
Nanopore-Based Direct RNA-Sequencing Reveals a High-Resolution Transcriptional Landscape of Porcine Reproductive and Respiratory Syndrome Virus

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
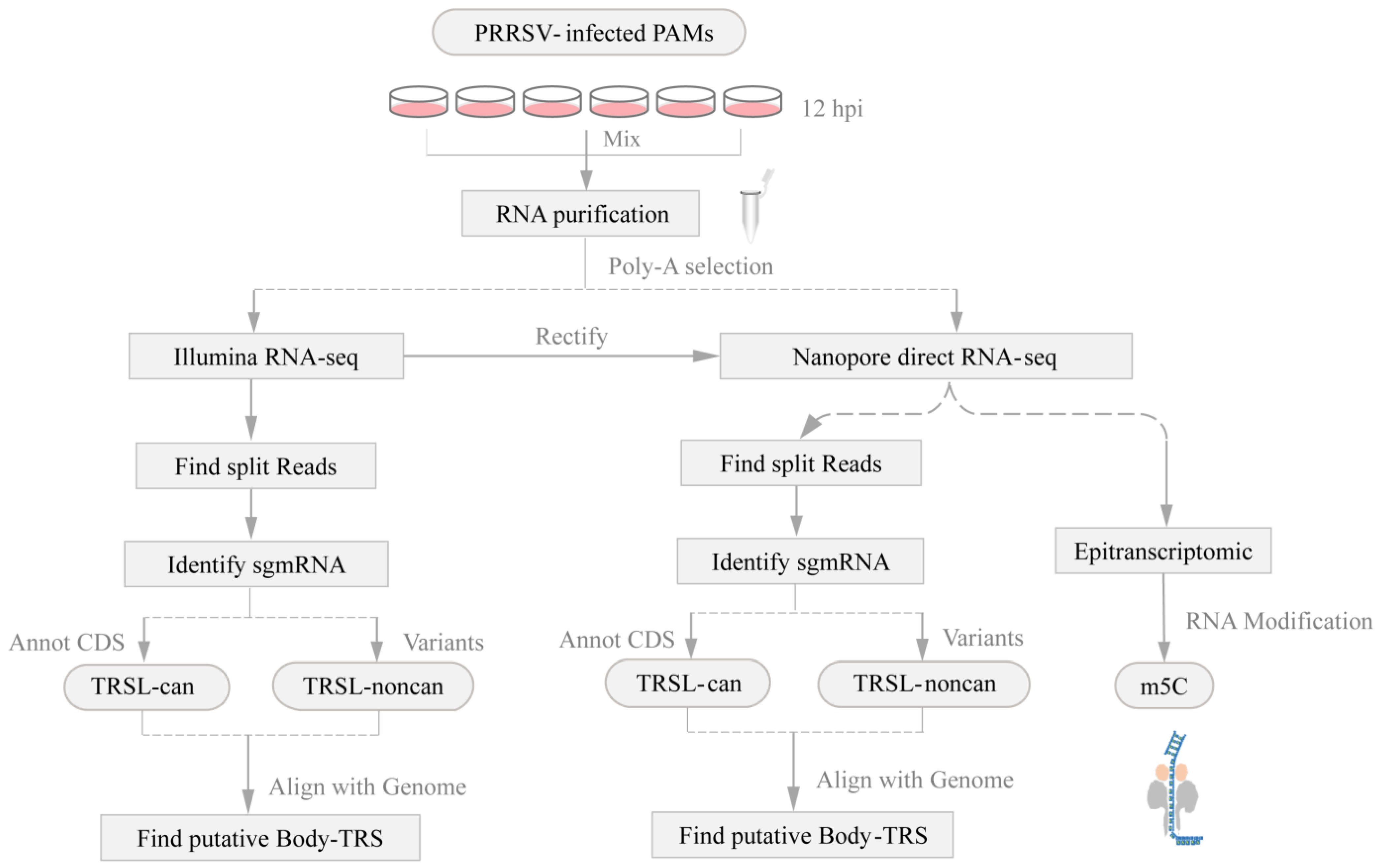
2. Materials and Methods
2.1. Sample Preparation and Total RNA Extraction
