden of HIV in Tissues Using Multiplexed Immunofluorescence and In Situ Hybridization: Methods for the Single-Cell Phenotypic Characterization of Cells Harboring HIV In Situ. J. Histochem. Cytochem. 2018, 66, 427–446. [Google Scholar] [CrossRef] [Green Version]
- Wonderlich, E.R.; Subramanian, K.; Cox, B.; Wiegand, A.; Lackman-Smith, C.; Bale, M.; Stone, M.; Hoh, R.; Kearney, M.; Maldarelli, F.; et al. Effector memory differentiation increases detection of replication-competent HIV-l in resting CD4+ T cells from virally suppressed individuals. PLoS Pathog. 2019, 15, e1008074. [Google Scholar] [CrossRef] [PubMed]
- Bruner, K.M.; Wang, Z.; Simonetti, F.R.; Bender, A.M.; Kwon, K.J.; Sengupta, S.; Fray, E.J.; Beg, S.A.; Antar, A.A.R.; Jenike, K.M.; et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 2019, 566, 120–125. [Google Scholar] [CrossRef]
- Antar, A.; Jenike, K.M.; Jang, S.; Rigau, D.N.; Reeves, D.B.; Hoh, R.; Krone, M.R.; Keruly, J.C.; Moore, R.D.; Schiffer, J.T.; et al. Longitudinal study reveals HIV-1–infected CD4+ T cell dynamics during long-term antiretroviral therapy. J. Clin. Investig. 2020, 130, 3543–3559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, S.H. Reverse Transcription of Retroviruses and LTR Retrotransposons. Microbiol. Spectr. 2015, 3, 1051–1077. [Google Scholar] [CrossRef] [PubMed]
- Bale, M.J.; Kearney, M. Review: HIV-1 phylogeny during suppressive antiretroviral therapy. Curr. Opin. HIV AIDS 2019, 14, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Maldarelli, F.; Kearney, M.; Palmer, S.; Stephens, R.; Mican, J.; Polis, M.A.; Davey, R.T.; Kovacs, J.; Shao, W.; Rock-Kress, D.; et al. HIV Populations Are Large and Accumulate High Genetic Diversity in a Nonlinear Fashion. J. Virol. 2013, 87, 10313–10323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzo-Redondo, R.; Fryer, H.R.; Bedford, T.; Kim, E.Y.; Archer, J.; Pond, S.L.K.; Chung, Y.S.; Penugonda, S.; Chipman, J.G.; Fletcher, C.V.; et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature 2016, 530, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.R.; Miller, R.L.; Kinloch, N.N.; Tsai, O.; Rigsby, H.; Sudderuddin, H.; Shahid, A.; Ganase, B.; Brumme, C.J.; Harris, M.; et al. Genetic Diversity, Compartmentalization, and Age of HIV Proviruses Persisting in CD4 + T Cell Subsets during Long-Term Combination Antiretroviral Therapy. J. Virol. 2020, 94, e01786-19. [Google Scholar] [CrossRef]
- Josefsson, L.; Palmer, S.; Faria, N.R.; Lemey, P.; Casazza, J.; Ambrozak, D.; Kearney, M.; Shao, W.; Kottilil, S.; Sneller, M.; et al. Single Cell Analysis of Lymph Node Tissue from HIV-1 Infected Patients Reveals that the Majority of CD4+ T-cells Contain One HIV-1 DNA Molecule. PLoS Pathog. 2013, 9, e1003432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delviks-Frankenberry, K.; Galli, A.; Nikolaitchik, O.; Mens, H.; Pathak, V.K.; Hu, W.-S. Mechanisms and Factors that Influence High Frequency Retroviral Recombination. Viruses 2011, 3, 1650–1680. [Google Scholar] [CrossRef] [Green Version]
- Wagner, T.A.; McLaughlin, S.; Garg, K.; Cheung, C.Y.K.; Larsen, B.B.; Styrchak, S.; Huang, H.C.; Edlefsen, P.T.; Mullins, J.I.; Frenkel, L.M. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 2014, 345, 570–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohn, L.B.; Silva, I.; Oliveira, T.; Rosales, R.; Parrish, E.H.; Learn, G.H.; Hahn, B.; Czartoski, J.L.; McElrath, M.J.; Lehmann, C.; et al. HIV-1 Integration Landscape during Latent and Active Infection. Cell 2015, 160, 420–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, R.-C.; Prodger, J.L.; Redd, A.D.; Poon, A.F.Y. Quantifying the clonality and dynamics of the within-host HIV-1 latent reservoir. Virus Evol. 2021, 7, veaa104. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, S.; Kirchner, R.; Amr, S.S.; Mansur, L.; Shakhbatyan, R.; Kim, M.; Bosque, A.; Siliciano, R.F.; Planelles, V.; Hofmann, O.; et al. HIV Integration Site Analysis of Cellular Models of HIV Latency with a Probe-Enriched Next-Generation Sequencing Assay. J. Virol. 2016, 90, 4511–4519. [Google Scholar] [CrossRef] [Green Version]
- Bedwell, G.J.; Jang, S.; Li, W.; Singh, P.K.; Engelman, A.N. rigrag: High-resolution mapping of genic targeting preferences during HIV-1 integration in vitro and in vivo. Nucleic Acids Res. 2021, 49, 7330–7346. [Google Scholar] [CrossRef] [PubMed]
- Einkauf, K.B.; Lee, G.Q.; Gao, C.; Sharaf, R.; Sun, X.; Hua, S.; Chen, S.M.; Jiang, C.; Lian, X.; Chowdhury, F.Z.; et al. Intact HIV-1 proviruses accumulate at distinct chromosomal positions during prolonged antiretroviral therapy. J. Clin. Investig. 2019, 129, 988–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patro, S.C.; Brandt, L.D.; Bale, M.; Halvas, E.K.; Joseph, K.W.; Shao, W.; Wu, X.; Guo, S.; Murrell, B.; Wiegand, A.; et al. Combined HIV-1 sequence and integration site analysis informs viral dynamics and allows reconstruction of replicating viral ancestors. Proc. Natl. Acad. Sci. USA 2019, 116, 25891–25899. [Google Scholar] [CrossRef]
- Pierson, T.; McArthur, J.; Siliciano, R.F. Reservoirs for HIV-1: Mechanisms for Viral Persistence in the Presence of Antiviral Immune Responses and Antiretroviral Therapy. Annu. Rev. Immunol. 2000, 18, 665–708. [Google Scholar] [CrossRef]
- Hermankova, M.; Ray, S.C.; Ruff, C.; Powell-Davis, M.; Ingersoll, R.; D’Aquila, R.T.; Quinn, T.C.; Siliciano, J.D.; Siliciano, R.F.; Persaud, D. HIV-1 Drug Resistance Profiles in Children and Adults with Viral Load of <50 copies/mL receiving combination therapy. JAMA 2001, 286, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Von Wyl, V.; Gianella, S.; Fischer, M.; Niederoest, B.; Kuster, H.; Battegay, M.; Bernasconi, E.; Cavassini, M.; Rauch, A.; Hirschel, B.; et al. Early Antiretroviral Therapy During Primary HIV-1 Infection Results in a Transient Reduction of the Viral Setpoint upon Treatment Interruption. PLoS ONE 2011, 6, e27463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianella, S.; von Wyl, V.; Fischer, M.; Niederoest, B.; Battegay, M.; Bernasconi, E.; Cavassini, M.; Rauch, A.; Hirschel, B.; Vernazza, P.; et al. Effect of early antiretroviral therapy during primary HIV-1 infection on cell-associated HIV-1 DNA and plasma HIV-1 RNA. Antivir. Ther. 2011, 16, 535–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ananworanich, J.; Schuetz, A.; Vandergeeten, C.; Sereti, I.; De Souza, M.; Rerknimitr, R.; Dewar, R.; Marovich, M.; Van Griensven, F.; Sekaly, R.; et al. Impact of Multi-Targeted Antiretroviral Treatment on Gut T Cell Depletion and HIV Reservoir Seeding during Acute HIV Infection. PLoS ONE 2012, 7, e33948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornhill, J.; Inshaw, J.; Kaleebu, P.; Cooper, D.; Ramjee, G.; Schechter, M.; Tambussi, G.; Fox, J.; Samuel, M.; Miro, J.M.; et al. Brief Report: Enhanced Normalization of CD4/CD8 Ratio with Earlier Antiretroviral Therapy at Primary HIV Infection. JAIDS J. Acquir. Immune Defic. Syndr. 2016, 73, 69–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, C.; Bachmann, N.; Mitov, V.; Blanquart, F.; Céspedes, S.P.; Turk, T.; Neumann, K.; Beerenwinkel, N.; Bogojeska, J.; Fellay, J.; et al. Heritability of the HIV-1 reservoir size and decay under long-term suppressive ART. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Thorball, C.W.; Borghesi, A.; Bachmann, N.; Von Siebenthal, C.; Vongrad, V.; Turk, T.; Neumann, K.; Beerenwinkel, N.; Bogojeska, J.; Roth, V.; et al. Host Genomics of the HIV-1 Reservoir Size and Its Decay Rate During Suppressive Antiretroviral Treatment. JAIDS J. Acquir. Immune Defic. Syndr. 2020, 85, 517–524. [Google Scholar] [CrossRef]
- Bachmann, N.; Von Siebenthal, C.; Vongrad, V.; Turk, T.; Neumann, K.; Beerenwinkel, N.; Bogojeska, J.; Fellay, J.; Roth, V.; The Swiss HIV Cohort Study; et al. Determinants of HIV-1 reservoir size and long-term dynamics during suppressive ART. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, E.M.; Simonetti, F.R.; Gorelick, R.J.; Hill, S.; Gouzoulis, M.A.; Bell, J.; Rehm, C.; Pérez, L.; Boritz, E.; Wu, X.; et al. Dynamic Shifts in the HIV Proviral Landscape During Long Term Combination Antiretroviral Therapy: Implications for Persistence and Control of HIV Infections. Viruses 2020, 12, 136. [Google Scholar] [CrossRef] [Green Version]
- Peluso, M.J.; Bacchetti, P.; Ritter, K.D.; Beg, S.; Lai, J.; Martin, J.N.; Hunt, P.W.; Henrich, T.J.; Siliciano, J.D.; Siliciano, R.F.; et al. Differential decay of intact and defective proviral DNA in HIV-1–infected individuals on suppressive antiretroviral therapy. JCI Insight 2020, 5, e13297. [Google Scholar] [CrossRef] [Green Version]
- Maldarelli, F. Targeting viral reservoirs: Ability of antiretroviral therapy to stop viral replication. Curr. Opin. HIV AIDS 2011, 6, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Buzon, M.J.; Massanella, M.; Llibre, J.M.; Esteve, A.; Dahl, V.; Puertas, M.C.; Gatell, J.M.; Domingo, P.; Paredes, R.; Sharkey, M.; et al. HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects. Nat. Med. 2010, 16, 460–465. [Google Scholar] [CrossRef]
- Thierry, E.; Deprez, E.; Delelis, O. Different Pathways Leading to Integrase Inhibitors Resistance. Front. Microbiol. 2017, 7, 2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Picado, J.; Zurakowski, R.; Buzón, M.J.; Stevenson, M. Episomal HIV-1 DNA and its relationship to other markers of HIV-1 persistence. Retrovirology 2018, 15, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Huertas, M.R.; Gutiérrez, C.; Madrid-Elena, N.; Hernández-Novoa, B.; Olalla-Sierra, J.; Plana, M.; Delgado, R.; Rubio, R.; Muñoz-Fernández, M.; Moreno, S. Prolonged administration of maraviroc reactivates latent HIV in vivo but it does not prevent antiretroviral-free viral rebound. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kityo, C.; Szubert, A.J.; Siika, A.; Heyderman, R.; Bwakura-Dangarembizi, M.; Lugemwa, A.; Mwaringa, S.; Griffiths, A.; Nkanya, I.; Kabahenda, S.; et al. Raltegravir-intensified initial antiretroviral therapy in advanced HIV disease in Africa: A randomised controlled trial. PLoS Med. 2018, 15, e1002706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaillon, A.; Gianella, S.; Lada, S.M.; Perez-Santiago, J.; Jordan, P.; Ignacio, C.; Karris, M.; Richman, D.D.; Mehta, S.R.; Little, S.J.; et al. Size, Composition, and Evolution of HIV DNA Populations during Early Antiretroviral Therapy and Intensification with Maraviroc. J. Virol. 2018, 92, e01589-17. [Google Scholar] [CrossRef] [Green Version]
- Henrich, T.J. Dolutegravir intensification and HIV persistence: 3 + 1 = 3. Lancet HIV 2018, 5, e201–e202. [Google Scholar] [CrossRef]
- Puertas, M.C.; Gómez-Mora, E.; Santos, J.R.; Molto, J.; Urrea, V.; Moron-Lopez, S.; Hernandez-Rodriguez, A.; Marfil, S.; Martínez-Bonet, M.; Matas, L.; et al. Impact of intensification with raltegravir on HIV-1-infected individuals receiving monotherapy with boosted PIs. J. Antimicrob. Chemother. 2018, 73, 1940–1948. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.J.; Rousseau, R.; Huibner, S.; Kovacs, C.; Benko, E.; Shahabi, K.; Kandel, G.; Ostrowski, M.; Kaul, R. Impact of intensified antiretroviral therapy during early HIV infection on gut immunology and inflammatory blood biomarkers. AIDS 2017, 31, 1529–1534. [Google Scholar] [CrossRef]
- Wang, X.; Mink, G.; Lin, D.; Song, X.; Rong, L. Influence of raltegravir intensification on viral load and 2-LTR dynamics in HIV patients on suppressive antiretroviral therapy. J. Theor. Biol. 2017, 416, 16–27. [Google Scholar] [CrossRef]
- Lafeuillade, A.; Assi, A.; Poggi, C.; Bresson-Cuquemelle, C.; Jullian, E.; Tamalet, C. Failure of combined antiretroviral therapy intensification with maraviroc and raltegravir in chronically HIV-1 infected patients to reduce the viral reservoir: The IntensHIV randomized trial. AIDS Res. Ther. 2014, 11, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puertas, M.C.; Massanella, M.; Llibre, J.M.; Ballestero, M.; Buzon, M.J.; Ouchi, D.; Esteve, A.; Boix, J.; Manzardo, C.; Miró, J.M.; et al. Intensification of a raltegravir-based regimen with maraviroc in early HIV-1 infection. AIDS 2014, 28, 325–334. [Google Scholar] [CrossRef]
- Gutiérrez, C.; Hernández-Novoa, B.; Vallejo, A.; Serrano-Villar, S.; Abad-Fernández, M.; Madrid, N.; Díaz, L.; Moreno, A.; Dronda, F.; Zamora, J.; et al. Dynamics of the HIV-1 latent reservoir after discontinuation of the intensification of antiretroviral treatment: Results of two clinical trials. AIDS 2013, 27, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Negredo, E.; Massanella, M.; Puertas, M.C.; Buzon, M.J.; Puig, J.; Pérez-Alvárez, N.; Pérez-Santiago, J.; Bonjoch, A.; Moltó, J.; Jou, A.; et al. Early but limited effects of raltegravir intensification on CD4 T cell reconstitution in HIV-infected patients with an immunodiscordant response to antiretroviral therapy. J. Antimicrob. Chemother. 2013, 68, 2358–2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharkey, M. Tracking episomal HIV DNA: Implications for viral persistence and eradication of HIV. Curr. Opin. HIV AIDS 2013, 8, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Chege, D.; Kovacs, C.; La Porte, C.; Ostrowski, M.; Raboud, J.; Su, D.; Kandel, G.; Brunetta, J.; Kim, C.J.; Sheth, P.M.; et al. Effect of raltegravir intensification on HIV proviral DNA in the blood and gut mucosa of men on long-term therapy: A randomized controlled trial. AIDS 2012, 26, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Hatano, H.; Scherzer, R.; Wu, Y.; Harvill, K.; Maka, K.; Hoh, R.; Sinclair, E.; Palmer, S.; Martin, J.N.; Busch, M.P.; et al. A Randomized Controlled Trial Assessing the Effects of Raltegravir Intensification on Endothelial Function in Treated HIV Infection. JAIDS J. Acquir. Immune Defic. Syndr. 2012, 61, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Llibre, J.M.; Buzón, M.J.; Massanella, M.; Esteve, A.; Dahl, V.; Puertas, M.C.; Domingo, P.; Gatell, J.M.; Larrouse, M.; Gutierrez, M.; et al. Treatment intensification with raltegravir in subjects with sustained HIV-1 viraemia suppression: A randomized 48-week study. Antivir. Ther. 2011, 17, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Buzon, M.J.; Codoñer, F.M.; Frost, S.D.W.; Pou, C.; Puertas, M.C.; Massanella, M.; Dalmau, J.; Llibre, J.M.; Stevenson, M.; Blanco, J.; et al. Deep Molecular Characterization of HIV-1 Dynamics under Suppressive HAART. PLoS Pathog. 2011, 7, e1002314. [Google Scholar] [CrossRef] [Green Version]
- Dahl, V.; Lee, E.; Peterson, J.; Spudich, S.S.; Leppla, I.; Sinclair, E.; Fuchs, D.; Palmer, S.; Price, R.W. Raltegravir Treatment Intensification Does Not Alter Cerebrospinal Fluid HIV-1 Infection or Immunoactivation in Subjects on Suppressive Therapy. J. Infect. Dis. 2011, 204, 1936–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archin, N.M.; Cheema, M.; Parker, D.; Wiegand, A.; Bosch, R.J.; Coffin, J.M.; Eron, J.; Cohen, M.; Margolis, D.M. Antiretroviral Intensification and Valproic Acid Lack Sustained Effect on Residual HIV-1 Viremia or Resting CD4+ Cell Infection. PLoS ONE 2010, 5, e9390. [Google Scholar] [CrossRef]
- McMahon, D.; Jones, J.; Wiegand, A.; Gange, S.; Kearney, M.; Palmer, S.; McNulty, S.; Metcalf, J.A.; Acosta, E.; Rehm, C.; et al. Short-Course Raltegravir Intensification Does Not Reduce Persistent Low-Level Viremia in Patients with HIV-1 Suppression during Receipt of Combination Antiretroviral Therapy. Clin. Infect. Dis. 2010, 50, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, A.; Verhofstede, C.; D’Avolio, A.; Watson, V.; Hagberg, L.; Fuchs, D.; Svennerholm, B.; Gisslén, M. Treatment Intensification Has no Effect on the HIV-1 Central Nervous System Infection in Patients on Suppressive Antiretroviral Therapy. JAIDS J. Acquir. Immune Defic. Syndr. 2010, 55, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Dinoso, J.B.; Kim, S.Y.; Wiegand, A.M.; Palmer, S.E.; Gange, S.; Cranmer, L.; O’Shea, A.; Callender, M.; Spivak, A.; Brennan, T.; et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 9403–9408. [Google Scholar] [CrossRef] [Green Version]
- Havlir, D.V.; Strain, M.C.; Clerici, M.; Ignacio, C.; Trabattoni, D.; Ferrante, P.; Wong, J.K. Productive Infection Maintains a Dynamic Steady State of Residual Viremia in Human Immunodeficiency Virus Type 1-Infected Persons Treated with Suppressive Antiretroviral Therapy for Five Years. J. Virol. 2003, 77, 11212–11219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ait- ar, A.; Kula, A.; Darcis, G.; Verdikt, R.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Rohr, O.; Van Lint, C. Current Status of Latency Reversing Agents Facing the Heterogeneity of HIV-1 Cellular and Tissue Reservoirs. Front. Microbiol. 2020, 10, 3060. [Google Scholar] [CrossRef] [Green Version]
- Sgarbanti, M.; Battistini, A. Therapeutics for HIV-1 reactivation from latency. Curr. Opin. Virol. 2013, 3, 394–401. [Google Scholar] [CrossRef]
- Mok, H.P.; Lever, A. Waking Up the Sleepers: HIV Latency and Reactivation. J. Formos. Med Assoc. 2008, 107, 909–914. [Google Scholar] [CrossRef] [Green Version]
- Dahl, V.; Josefsson, L.; Palmer, S. HIV reservoirs, latency, and reactivation: Prospects for eradication. Antivir. Res. 2010, 85, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Gulick, R.M. Structured Treatment Interruption in Patients Infected with HIV: A new approach to therapy. Drugs 2002, 62, 245–253. [Google Scholar] [CrossRef]
- Davey, R.T.; Bhat, N.; Yoder, C.; Chun, T.-W.; Metcalf, J.A.; Dewar, R.; Natarajan, V.; Lempicki, R.; Adelsberger, J.W.; Miller, K.D.; et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA 1999, 96, 15109–15114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, G.E.; Frater, J. Post-treatment and spontaneous HIV control. Curr. Opin. HIV AIDS 2018, 13, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Joos, B.; Fischer, M.; Kuster, H.; Pillai, S.K.; Wong, J.K.; Böni, J.; Hirschel, B.; Weber, R.; Trkola, A.; Günthard, H.F.; et al. HIV rebounds from latently infected cells, rather than from continuing low-level replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16725–16730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothenberger, M.K.; Keele, B.F.; Wietgrefe, S.W.; Fletcher, C.V.; Beilman, G.; Chipman, J.; Khoruts, A.; Estes, J.D.; Anderson, J.; Callisto, S.P.; et al. Large number of rebounding/founder HIV variants emerge from multifocal infection in lymphatic tissues after treatment interruption. Proc. Natl. Acad. Sci. USA 2015, 112, E1126–E1134. [Google Scholar] [CrossRef] [Green Version]