2.2. Library Preparation and Sequencing
2.3. Genome-Wide PRRSV Phylogenetic Analysis
2.4. Data Analysis
2.5. Identification of 5mC Methylation
3. Results
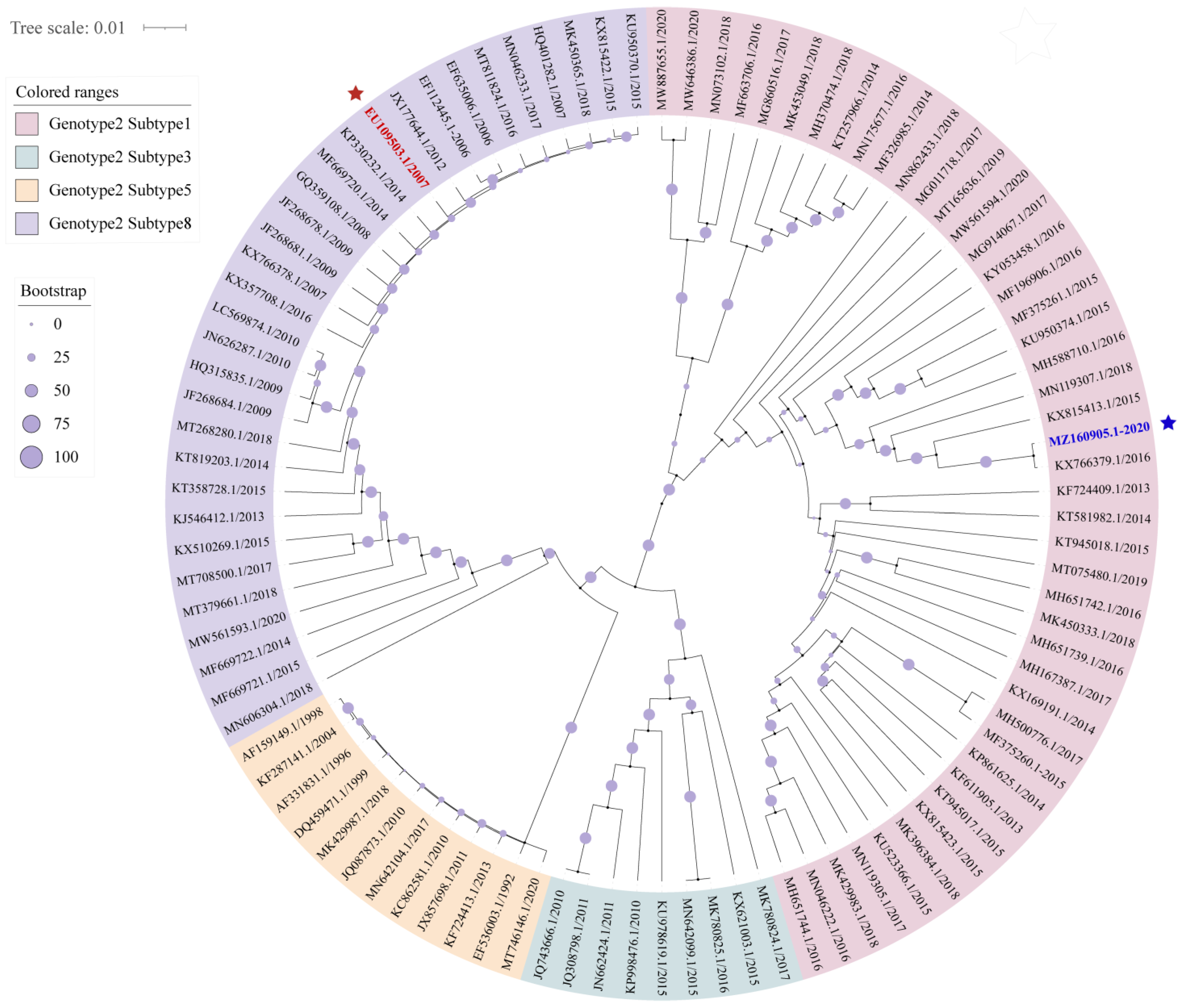