- Bednar, M.M.; Hauser, B.; Zhou, S.; Jacobson, J.M.; Eron, J.J.; Frank, I.; Swanstrom, R. Diversity and Tropism of HIV-1 Rebound Virus Populations in Plasma Level After Treatment Discontinuation. J. Infect. Dis. 2016, 214, 403–407. [Google Scholar] [CrossRef] [Green Version]
- Vibholm, L.K.; Lorenzi, J.C.C.; Pai, J.A.; Cohen, Y.Z.; Oliveira, T.; Barton, J.P.; Noceda, M.G.; Lu, C.-L.; Ablanedo-Terrazas, Y.; Estrada, P.M.D.R.; et al. Characterization of Intact Proviruses in Blood and Lymph Node from HIV-Infected Individuals Undergoing Analytical Treatment Interruption. J. Virol. 2019, 93, e01920-18. [Google Scholar] [CrossRef] [Green Version]
- De Scheerder, M.-A.; Vrancken, B.; Dellicour, S.; Schlub, T.; Lee, E.; Shao, W.; Rutsaert, S.; Verhofstede, C.; Kerre, T.; Malfait, T.; et al. HIV Rebound Is Predominantly Fueled by Genetically Identical Viral Expansions from Diverse Reservoirs. Cell Host Microbe 2019, 26, 347–358.e7. [Google Scholar] [CrossRef] [Green Version]
- Kearney, M.F.; Wiegand, A.; Shao, W.; Coffin, J.M.; Mellors, J.W.; Lederman, M.; Gandhi, R.T.; Keele, B.F.; Li, J.Z. Origin of Rebound Plasma HIV Includes Cells with Identical Proviruses That Are Transcriptionally Active before Stopping of Antiretroviral Therapy. J. Virol. 2016, 90, 1369–1376. [Google Scholar] [CrossRef] [Green Version]
- Fehér, C.; Leal, L.; Plana, M.; Climent, N.; Guardo, A.C.; Martínez, E.; Castro, P.; Díaz-Brito, V.; Mothe, B.; De Quirós, J.C.L.B.; et al. Virological Outcome Measures During Analytical Treatment Interruptions in Chronic HIV-1-Infected Patients. Open Forum Infect. Dis. 2019, 6, ofz485. [Google Scholar] [CrossRef]
- Fajnzylber, J.; Sharaf, R.; Hutchinson, J.N.; Aga, E.; Bosch, R.J.; Hartogensis, W.; Jacobson, J.M.; Connick, E.; Volberding, P.; Skiest, D.J.; et al. Frequency of Post Treatment Control Varies by ART Restart and Viral Load Criteria. AIDS 2021, 35, 2225–2227. [Google Scholar] [CrossRef]
- Li, J.Z.; Etemad, B.; Ahmed, H.; Aga, E.; Bosch, R.J.; Mellors, J.W.; Kuritzkes, D.R.; Lederman, M.M.; Para, M.; Gandhi, R.T. The size of the expressed HIV reservoir predicts timing of viral rebound after treatment interruption. AIDS 2015, 30, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Bar, K.J.; Sneller, M.C.; Harrison, L.J.; Justement, J.S.; Overton, E.T.; Petrone, M.E.; Salantes, D.B.; Seamon, C.A.; Scheinfeld, B.; Kwan, R.W.; et al. Effect of HIV Antibody VRC01 on Viral Rebound after Treatment Interruption. N. Engl. J. Med. 2016, 375, 2037–2050. [Google Scholar] [CrossRef]
- Li, J.Z.; Aga, E.; Bosch, R.J.; Pilkinton, M.; Kroon, E.; MacLaren, L.; Keefer, M.; Fox, L.; Barr, L.; Acosta, E.; et al. Time to Viral Rebound After Interruption of Modern Antiretroviral Therapies. Clin. Infect. Dis. 2021, ciab541. [Google Scholar] [CrossRef] [PubMed]
- Clarridge, K.E.; Blazkova, J.; Einkauf, K.; Petrone, M.; Refsland, E.W.; Justement, J.S.; Shi, V.; Huiting, E.D.; Seamon, C.A.; Lee, G.Q.; et al. Effect of analytical treatment interruption and reinitiation of antiretroviral therapy on HIV reservoirs and immunologic parameters in infected individuals. PLoS Pathog. 2018, 14, e1006792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Simonetti, F.R.; Ho, Y.-C. The forces driving clonal expansion of the HIV-1 latent reservoir. Virol. J. 2020, 17, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzon, M.J.; Martin-Gayo, E.; Pereyra, F.; Ouyang, Z.; Sun, H.; Li, J.Z.; Piovoso, M.; Shaw, A.; Dalmau, J.; Zangger, N.; et al. Long-Term Antiretroviral Treatment Initiated at Primary HIV-1 Infection Affects the Size, Composition, and Decay Kinetics of the Reservoir of HIV-1-Infected CD4 T Cells. J. Virol. 2014, 88, 10056–10065. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Broncano, P.; Maddali, S.; Einkauf, K.B.; Jiang, C.; Gao, C.; Chevalier, J.; Chowdhury, F.Z.; Maswabi, K.; Ajibola, G.; Moyo, S.; et al. Early antiretroviral therapy in neonates with HIV-1 infection restricts viral reservoir size and induces a distinct innate immune profile. Sci. Transl. Med. 2019, 11, eaax7350. [Google Scholar] [CrossRef] [PubMed]
- Woldemeskel, B.A.; Kwaa, A.K.; Blankson, J.N. Viral reservoirs in elite controllers of HIV-1 infection: Implications for HIV cure strategies. EBioMedicine 2020, 62, 103118. [Google Scholar] [CrossRef] [PubMed]
- Besson, G.J.; Lalama, C.M.; Bosch, R.J.; Gandhi, R.T.; Bedison, M.A.; Aga, E.; Riddler, S.A.; McMahon, D.K.; Hong, F.; Mellors, J.W. HIV-1 DNA Decay Dynamics in Blood During More Than a Decade of Suppressive Antiretroviral Therapy. Clin. Infect. Dis. 2014, 59, 1312–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vider-Shalit, T.; Almani, M.; Sarid, R.; Louzoun, Y. The HIV hide and seek game: An immunogenomic analysis of the HIV epitope repertoire. AIDS 2009, 23, 1311–1318. [Google Scholar] [CrossRef]
- Garcia-Bates, T.M.; Palma, M.L.; Anderko, R.R.; Hsu, D.C.; Ananworanich, J.; Korber, B.T.; Gaiha, G.D.; Phanuphak, N.; Thomas, R.; Tovanabutra, S.; et al. Dendritic cells focus CTL responses toward highly conserved and topologically important HIV-1 epitopes. EBioMedicine 2021, 63, 103175. [Google Scholar] [CrossRef] [PubMed]
- Murakoshi, H.; Zou, C.; Kuse, N.; Akahoshi, T.; Chikata, T.; Gatanaga, H.; Oka, S.; Hanke, T.; Takiguchi, M. CD8+ T cells specific for conserved, cross-reactive Gag epitopes with strong ability to suppress HIV-1 replication. Retrovirology 2018, 15, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graf, E.H.; Pace, M.; Peterson, B.A.; Lynch, L.J.; Chukwulebe, S.B.; Mexas, A.M.; Shaheen, F.; Martin, J.N.; Deeks, S.G.; Connors, M.; et al. Gag-Positive Reservoir Cells Are Susceptible to HIV-Specific Cytotoxic T Lymphocyte Mediated Clearance In Vitro and Can Be Detected In Vivo. PLoS ONE 2013, 8, e71879. [Google Scholar] [CrossRef]
- Coffin, J.M.; Wells, D.W.; Zerbato, J.M.; Kuruc, J.D.; Guo, S.; Luke, B.T.; Eron, J.J.; Bale, M.; Spindler, J.; Simonetti, F.R.; et al. Clones of infected cells arise early in HIV-infected individuals. JCI Insight 2019, 4, e128432. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.H.; Coffin, J.M. What Integration Sites Tell Us about HIV Persistence. Cell Host Microbe 2016, 19, 588–598. [Google Scholar] [CrossRef] [Green Version]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, A.F.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.-R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef]
- Wang, Z.; Gurule, E.E.; Brennan, T.P.; Gerold, J.M.; Kwon, K.J.; Hosmane, N.N.; Kumar, M.; Beg, S.A.; Capoferri, A.A.; Ray, S.C.; et al. Expanded cellular clones carrying replication-competent HIV-1 persist, wax, and wane. Proc. Natl. Acad. Sci. USA 2018, 115, E2575–E2584. [Google Scholar] [CrossRef] [Green Version]
- Simonetti, F.R.; Zhang, H.; Soroosh, G.P.; Duan, J.; Rhodehouse, K.; Hill, A.L.; Beg, S.A.; McCormick, K.; Raymond, H.E.; Nobles, C.L.; et al. Antigen-driven clonal selection shapes the persistence of HIV-1–infected CD4+ T cells in vivo. J. Clin. Investig. 2021, 131, e145254. [Google Scholar] [CrossRef]
- Mendoza, P.; Jackson, J.R.; Oliveira, T.Y.; Gaebler, C.; Ramos, V.; Caskey, M.; Jankovic, M.; Nussenzweig, M.C.; Cohn, L.B. Antigen-responsive CD4+ T cell clones contribute to the HIV-1 latent reservoir. J. Exp. Med. 2020, 217, e20200051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferdin, J.; Goričar, K.; Dolžan, V.; Plemenitaš, A.; Martin, J.N.; Peterlin, B.M.; Deeks, S.; Lenassi, M. Viral protein Nef is detected in plasma of half of HIV-infected adults with undetectable plasma HIV RNA. PLoS ONE 2018, 13, e0191613. [Google Scholar] [CrossRef]
- Imamichi, H.; Smith, M.; Adelsberger, J.W.; Izumi, T.; Scrimieri, F.; Sherman, B.T.; Rehm, C.A.; Imamichi, T.; Pau, A.; Catalfamo, M.; et al. Defective HIV-1 proviruses produce viral proteins. Proc. Natl. Acad. Sci. USA 2020, 117, 3704–3710. [Google Scholar] [CrossRef] [Green Version]
- Grund, B.; Baker, J.V.; Deeks, S.; Wolfson, J.; Wentworth, D.; Cozzi-Lepri, A.; Cohen, C.J.; Phillips, A.; Lundgren, J.D.; Neaton, J.D.; et al. Relevance of Interleukin-6 and D-Dimer for Serious Non-AIDS Morbidity and Death among HIV-Positive Adults on Suppressive Antiretroviral Therapy. PLoS ONE 2016, 11, e0155100. [Google Scholar] [CrossRef]
- Sandler, N.G.; Wand, H.; Roque, A.; Law, M.; Nason, M.C.; Nixon, D.E.; Pedersen, C.; Ruxrungtham, K.; Lewin, S.R.; Emery, S.; et al. Plasma Levels of Soluble CD14 Independently Predict Mortality in HIV Infection. J. Infect. Dis. 2011, 203, 780–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasi, M.; De Biasi, S.; Gibellini, L.; Bianchini, E.; Pecorini, S.; Bacca, V.; Guaraldi, G.; Mussini, C.; Pinti, M.; Cossarizza, A. Ageing and inflammation in patients with HIV infection. Clin. Exp. Immunol. 2016, 187, 44–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marconi, V.C.; Moser, C.; Gavegnano, C.; Deeks, S.G.; Lederman, M.M.; Overton, E.T.; Tsibris, A.; Hunt, P.W.; Kantor, A.; Sekaly, R.-P.; et al. Randomized Trial of Ruxolitinib in Antiretroviral-Treated Adults with HIV. Clin. Infect. Dis. 2021, ciab212. [Google Scholar] [CrossRef] [PubMed]
- Adan, M.; Lau, C.-Y.; Dewar, R.; Higgins, J.; Rehm, C.; Ganesan, A.; McMahon, D.; Anderson, E.; Maldarelli, F. Role of Host Immune Activation in Enrichment in Proportion of Deleted Proviruses During ART. In Proceedings of the Retroviruses at Cold Spring Harbor Laboratory, Laurel Hollow, NY, USA, 22–27 May 2017. [Google Scholar]
- Katlama, C.; Lambert-Niclot, S.; Assoumou, L.; Papagno, L.; Lecardonnel, F.; Zoorob, R.; Tambussi, G.; Clotet, B.; Youle, M.; Achenbach, C.; et al. Treatment intensification followed by interleukin-7 reactivates HIV without reducing total HIV DNA: A randomized trial. AIDS 2016, 30, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Vandergeeten, C.; Fromentin, R.; DaFonseca, S.; Lawani, M.B.; Sereti, I.; Lederman, M.M.; Ramgopal, M.; Routy, J.-P.; Sékaly, R.-P.; Chomont, N. Interleukin-7 promotes HIV persistence during antiretroviral therapy. Blood 2013, 121, 4321–4329. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Purton, J.F.; Surh, C.D.; Sprent, J. Cytokines and T-cell homeostasis. Curr. Opin. Immunol. 2007, 19, 320–326. [Google Scholar] [CrossRef]
- Bosque, A.; Famiglietti, M.; Weyrich, A.S.; Goulston, C.; Planelles, V. Homeostatic Proliferation Fails to Efficiently Reactivate HIV-1 Latently Infected Central Memory CD4+ T Cells. PLoS Pathog. 2011, 7, e1002288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosinger, S.E.; Sodora, D.L.; Silvestri, G. Generalized immune activation and innate immune responses in simian immunodeficiency virus infection. Curr. Opin. HIV AIDS 2011, 6, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Klatt, N.R.; Chomont, N.; Douek, D.C.; Deeks, S.G. Immune activation and HIV persistence: Implications for curative approaches to HIV infection. Immunol. Rev. 2013, 254, 326–342. [Google Scholar] [CrossRef] [Green Version]
- Bushman, F.D. Retroviral Insertional Mutagenesis in Humans: Evidence for Four Genetic Mechanisms Promoting Expansion of Cell Clones. Mol. Ther. 2020, 28, 352–356. [Google Scholar] [CrossRef]
- Stevenson, E.M.; Ward, A.R.; Truong, R.; Thomas, A.S.; Huang, S.-H.; Dilling, T.R.; Terry, S.; Bui, J.K.; Mota, T.M.; Danesh, A.; et al. HIV-specific T cell responses reflect substantive in vivo interactions with antigen despite long-term therapy. JCI Insight 2021, 6, e142640. [Google Scholar] [CrossRef] [PubMed]
- Zaunders, J.; Munier, C.M.L.; McGuire, H.M.; Law, H.; Howe, A.; Xu, Y.; Groth, B.F.D.S.; Schofield, P.; Christ, D.; Milner, B.; et al. Mapping the extent of heterogeneity of human CCR5+ CD4+ T cells in peripheral blood and lymph nodes. AIDS 2020, 34, 833–848. [Google Scholar] [CrossRef]
- Leong, Y.A.; Atnerkar, A.; Yu, D. Human Immunodeficiency Virus Playing Hide-and-Seek: Understanding the TFH Cell Reservoir and Proposing Strategies to Overcome the Follicle Sanctuary. Front. Immunol. 2017, 8, 622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodham, A.W.; Skeate, J.G.; Sanna, A.M.; Taylor, J.R.; Da Silva, D.M.; Cannon, P.M.; Kast, W.M. Human Immunodeficiency Virus Immune Cell Receptors, Coreceptors, and Cofactors: Implications for Prevention and Treatment. AIDS Patient Care STDs 2016, 30, 291–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppensteiner, H.; Brack-Werner, R.; Schindler, M. Macrophages and their relevance in Human Immunodeficiency Virus Type I infection. Retrovirology 2012, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppensteiner, H.; Banning, C.; Schneider, C.; Hohenberg, H.; Schindler, M. Macrophage Internal HIV-1 Is Protected from Neutralizing Antibodies. J. Virol. 2011, 86, 2826–2836. [Google Scholar] [CrossRef] [Green Version]
- Martin-Gayo, E.; Yu, X.G. Role of Dendritic Cells in Natural Immune Control of HIV-1 Infection. Front. Immunol. 2019, 10, 1306. [Google Scholar] [CrossRef] [PubMed]
- Preza, G.C.; Tanner, K.; Elliott, J.; Yang, O.O.; Anton, P.A.; Ochoa, M.-T. Antigen-Presenting Cell Candidates for HIV-1 Transmission in Human Distal Colonic Mucosa Defined by CD207 Dendritic Cells and CD209 Macrophages. AIDS Res. Hum. Retroviruses 2014, 30, 241–249. [Google Scholar] [CrossRef] [Green Version]
- De Witte, L.; Nabatov, A.; Pion, M.; Fluitsma, D.; Jong, M.A.W.P.D.; De Gruijl, T.; Piguet, V.; Van Kooyk, Y.; Geijtenbeek, T.B.H. Langerin is a natural barrier to HIV-1 transmission by Langerhans cells. Nat. Med. 2007, 13, 367–371. [Google Scholar] [CrossRef] [PubMed]
- West, H.C.; Bennett, C.L. Redefining the Role of Langerhans Cells as Immune Regulators within the Skin. Front. Immunol. 2018, 8, 1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayr, L.; Su, B.; Moog, C. Langerhans Cells: The ‘Yin and Yang’ of HIV Restriction and Transmission. Trends Microbiol. 2017, 25, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.M.; Sarrami-Forooshani, R.; Geijtenbeek, T.B. HIV-1 border patrols: Langerhans cells control antiviral responses and viral transmission. Futur. Virol. 2015, 10, 1231–1243. [Google Scholar] [CrossRef]
- Wacleche, V.S.; Tremblay, C.L.; Routy, J.-P.; Ancuta, P. The Biology of Monocytes and Dendritic Cells: Contribution to HIV Pathogenesis. Viruses 2018, 10, 65. [Google Scholar] [CrossRef] [Green Version]
- Cavrois, M.; Neidleman, J.; Kreisberg, J.; Greene, W.C. In Vitro Derived Dendritic Cells trans-Infect CD4 T Cells Primarily with Surface-Bound HIV-1 Virions. PLoS Pathog. 2007, 3, e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappocciolo, G.; Sluis-Cremer, N.; Rinaldo, C.R. Efficient HIV-1 Trans Infection of CD4+ T Cells Occurs in the Presence of Antiretroviral Therapy. In Open Forum Infectious Diseases; Oxford University Press: Oxford, UK, 2019; p. ofz253. [Google Scholar] [CrossRef]
- Wu, L.; KewalRamani, V.N. Dendritic-cell interactions with HIV: Infection and viral dissemination. Nat. Rev. Immunol. 2006, 6, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Jiang, A.-P.; Jiang, J.-F.; Guo, M.-G.; Jin, Y.-M.; Li, Y.-Y.; Wang, J.-H. Human Blood-Circulating Basophils Capture HIV-1 and Mediate Viral trans -Infection of CD4 + T Cells. J. Virol. 2015, 89, 8050–8062. [Google Scholar] [CrossRef] [Green Version]
- Jiang, A.-P.; Jiang, J.-F.; Wei, J.-F.; Guo, M.-G.; Qin, Y.; Guo, Q.-Q.; Ma, L.; Liu, B.-C.; Wang, X.; Veazey, R.S.; et al. Human Mucosal Mast Cells Capture HIV-1 and Mediate Viral trans -Infection of CD4 + T Cells. J. Virol. 2016, 90, 2928–2937. [Google Scholar] [CrossRef] [Green Version]
- DeLucia, D.C.; Rinaldo, C.R.; Rappocciolo, G. Inefficient HIV-1 trans Infection of CD4 + T Cells by Macrophages from HIV-1 Nonprogressors Is Associated with Altered Membrane Cholesterol and DC-SIGN. J. Virol. 2018, 92, e00092-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, P.; Prevedel, L.; Eugenin, E.A. HIV-infected macrophages and microglia that survive acute infection become viral reservoirs by a mechanism involving Bim. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Herskovitz, J.; Gendelman, H.E. HIV and the Macrophage: From Cell Reservoirs to Drug Delivery to Viral Eradication. J. Neuroimmune Pharmacol. 2018, 14, 52–67. [Google Scholar] [CrossRef]
- Hertoghs, N.; Nijmeijer, B.M.; Van Teijlingen, N.H.; Fenton-May, A.E.; Kaptein, T.M.; Van Hamme, J.L.; Kappes, J.C.; Kootstra, N.A.; Hahn, B.H.; Borrow, P.; et al. Sexually transmitted founder HIV-1 viruses are relatively resistant to Langerhans cell-mediated restriction. PLoS ONE 2019, 14, e0226651. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Yukl, S.A. Tissue reservoirs of HIV. Curr Opin HIV AIDS. 2016, 11, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Chaillon, A.; Gianella, S.; Dellicour, S.; Rawlings, S.A.; Schlub, T.E.; de Oliveira, M.F.; Ignacio, C.; Porrachia, M.; Vrancken, B.; Smith, D.M. HIV persists throughout deep tissues with repopulation from multiple anatomical sources. J. Clin. Investig. 2020, 130, 1699–1712. [Google Scholar] [CrossRef] [Green Version]
- Spudich, S.; González-Scarano, F. HIV-1-Related Central Nervous System Disease: Current Issues in Pathogenesis, Diagnosis, and Treatment. Cold Spring Harb. Perspect. Med. 2012, 2, a007120. [Google Scholar] [CrossRef] [Green Version]
- Dahl, V.; Peterson, J.; Spudich, S.; Lee, E.; Shacklett, B.L.; Price, R.W.; Palmer, S. Single-copy assay quantification of HIV-1 RNA in paired cerebrospinal fluid and plasma samples from elite controllers. AIDS 2013, 27, 1145–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winston, A.; Antinori, A.; Cinque, P.; Fox, H.S.; Gisslen, M.; Henrich, T.J.; Letendre, S.; Persaud, D.; Price, R.W.; Spudich, S. Defining cerebrospinal fluid HIV RNA escape. AIDS 2019, 33, S107–S111. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; Kincer, L.P.; Bowman, N.M.; Evans, C.; Vinikoor, M.J.; Lippincott, C.K.; Gisslén, M.; Spudich, S.; Menezes, P.; Robertson, K.; et al. Human Immunodeficiency Virus Type 1 RNA Detected in the Central Nervous System (CNS) After Years of Suppressive Antiretroviral Therapy Can Originate from a Replicating CNS Reservoir or Clonally Expanded Cells. Clin. Infect. Dis. 2018, 69, 1345–1352. [Google Scholar] [CrossRef]