3.1. The Prevalent Status and Genetic Diversity of PRRSV-2 in China
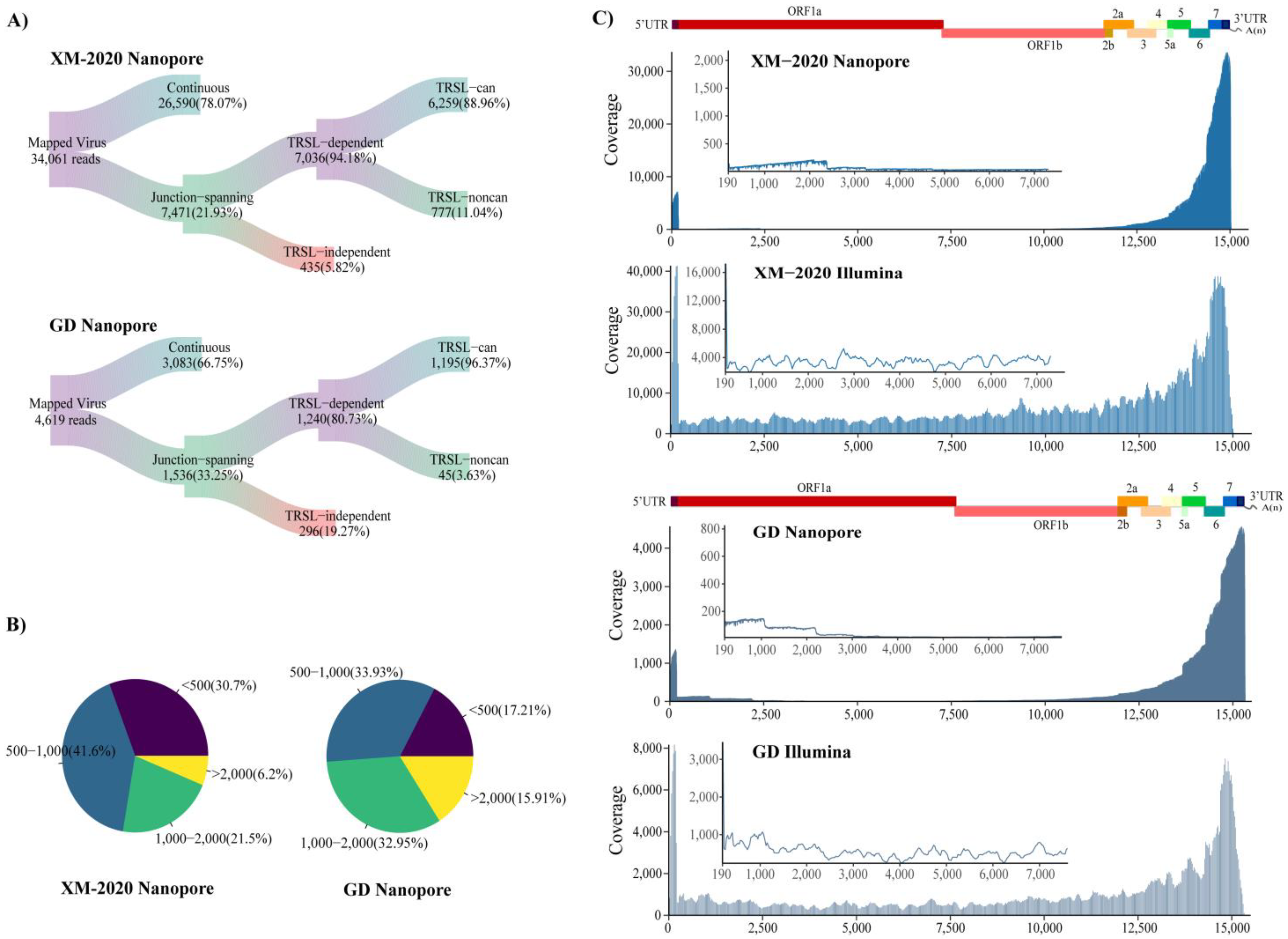
3.2. NADC30-Like PRRSV and HP-PRRSV