- Joseph, S.B.; Arrildt, K.T.; Sturdevant, C.B.; Swanstrom, R. HIV-1 target cells in the CNS. J. Neurovirol. 2014, 21, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Schnell, G.; Joseph, S.; Spudich, S.; Price, R.W.; Swanstrom, R. HIV-1 Replication in the Central Nervous System Occurs in Two Distinct Cell Types. PLoS Pathog. 2011, 7, e1002286. [Google Scholar] [CrossRef] [Green Version]
- Sturdevant, C.B.; Joseph, S.B.; Schnell, G.; Price, R.W.; Swanstrom, R.; Spudich, S. Compartmentalized Replication of R5 T Cell-Tropic HIV-1 in the Central Nervous System Early in the Course of Infection. PLoS Pathog. 2015, 11, e1004720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutgen, V.; Narasipura, S.D.; Barbian, H.J.; Richards, M.; Wallace, J.; Razmpour, R.; Buzhdygan, T.; Ramirez, S.; Prevedel, L.; Eugenin, E.A.; et al. HIV infects astrocytes in vivo and egresses from the brain to the periphery. PLoS Pathog. 2020, 16, e1008381. [Google Scholar] [CrossRef] [PubMed]
- Maldarelli, F. The gift of a lifetime: Analysis of HIV at autopsy. J. Clin. Investig. 2020, 130, 1611–1614. [Google Scholar] [CrossRef]
- Eggers, C.; Arendt, G.; Hahn, K.; Husstedt, I.W.; Maschke, M.; Neuen-Jacob, E.; Obermann, M.; Rosenkranz, T.; Schielke, E.; For the German Association of Neuro-AIDS und Neuro-Infectiology (DGNANI); et al. HIV-1-associated neurocognitive disorder: Epidemiology, pathogenesis, diagnosis, and treatment. J. Neurol. 2017, 264, 1715–1727. [Google Scholar] [CrossRef] [PubMed]
- Hellmuth, J.; Valcour, V.; Spudich, S. CNS reservoirs for HIV: Implications for eradication. J. Virus Erad. 2015, 1, 67–71. [Google Scholar] [CrossRef]
- Henderson, L.J.; Johnson, T.P.; Smith, B.R.; Reoma, L.; Santamaria, U.A.; Bachani, M.; DeMarino, C.; Barclay, R.A.; Snow, J.; Sacktor, N.; et al. Presence of Tat and transactivation response element in spinal fluid despite antiretroviral therapy. AIDS 2019, 33, S145–S157. [Google Scholar] [CrossRef]
- Marino, J.; Maubert, M.E.; Mele, A.R.; Spector, C.; Wigdahl, B.; Nonnemacher, M.R. Functional impact of HIV-1 Tat on cells of the CNS and its role in HAND. Cell. Mol. Life Sci. 2020, 77, 5079–5099. [Google Scholar] [CrossRef]
- Marino, J.; Wigdahl, B.; Nonnemacher, M.R. Extracellular HIV-1 Tat Mediates Increased Glutamate in the CNS Leading to Onset of Senescence and Progression of HAND. Front. Aging Neurosci. 2020, 12, 168. [Google Scholar] [CrossRef]
- Cowley, D.; Gray, L.R.; Wesselingh, S.L.; Gorry, P.R.; Churchill, M.J. Genetic and functional heterogeneity of CNS-derived tat alleles from patients with HIV-associated dementia. J. NeuroVirology 2010, 17, 70–81. [Google Scholar] [CrossRef]
- Kearney, M.F.; Wiegand, A.; Shao, W.; McManus, W.; Bale, M.; Luke, B.; Maldarelli, F.; Mellors, J.W.; Coffin, J.M. Ongoing HIV Replication During ART Reconsidered. Open Forum Infect. Dis. 2017, 4, ofx173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boritz, E.A.; Darko, S.; Swaszek, L.; Wolf, G.; Wells, D.; Wu, X.; Henry, A.R.; Laboune, F.; Hu, J.; Ambrozak, D.; et al. Multiple Origins of Virus Persistence during Natural Control of HIV Infection. Cell 2016, 166, 1004–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guadalupe, M.; Reay, E.; Sankaran, S.; Prindiville, T.; Flamm, J.; McNeil, A.; Dandekar, S. Severe CD4 + T-Cell Depletion in Gut Lymphoid Tissue during Primary Human Immunodeficiency Virus Type 1 Infection and Substantial Delay in Restoration following Highly Active Antiretroviral Therapy. J. Virol. 2003, 77, 11708–11717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenchley, J.M.; Douek, D.C. HIV infection and the gastrointestinal immune system. Mucosal Immunol. 2007, 1, 23–30. [Google Scholar] [CrossRef]
- Macal, M.; Sankaran, S.; Chun, T.-W.; Reay, E.; Flamm, J.; Prindiville, T.J.; Dandekar, S. Effective CD4+ T-cell restoration in gut-associated lymphoid tissue of HIV-infected patients is associated with enhanced Th17 cells and polyfunctional HIV-specific T-cell responses. Mucosal Immunol. 2008, 1, 475–488. [Google Scholar] [CrossRef]
- Imamichi, H.; DeGray, G.; Dewar, R.L.; Mannon, P.; Yao, M.; Chairez, C.; Sereti, I.; Kovacs, J.A. Lack of Compartmentalization of HIV-1 Quasispecies Between the Gut and Peripheral Blood Compartments. J. Infect. Dis. 2011, 204, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moron-Lopez, S.; Puertas, M.C.; Gálvez, C.; Navarro, J.; Carrasco, A.; Esteve, M.; Manyé, J.; Crespo, M.; Salgado, M.; Martinez-Picado, J. Sensitive quantification of the HIV-1 reservoir in gut-associated lymphoid tissue. PLoS ONE 2017, 12, e0175899. [Google Scholar] [CrossRef] [Green Version]
- Bull, M.E.; McKernan, J.L.; Styrchak, S.; Kraft, K.; Hitti, J.; Cohn, S.E.; Tapia, K.; Deng, W.; Holte, S.; Mullins, J.I.; et al. Phylogenetic Analyses Comparing HIV Sequences from Plasma at Virologic Failure to Cervix Versus Blood Sequences from Antecedent Antiretroviral Therapy Suppression. AIDS Res. Hum. Retroviruses 2019, 35, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Mabvakure, B.M.; Lambson, B.E.; Ramdayal, K.; Masson, L.; Kitchin, D.; Allam, M.; Karim, S.A.; Williamson, C.; Passmore, J.-A.; Martin, D.P.; et al. Evidence for both Intermittent and Persistent Compartmentalization of HIV-1 in the Female Genital Tract. J. Virol. 2019, 93, e00311-19. [Google Scholar] [CrossRef] [Green Version]
- Bull, M.; Learn, G.; Genowati, I.; McKernan, J.; Hitti, J.; Lockhart, D.; Tapia, K.; Holte, S.; Dragavon, J.; Coombs, R.; et al. Compartmentalization of HIV-1 within the Female Genital Tract Is Due to Monotypic and Low-Diversity Variants Not Distinct Viral Populations. PLoS ONE 2009, 4, e7122. [Google Scholar] [CrossRef] [Green Version]
- Klein, K.; Nickel, G.; Nankya, I.; Kyeyune, F.; Demers, K.; Ndashimye, E.; Kwok, C.; Chen, P.-L.; Rwambuya, S.; Poon, A.; et al. Higher sequence diversity in the vaginal tract than in blood at early HIV-1 infection. PLoS Pathog. 2018, 14, e1006754. [Google Scholar] [CrossRef] [Green Version]
- Kariuki, S.M.; Selhorst, P.; Anthony, C.; Matten, D.; Abrahams, M.-R.; Martin, D.P.; Ariën, K.K.; Rebe, K.; Williamson, C.; Dorfman, J.R. Compartmentalization and Clonal Amplification of HIV-1 in the Male Genital Tract Characterized Using Next-Generation Sequencing. J. Virol. 2020, 94, e00229-20. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, A.; Wasserman, S.S.; Burns, D.; Wright, D.J.; Cohn, J.; Landay, A.; Weber, K.; Cohen, M.; Levine, A.; Minkoff, H.; et al. Determinants of HIV-1 shedding in the genital tract of women. Lancet 2001, 358, 1593–1601. [Google Scholar] [CrossRef]
- Zaikos, T.D.; Terry, V.H.; Kettinger, N.T.S.; Lubow, J.; Painter, M.M.; Virgilio, M.; Neevel, A.; Taschuk, F.; Onafuwa-Nuga, A.; McNamara, L.A.; et al. Hematopoietic Stem and Progenitor Cells Are a Distinct HIV Reservoir that Contributes to Persistent Viremia in Suppressed Patients. Cell Rep. 2018, 25, 3759–3773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNamara, L.; Collins, K.L. Hematopoietic stem/precursor cells as HIV reservoirs. Curr. Opin. HIV AIDS 2011, 6, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, N.T.; Zaikos, T.D.; Terry, V.; Taschuk, F.; McNamara, L.A.; Onafuwa-Nuga, A.; Yucha, R.; Signer, R.A.J.; Iv, J.R.; Bixby, D.; et al. CD4 is expressed on a heterogeneous subset of hematopoietic progenitors, which persistently harbor CXCR4 and CCR5-tropic HIV proviral genomes in vivo. PLoS Pathog. 2017, 13, e1006509. [Google Scholar] [CrossRef] [Green Version]
- Durand, C.M.; Ghiaur, G.; Siliciano, J.D.; Rabi, S.A.; Eisele, E.E.; Salgado, M.; Shan, L.; Lai, J.F.; Zhang, H.; Margolick, J.; et al. HIV-1 DNA Is Detected in Bone Marrow Populations Containing CD4+ T Cells but Is not Found in Purified CD34+ Hematopoietic Progenitor Cells in Most Patients on Antiretroviral Therapy. J. Infect. Dis. 2012, 205, 1014–1018. [Google Scholar] [CrossRef]
- Alexaki, A.; Wigdahl, B. HIV-1 Infection of Bone Marrow Hematopoietic Progenitor Cells and Their Role in Trafficking and Viral Dissemination. PLoS Pathog. 2008, 4, e1000215. [Google Scholar] [CrossRef]
- Henrich, T.J.; Beckford-Vera, D.; Martinez-Ortiz, E.; Aslam, M.; Thanh, C.; Kumar, S.; Wu, I.-W.; Hoh, R.; Flavell, R.; Seo, Y.; et al. Whole-body PET imaging of the HIV reservoir using radiolabeled VRC01. In Proceedings of the Conference on Retroviruses and Opportunistic Infections, Boston, MA, USA, 8–11 March 2020. [Google Scholar]
- Aamer, H.A.; McClure, J.; Ko, D.; Maenza, J.; Collier, A.C.; Coombs, R.W.; Mullins, J.I.; Frenkel, L.M. Cells producing residual viremia during antiretroviral treatment appear to contribute to rebound viremia following interruption of treatment. PLoS Pathog. 2020, 16, e1008791. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Ruggiero, A.; Paxton, W.A.; Pollakis, G. Measuring the Success of HIV-1 Cure Strategies. Front. Cell. Infect. Microbiol. 2020, 10, 134. [Google Scholar] [CrossRef]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.-C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F. Stimulation of HIV-1-Specific Cytolytic T Lymphocytes Facilitates Elimination of Latent Viral Reservoir after Virus Reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Maina, E.K.; Adan, A.A.; Mureithi, H.; Muriuki, J.; Lwembe, R.M. A Review of Current Strategies Towards the Elimination of Latent HIV-1 and Subsequent HIV-1 Cure. Curr. HIV Res. 2021, 19, 14–26. [Google Scholar] [CrossRef]
- McBrien, J.B.; Mavigner, M.; Franchitti, L.; Smith, S.A.; White, E.; Tharp, G.K.; Walum, H.; Busman-Sahay, K.; Aguilera-Sandoval, C.R.; Thayer, W.O.; et al. Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8+ cells. Nature 2020, 578, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Nixon, C.C.; Mavigner, M.; Sampey, G.C.; Brooks, A.D.; Spagnuolo, R.A.; Irlbeck, D.M.; Mattingly, C.; Ho, P.T.; Schoof, N.; Cammon, C.G.; et al. Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nature 2020, 578, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Abner, E.; Jordan, A. HIV "shock and kill" therapy: In need of revision. Antiviral Res. 2019, 166, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Lichterfeld, M. Reactivation of latent HIV moves shock-and-kill treatments forward. Nature 2020, 578, 42–43. [Google Scholar] [CrossRef] [PubMed]
- Clutton, G.T.; Jones, R.B. Diverse Impacts of HIV Latency-Reversing Agents on CD8+ T-Cell Function: Implications for HIV Cure. Front. Immunol. 2018, 9, 1452. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; De Crignis, E.; Rokx, C.; Verbon, A.; van Gelder, T.; Mahmoudi, T.; Katsikis, P.D.; Mueller, Y.M. T cell toxicity of HIV latency reversing agents. Pharmacol. Res. 2018, 139, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.B.; O’Connor, R.; Mueller, S.; Foley, M.H.; Szeto, G.; Karel, D.; Lichterfeld, M.; Kovacs, C.; Ostrowski, M.A.; Trocha, A.; et al. Histone Deacetylase Inhibitors Impair the Elimination of HIV-Infected Cells by Cytotoxic T-Lymphocytes. PLoS Pathog. 2014, 10, e1004287. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef] [Green Version]
- Sarabia, I.; Bosque, A. HIV-1 Latency and Latency Reversal: Does Subtype Matter? Viruses 2019, 11, 1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, H.; Schorn, A.J. Endogenous Retroviruses Walk a Fine Line between Priming and Silencing. Viruses 2020, 12, 792. [Google Scholar] [CrossRef] [PubMed]
- Ahlenstiel, C.L.; Symonds, G.; Kent, S.J.; Kelleher, A.D. Block and Lock HIV Cure Strategies to Control the Latent Reservoir. Front. Cell. Infect. Microbiol. 2020, 10, 424. [Google Scholar] [CrossRef] [PubMed]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.; Marchand, D.; Bardiot, D.; Van Der Veken, N.J.; Van Remoortel, B.; Strelkov, S.V.; et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Debyser, Z.; VanSant, G.; Bruggemans, A.; Janssens, J.; Christ, F. Insight in HIV Integration Site Selection Provides a Block-and-Lock Strategy for a Functional Cure of HIV Infection. Viruses 2018, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- Orphanides, G.; LeRoy, G.; Chang, C.-H.; Luse, D.S.; Reinberg, D. FACT, a Factor that Facilitates Transcript Elongation through Nucleosomes. Cell 1998, 92, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.-W.; Valieva, M.E.; Safina, A.; Chereji, R.V.; Wang, J.; Kulaeva, O.I.; Morozov, A.V.; Kirpichnikov, M.P.; Feofanov, A.V.; Gurova, K.V.; et al. Mechanism of FACT removal from transcribed genes by anticancer drugs curaxins. Sci. Adv. 2018, 4, eaav2131. [Google Scholar] [CrossRef] [Green Version]
- Joshi, P.; Stoddart, C.A. Impaired Infectivity of Ritonavir-resistant HIV Is Rescued by Heat Shock Protein 90AB1. J. Biol. Chem. 2011, 286, 24581–24592. [Google Scholar] [CrossRef] [Green Version]
- Gavegnano, C.; Detorio, M.; Montero, C.; Bosque, A.; Planelles, V.; Schinazi, R.F. Ruxolitinib and Tofacitinib Are Potent and Selective Inhibitors of HIV-1 Replication and Virus Reactivation In Vitro. Antimicrob. Agents Chemother. 2014, 58, 1977–1986. [Google Scholar] [CrossRef] [Green Version]
- Noël, N.; Jacquelin, B.; Huot, N.; Goujard, C.; Lambotte, O.; Müller-Trutwin, M. Interferon-associated therapies toward HIV control: The back and forth. Cytokine Growth Factor Rev. 2018, 40, 99–112. [Google Scholar] [CrossRef]
- Utay, N.S.; Douek, D.C. Interferons and HIV Infection: The Good, the Bad, and the Ugly. Pathog. Immun. 2016, 1, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papasavvas, E.; Azzoni, L.; Kossenkov, A.V.; Dawany, N.; Morales, K.H.; Fair, M.; Ross, B.N.; Lynn, K.; Mackiewicz, A.; Mounzer, K.; et al. NK Response Correlates with HIV Decrease in Pegylated IFN-α2a–Treated Antiretroviral Therapy–Suppressed Subjects. J. Immunol. 2019, 203, 705–717. [Google Scholar] [CrossRef]
- Azzoni, L.; Foulkes, A.S.; Papasavvas, E.; Mexas, A.M.; Lynn, K.M.; Mounzer, K.; Tebas, P.; Jacobson, J.M.; Frank, I.; Busch, M.P.; et al. Pegylated Interferon Alfa-2a Monotherapy Results in Suppression of HIV Type 1 Replication and Decreased Cell-Associated HIV DNA Integration. J. Infect. Dis. 2012, 207, 213–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papasavvas, E.; Azzoni, L.; Pagliuzza, M.A.; Abdel-Mohsen, M.; Ross, B.N.; Fair, M.; Howell, B.J.; Hazuda, D.J.; Chomont, N.; Li, Q.; et al. Safety, Immune, and Antiviral Effects of Pegylated Interferon Alpha 2b Administration in Antiretroviral Therapy-Suppressed Individuals: Results of Pilot Clinical Trial. AIDS Res. Hum. Retroviruses 2021, 37, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Juárez, A.; Frias, M.; Rivero, A. Current views on interferon therapy for HIV. Expert Opin. Biol. Ther. 2016, 16, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, W.; Sun, M.; Li, T. Broadly neutralizing antibodies for HIV-1: Efficacies, challenges and opportunities. Emerg. Microbes Infect. 2020, 9, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Martin, M.A. Of Mice, Macaques, and Men: Broadly Neutralizing Antibody Immunotherapy for HIV-1. Cell Host Microbe 2017, 22, 207–216. [Google Scholar] [CrossRef]
- Das, A.T.; Binda, C.S.; Berkhout, B. Elimination of infectious HIV DNA by CRISPR–Cas9. Curr. Opin. Virol. 2019, 38, 81–88. [Google Scholar] [CrossRef]
- Nerys-Junior, A.; Braga-Dias, L.P.; Pezzuto, P.; Cotta-de-Almeida, V.; Tanuri, A. Comparison of the editing patterns and editing efficiencies of TALEN and CRISPR-Cas9 when targeting the human CCR5 gene. Genet. Mol. Biol. 2018, 41, 167–179. [Google Scholar] [CrossRef]
- Tebas, P.; Jadlowsky, J.K.; Shaw, P.A.; Tian, L.; Esparza, E.; Brennan, A.L.; Kim, S.; Naing, S.Y.; Richardson, M.W.; Vogel, A.N.; et al. CCR5-edited CD4+ T cells augment HIV-specific immunity to enable post-rebound control of HIV replication. J. Clin. Investig. 2021, 131, e144486. [Google Scholar] [CrossRef]
- Sullivan, N.T.; Allen, A.G.; Atkins, A.J.; Chung, C.H.; Dampier, W.; Nonnemacher, M.R.; Wigdahl, B. Designing Safer CRISPR/Cas9 Therapeutics for HIV: Defining Factors That Regulate and Technologies Used to Detect Off-Target Editing. Front. Microbiol. 2020, 11, 1872. [Google Scholar] [CrossRef] [PubMed]
- Jilg, N.; Li, J.Z. On the Road to a HIV Cure: Moving Beyond Berlin and London. Infect. Dis. Clin. North Am. 2019, 33, 857–868. [Google Scholar] [CrossRef]
- Lopalco, L. CCR5: From Natural Resistance to a New Anti-HIV Strategy. Viruses 2010, 2, 574–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmet, K.; Van Wanzeele, F.; Demecheleer, E.; Dauwe, K.; Pelgrom, J.; Van Der Gucht, B.; Vogelaers, D.; Plum, J.; Stuyver, L.; Vandekerckhove, L.; et al. Impact of Δ32-CCR5 heterozygosity on HIV-1 genetic evolution and variability—A study of 4 individuals infected with closely related HIV-1 strains. Virology 2008, 379, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Hütter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Müssig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kücherer, C.; Blau, O.; et al. Long-Term Control of HIV byCCR5Delta32/Delta32 Stem-Cell Transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hütter, G.; Thiel, E. Allogeneic transplantation of CCR5-deficient progenitor cells in a patient with HIV infection: An update after 3 years and the search for patient no. 2. AIDS 2011, 25, 273–274. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Peppa, D.; Hill, A.L.; Gálvez, C.; Salgado, M.; Pace, M.; McCoy, E.L.; Griffith, A.S.; Thornhill, J.; Alrubayyi, A.; et al. Evidence for HIV-1 cure after CCR5Δ32/Δ32 allogeneic haemopoietic stem-cell transplantation 30 months post analytical treatment interruption: A case report. Lancet HIV 2020, 7, e340–e347. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.K.; Abdul-Jawad, S.; McCoy, L.E.; Mok, H.P.; Peppa, D.; Salgado, M.; Martinez-Picado, J.; Nijhuis, M.; Wensing, A.M.J.; Lee, H.; et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature 2019, 568, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Marban, C.; Forouzanfar, F.; Ait-Ammar, A.; Fahmi, F.; El Mekdad, H.; Daouad, F.; Rohr, O.; Schwartz, C. Targeting the Brain Reservoirs: Toward an HIV Cure. Front. Immunol. 2016, 7, 397. [Google Scholar] [CrossRef] [Green Version]
- Migueles, A.S.; Connors, M. Success and failure of the cellular immune response against HIV-1. Nat. Immunol. 2015, 16, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Etemad, B.; Esmaeilzadeh, E.; Li, J.Z. Learning from the Exceptions: HIV Remission in Post-treatment Controllers. Front. Immunol. 2019, 10, 1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalo-Gil, E.; Ikediobi, U.; Sutton, R.E. Mechanisms of Virologic Control and Clinical Characteristics of HIV+ Elite/Viremic Controllers. Yale J. Boil. Med. 2017, 90, 245–259. [Google Scholar]
- Uruena, A.; Cassetti, I.; Kashyap, N.; Deleage, C.; Estes, J.D.; Trindade, C.; Hammoud, A.D.; Burbelo, P.D.; Natarajan, V.; Dewar, R.; et al. Prolonged Posttreatment Virologic Control and Complete Seroreversion After Advanced Human Immunodeficiency Virus-1 Infection. Open Forum Infect. Dis. 2020, 8, ofaa613. [Google Scholar] [CrossRef] [PubMed]


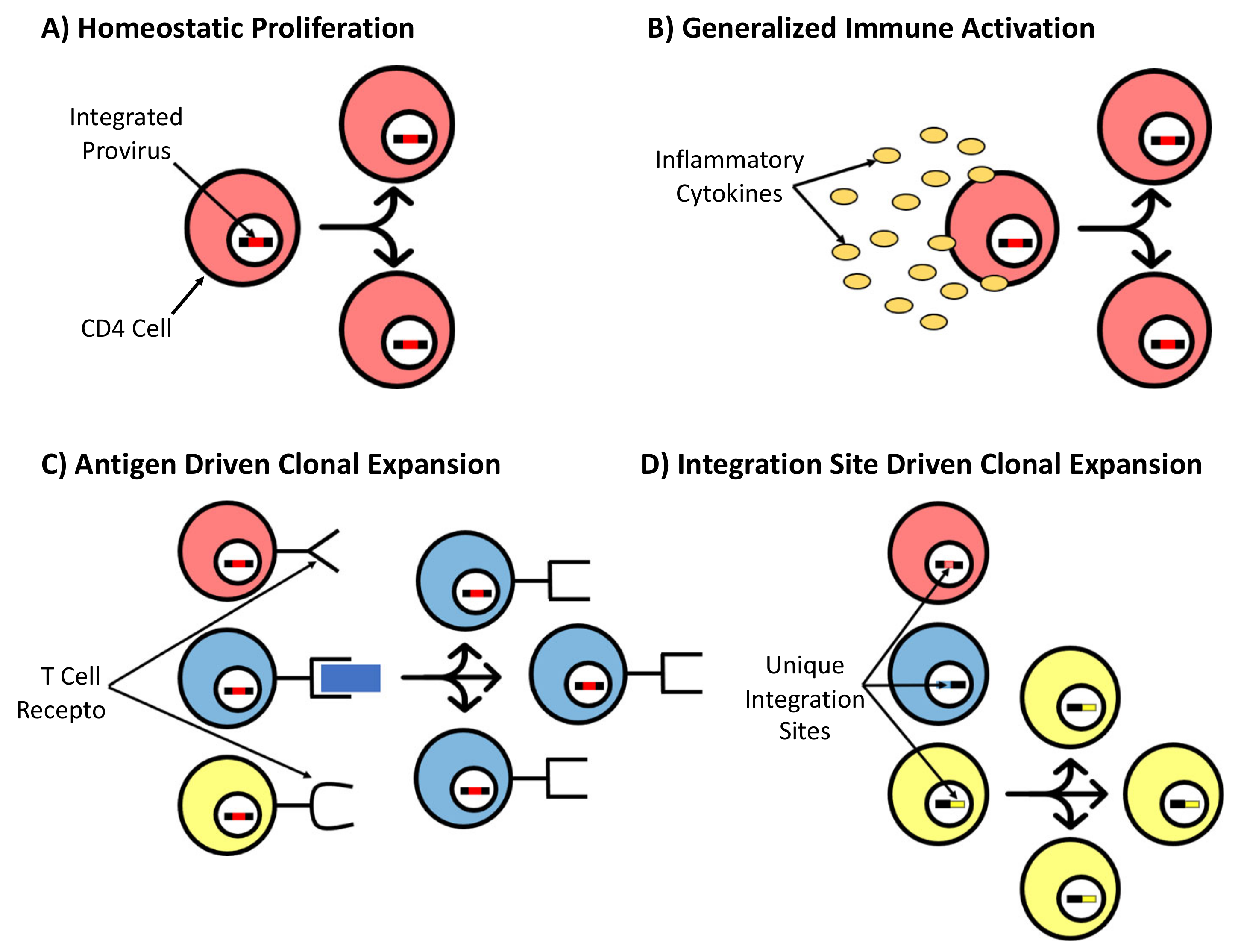



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| INSTI—1st Generation | Half-Life (T1/2) | Major Resistance Mutations | Less Common Resistance Mutations | |
|---|---|---|---|---|
| Shared | Specific | |||
| Raltegravir | ~9 h | T66K, E92Q, G118R, E138KAT, G140SAC, Q148HRK, N155H, R263K | T66AI, Y143RCH | (H51Y), (L74M), (T97A), F121Y, Y143KSGA, V151L, N155ST |
| Elvitegravir | ~13 h | T66AI, S147G | H51Y, E92VG, F121Y, P145S, Q146P, Q148N, V151LA, S153YF, N155ST | |
| INSTI—2nd generation | ||||
| Dolutegravir | ~14 h | (H51Y), (L74MF), (T97A), (G149A), V151L, S153YF, S230R, (M50I) | ||
| Bictegravir | ~17 h | (H51Y), S153YF | ||
| INSTI—Long acting | ||||
| Cabotegravir (injection) | ~40 days [86] | In vitro: E138K, Q148K, G140S Clinical trials: G118R, E138K, G140SA, Q148KR, N155H | In vitro: T124A, Q146L, S153Y, I162M, (C56S, V72I, L74M, V75A, T122N, G149A, M154I) Clinical trials: G140R, (T66K, L74M, E92Q. E138A, Y143H) | |
| Year | Authors | Title | Significance |
|---|---|---|---|
| 2020 | López-Huertas, M.R.; Gutiérrez, C.; Madrid-Elena, N.; Hernández-Novoa, B.; Olalla-Sierra, J.; Plana, M.; Delgado, R.; Rubio, R.; Muñoz-Fernández, M.Á.; Moreno, S. [134] | Prolonged administration of maraviroc reactivates latent HIV in vivo, but it does not prevent ARV-free viral rebound | MVC intensification for 48 weeks increased residual viremia and episomal 2-LTR DNA circles suggesting that maraviroc could reactivate latent HIV. MVC induced an increase in cell-associated HIV RNA. Rapid rebound of viremia occurred after ART discontinuation. MVC can reactivate latent HIV in vivo but does not reduce the reservoir. |
| 2018 | Kityo, C.; Szubert, A.J.; Siika, A.; Heyderman, R.; Bwakura-Dangarembizi, M.; Lugemwa, A.; Mwaringa, S.; Griffiths, A.; Nkanya, I.; Kabahenda, S.; Wachira, S.; Musoro, G.; Rajapakse, C.; Etyang, T.; Abach, J.; Spyer, M.J.; Wavamunno, P.; Nyondo-Mipando, L.; Chidziva, E.; Nathoo, K.; Klein, N.; Hakim, J.; Gibb, D.M.; Walker, A.S.; Pett, S.L.; REALITY trial team [135] | Raltegravir-intensified initial antiretroviral therapy in advanced HIV disease in Africa: A randomized controlled trial | 12 weeks of RAL intensification reduced HIV viremia significantly faster than standard triple-drug ART but did not reduce mortality or clinical events. There was no excess of IRIS-compatible events, suggesting that ISNTIs are safe in severe immunocompromise. |
| 2018 | Chaillon, A.; Gianella, S.; Lada, S.M.; Perez-Santiago, J.; Jordan, P.; Ignacio, C.; Karris, M.; Richman, D.D.; Mehta, S.R.; Little, S.J.; Wertheim, J.O.; Smith, D.M. [136] | Size, composition, and evolution of HIV DNA populations during early antiretroviral therapy and intensification with maraviroc | Low-level viremia persisted when standard ART +/− maraviroc intensification was started during acute infection. MVC did not impact viral evolution, diversity, or population structure over 90 weeks. This does not support propagation of infection as a source of viremia on ART. |
| 2018 | Henrich, T.J. [137] | Dolutegravir intensification and HIV persistence: 3 + 1 = 3 | There is little clinical support for intensifying existing regimens with additional drug classes. Three-drug ART remains the mainstay of therapy unless resistance requires otherwise. |
| 2018 | Martinez-Picado, J.; Zurakowski, R.; Buzon, M.J.; Stevenson, M. [133] | Episomal HIV-1 DNA and its relationship to other markers of HIV-1 persistence | A steady state of de novo infection in ART-suppressed individuals may drive immune activation and inflammation, reflecting residual viral reservoir activity during effective ART. |
| 2018 | Puertas, M.C.; Gómez-Mora, E.; Santos, J.R.; Molto, J.; Urrea, V.; Moron-Lopez, S.; Hernandez-Rodriguez, A.; Marfil, S.; Martínez-Bonet, M.; Matas, L.; Muñoz-Fernández, M.A.; Clotet, B.; Blanco, J.; Martinez-Picado, J. [138] | Impact of intensification with raltegravir on HIV-1-infected individuals receiving monotherapy with boosted PIs | ART intensification with 24 weeks of RAL, in patients receiving maintenance monotherapy with ritonavir-boosted DRV or LPV, transiently increased 2-LTR circles and did not change the proportion of patients with detectable residual viremia. |
| 2018 | Rasmussen, T.A.; McMahon, J.; Chang, J.J.; Audsley, J.; Rhodes, A.; Tennakoon, S.; Dantanarayana, A.; Spelman, T.; Schmidt, T.; Kent, S.J.; Morcilla, V.; Palmer, S.; Elliott, J.H.; Lewin, S.R. [11] | The effect of antiretroviral intensification with dolutegravir on residual virus replication in HIV-infected individuals: a randomized placebo-controlled double-blind trial | A total of 8 weeks of ART intensification with dolutegravir did not change 2-LTR circles. Given that the inhibition of active replication would increase 2-LTR circles, residual replication was not demonstrated. |
| 2017 | Kim, C.J.; Rousseau, R.; Huibner, S.; Kovacs, C.; Benko, E.; Shahabi, K.; Kandel, G.; Ostrowski, M.; Kaul, R. [139] | Impact of intensified antiretroviral therapy during early HIV infection on gut immunology and inflammatory blood biomarkers | Intensification of FTC/TDF + LPV/r, started in early HIV infection with RAL + MVC vs. placebo, did not result in differences in blood or gut immune parameters. Most parameters improved, but did not completely normalize, in both groups. |
| 2017 | Wang, X.; Mink, G.; Lin, D.; Song, X.; Rong, L. [140] | Influence of raltegravir intensification on viral load and 2-LTR dynamics in HIV patients on suppressive antiretroviral therapy | On the basis of multi-stage models, RAL intensification has a minor effect on viral load and 2-LTR in HIV patients on suppressive ART. |
| 2014 | Lafeuillade, A.; Assi, A.; Poggi, C.; Bresson-Cuquemelle, C.; Jullian, E.; Tamalet, C. [141] | Failure of combined antiretroviral therapy intensification with maraviroc and raltegravir in chronically HIV-1-infected patients to reduce the viral reservoir: the IntensHIV randomized trial | Intensification of protease-inhibitor-based ART with MVC and RAL does not impact blood proviral DNA but can decrease cell-associated HIV RNA and CD8 activation. |
| 2014 | Puertas, M.C.; Massanella, M.; Llibre, J.M.; Ballestero, M.; Buzon, M.J.; Ouchi, D.; Esteve, A.; Boix, J.; Manzardo, C.; Miró, J.M.; Gatell, J.M.; Clotet, B.; Blanco, J.; Martinez-Picado, J.; MaraviBoost Collaborative Group [142] | Intensification of a raltegravir-based regimen with maraviroc in early HIV-1 infection | Addition of MVC to TDF / FTC + RAL, started in early HIV-1 infection, results in modest reduction in reservoir size at 48 weeks. Plasma viremia decreased in both groups but remained detectable in several subjects. |
| 2013 | Gutiérrez, C.; Hernández-Novoa, B.; Vallejo, A.; Serrano-Villar, S.; Abad-Fernández, M.; Madrid, N.; Díaz, L.; Moreno, A.; Dronda, F.; Zamora, J.; Muñoz-Fernández, M.A.; Moreno, S. [143] | Dynamics of the HIV-1 latent reservoir after discontinuation of the intensification of antiretroviral treatment: results of two clinical trials | A total of 48 weeks of ART intensification, followed by a return to baseline ART for 24 weeks, found fewer latently infected memory CD4 T cells with replication-competent virus and 2-LTR circles. This suggests intensification has persistent effects after discontinuation but does not eliminate the reservoir. |
| 2013 | Negredo, E.; Massanella, M.; Puertas, M.C.; Buzon, M.J.; Puig, J.; Pérez-Alvárez, N.; Pérez-Santiago, J.; Bonjoch, A.; Moltó, J.; Jou, A.; Echeverría, P.; Llibre, J.M.; Martínez-Picado, J.; Clotet, B.; Blanco, J. [144] | Early but limited effects of raltegravir intensification on CD4 T-cell reconstitution in HIV-infected patients with an immunodiscordant response to antiretroviral therapy | RAL intensification in immunodiscordant patients (CD4 < 350 on suppressive ART) did not change proviral DNA levels; episomal DNA and ultrasensitive plasma viral load were barely detected; CD4 T-cell increases were limited. Residual viral replication is not the main cause of poor CD4 T-cell recovery in immunodiscordance. |
| 2013 | Sharkey, M. [145] | Tracking episomal HIV DNA: implications for viral persistence and eradication of HIV | The 2-LTR circles are surrogates for replication that can be used to monitor the effects of ART intensification, the sources of viral rebound, and viral variants contributing to treatment failure. |
| 2012 | Chege, D.; Kovacs, C.; La Porte, C.; Ostrowski, M.; Raboud, J.; Su, D.; Kandel, G.; Brunetta, J.; Kim, C.J.; Sheth, P.M.; Kaul, R.; Loutfy, M.R. [146] | Effect of raltegravir intensification on HIV proviral DNA in the blood and gut mucosa of men on long-term therapy: a randomized controlled trial | Intensification of suppressive ART with RAL was not associated with differences in blood or gut HIV proviral levels or CD4 T-cell increases in this double-blind randomized placebo-controlled study. |
| 2012 | Hatano, H.; Scherzer, R.; Wu, Y.; Harvill, K.; Maka, K.; Hoh, R.; Sinclair, E.; Palmer, S.; Martin, J.N.; Busch, M.P.; Deeks, S.G.; Hsue, P.Y. [147] | A randomized controlled trial assessing the effects of raltegravir intensification on endothelial function in treated HIV infection | Addition of RAL to suppressive ART did not affect the rate of change of the flow-mediated vasodilation of the brachial artery, a marker of endothelial function. |
| 2012 | Llibre, J.M.; Buzón, M.J.; Massanella, M.; Esteve, A.; Dahl, V.; Puertas, M.C.; Domingo, P.; Gatell, J.M.; Larrouse, M.; Gutierrez, M.; Palmer, S.; Stevenson, M.; Blanco, J.; Martinez-Picado, J.; Clotet, B. [148] | Treatment intensification with raltegravir in subjects with sustained HIV-1 viremia suppression: a randomized 48-week study | This prospective open-label randomized study of 48 weeks of RAL intensification in patients on suppressive ART did not show a change in total or integrated HIV DNA. Ultrasensitive VL remained stable. |
| 2011 | Buzon, M.J.; Codoñer, F.M.; Frost, S.D.W.; Pou, C.; Puertas, M.C.; Massanella, M.; Dalmau, J.; Llibre, J.M.; Stevenson, M.; Blanco, J.; Clotet, B.; Paredes, R.; Martinez-Picado, J. [149] | Deep molecular characterization of HIV-1 dynamics under suppressive HAART | RAL intensification of suppressive ART resulted in a transient increase in episomal DNA in most subjects. In subjects with episomal DNA increases, immune activation was higher at baseline and normalized with RAL intensification, suggesting that active replication may persist and drive immune activation. |
| 2011 | Dahl, V.; Lee, E.; Peterson, J.; Spudich, S.S.; Leppla, I.; Sinclair, E.; Fuchs, D.; Palmer, S.; Price, R.W. [150] | Raltegravir treatment intensification does not alter cerebrospinal fluid HIV-1 infection or immunoactivation in subjects on suppressive therapy | RAL intensification of suppressive ART did not reduce intrathecal immunoactivation or alter CSF viral load. Patients had very low CSF viral loads, regardless of intensification. |
| 2010 | Archin, N.M.; Cheema, M.; Parker, D.; Wiegand, A.; Bosch, R.J.; Coffin, J.M.; Eron, J.; Cohen, M.; Margolis, D.M. [151] | Antiretroviral intensification and valproic acid lack sustained effect on residual HIV-1 viremia or resting CD4 T-cell infection | Addition of valproic acid (HDAC inhibitor) and RAL, valproic acid and T20 or T20, failed to progressively reduce the frequency of resting CD4 T-cell infection or ablate low-level viremia. |
| 2010 | Buzon, M.J.; Massanella, M.; Llibre, J.M.; Esteve, A.; Dahl, V.; Puertas, M.C.; Gatell, J.M.; Domingo, P.; Paredes, R.; Sharkey, M.; Palmer, S.; Stevenson, M.; Clotet, B.; Blanco, J.; Martinez-Picado, J. [131] | HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects | RAL intensification of suppressive ART resulted in a transient increase in episomal DNA and normalization of immune activation. This suggests that replication persists in some infected individuals on ART and drives immune activation. |
| 2010 | Hammer, S.M.; Ribaudo, H.; Bassett, R.; Mellors, J.W.; Demeter, L.M.; Coombs, R.W.; Currier, J.; Morse, G.D.; Gerber, J.G.; Martinez, A.I.; Spreen, W.; Fischl, M.A.; Squires, K.E.; AIDS Clinical Trials Group (ACTG) 372A Study Team [1] | A randomized placebo-controlled trial of abacavir intensification in HIV-1-infected adults with virologic suppression on a protease-inhibitor-containing regimen | ABC intensification of IDV + AZT + 3TC in patients with plasma viral loads <500 copies/mL did not confer clinical or virologic benefit. Proportion of subjects with plasma viral loads < 50 copies/mL, CD4 T-cell increase, rates of intermittent viremia, suppression of plasma viral loads < 6 copies/mL, and HIV proviral DNA in peripheral blood mononuclear cells (PBMC) were not different. |
| 2010 | McMahon, D.; Jones, J.; Wiegand, A.; Gange, S.; Kearney, M.; Palmer, S.; McNulty, S.; Metcalf, J.A.; Acosta, E.; Rehm, C.; Coffin, J.M.; Mellors, J.W.; Maldarelli, F. [152] | Short-course raltegravir intensification does not reduce persistent low-level viremia in patients with HIV-1 suppression during receipt of combination antiretroviral therapy | The 4 weeks of ART intensification with RAL did not decrease persistent viremia in subjects receiving suppressive ART. This indicates that rapidly cycling HIV-infected cells were not present. |
| 2010 | Yilmaz, A.; Verhofstede, C.; D’Avolio, A.; Watson, V.; Hagberg, L.; Fuchs, D.; Svennerholm, B.; Gisslén, M. [153] | Treatment intensification has no effect on the HIV-1 central nervous system infections in patients on suppressive antiretroviral therapy | Switch-over intensification with 4 weeks of MVC or LPV/r (good CNS penetration) and 4 weeks with T20 (poor CNS penetration) did not change residual CSF HIV RNA or intrathecal immunoactivation in patients on ART. This does not support ongoing viral replication in the CNS. |
| 2009 | Dinoso, J.B.; Kim, S.Y.; Wiegand, A.M.; Palmer, S.E.; Gange, S.; Cranmer, L.; O’Shea, A.; Callender, M.; Spivak, A.; Brennan, T.; Kearney, M.F.; Proschan, M.A.; Mican, J.M.; Rehm, C.A.; Coffin, J.M.; Mellors, J.W.; Siliciano, R.F.; Maldarelli, F. [154] | Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy | ART intensification with EFV, LPV/r, or ATV/r did not decrease viremia. Lack of response was not associated with the drug-resistant virus or suboptimal drug concentrations. Viremia likely due to output from stable reservoirs, not ongoing cycles of replication. |
| 2003 | Havlir, D.V.; Strain, M.C.; Clerici, M.; Ignacio, C.; Trabattoni, D.; Ferrante, P.; Wong, J.K. [155] | Productive infection maintains a dynamic steady state of residual viremia in people infected with human immunodeficiency virus type 1 treated with suppressive antiretroviral therapy for 5 years | Addition of ABC in patients suppressed on IDV and EFV decreased HIV viral load but residual viremia plateaued at 3.2 to 23 copies/mL. Residual viremia level was established by 9 months, predicted by baseline proviral DNA, and remained constant for 5 years. |
| Cell Type | Ligands on HIV Envelope | Primary Receptor | Coreceptor | Cofactors | Special Considerations |
|---|---|---|---|---|---|
| CD4+ T Cells | ICAM-1, gp41, gp120 | CD4 | CCR5/CXCR4 | α4β7 Integrin, CD26, LFA-1 | Primary target for HIV cure. |
| Macrophages | MHC II, PS, ICAM-1, gp41, gp120 | CD4 | CCR5/CXCR4 | Syndecans, integrins, alternate chemokine cytokine receptors *, Annexin A2, MMR, LFA-1 | Present in key anatomic locations, such as lymph nodes, central nervous system (microglia), and gut [223,224]. |
| Antigen-Presenting Cells (e.g., dendritic cells, Langerhans cells) | gp41, gp120 | CD4 (productive infection); DC-SIGN, DCIR, Langerin (non-productive infection) ** | CCR5/CXCR4 (for infections occurring via CD4 primary receptor) | N/A | There is some debate whether Langerhans cells are susceptible to productive HIV infection [212,214,215,225]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lau, C.-Y.; Adan, M.A.; Maldarelli, F. Why the HIV Reservoir Never Runs Dry: Clonal Expansion and the Characteristics of HIV-Infected Cells Challenge Strategies to Cure and Control HIV Infection. Viruses 2021, 13, 2512. https://doi.org/10.3390/v13122512
Lau C-Y, Adan MA, Maldarelli F. Why the HIV Reservoir Never Runs Dry: Clonal Expansion and the Characteristics of HIV-Infected Cells Challenge Strategies to Cure and Control HIV Infection. Viruses. 2021; 13(12):2512. https://doi.org/10.3390/v13122512
Chicago/Turabian StyleLau, Chuen-Yen, Matthew A. Adan, and Frank Maldarelli. 2021. "Why the HIV Reservoir Never Runs Dry: Clonal Expansion and the Characteristics of HIV-Infected Cells Challenge Strategies to Cure and Control HIV Infection" Viruses 13, no. 12: 2512. https://doi.org/10.3390/v13122512
APA StyleLau, C. -Y., Adan, M. A., & Maldarelli, F. (2021). Why the HIV Reservoir Never Runs Dry: Clonal Expansion and the Characteristics of HIV-Infected Cells Challenge Strategies to Cure and Control HIV Infection. Viruses, 13(12), 2512. https://doi.org/10.3390/v13122512