Induce Different Transcriptional Activity during Infection in Susceptible PAM Cells
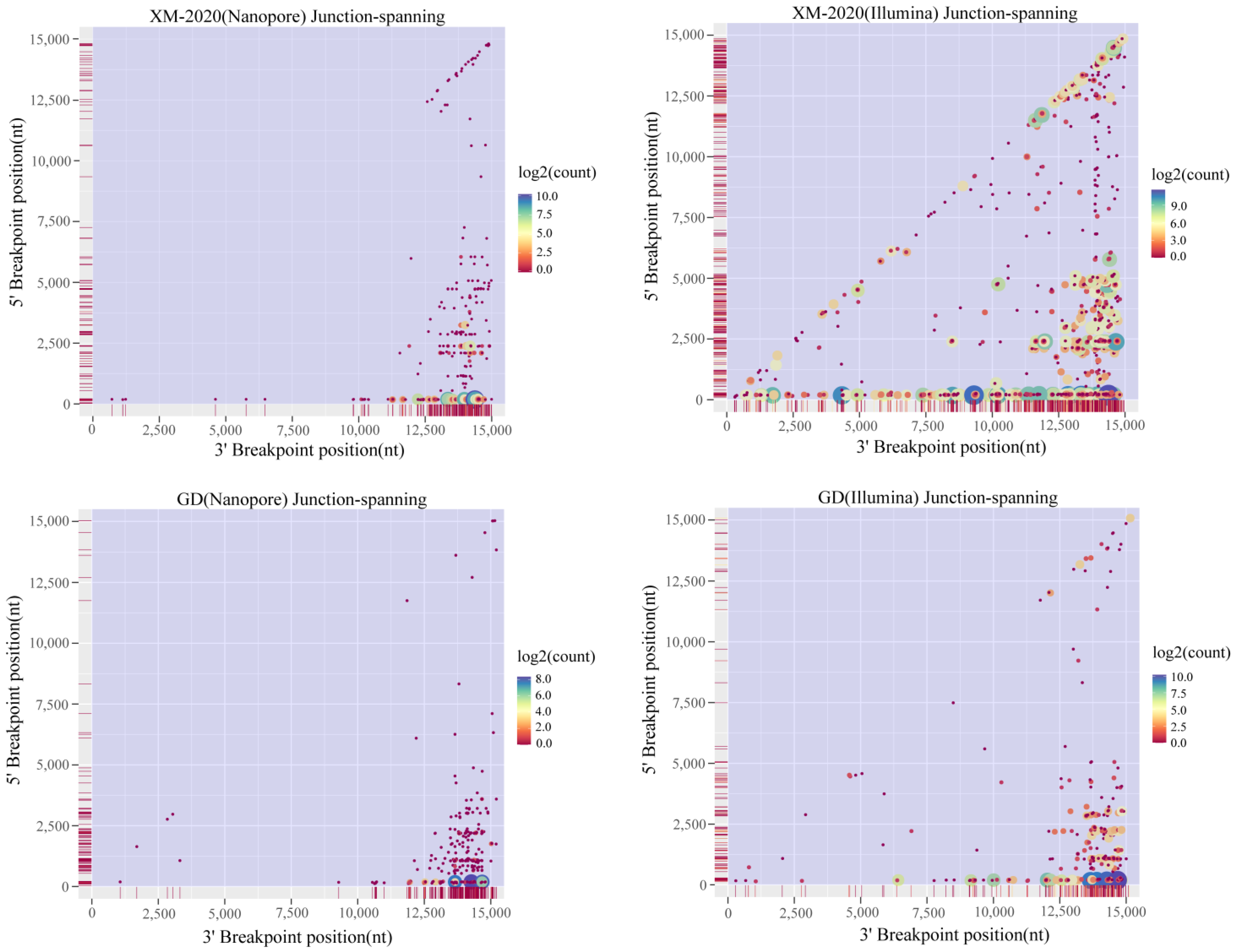
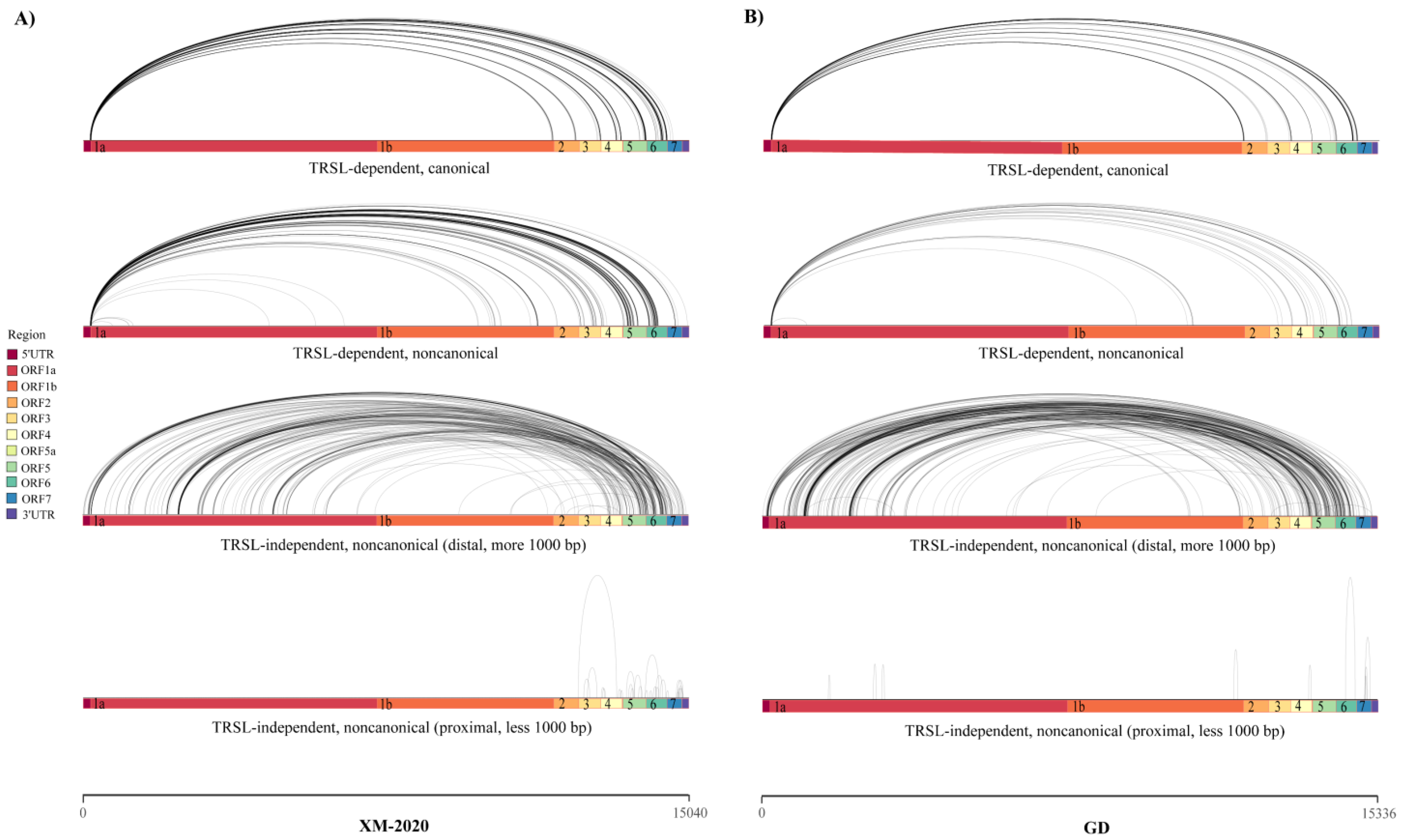
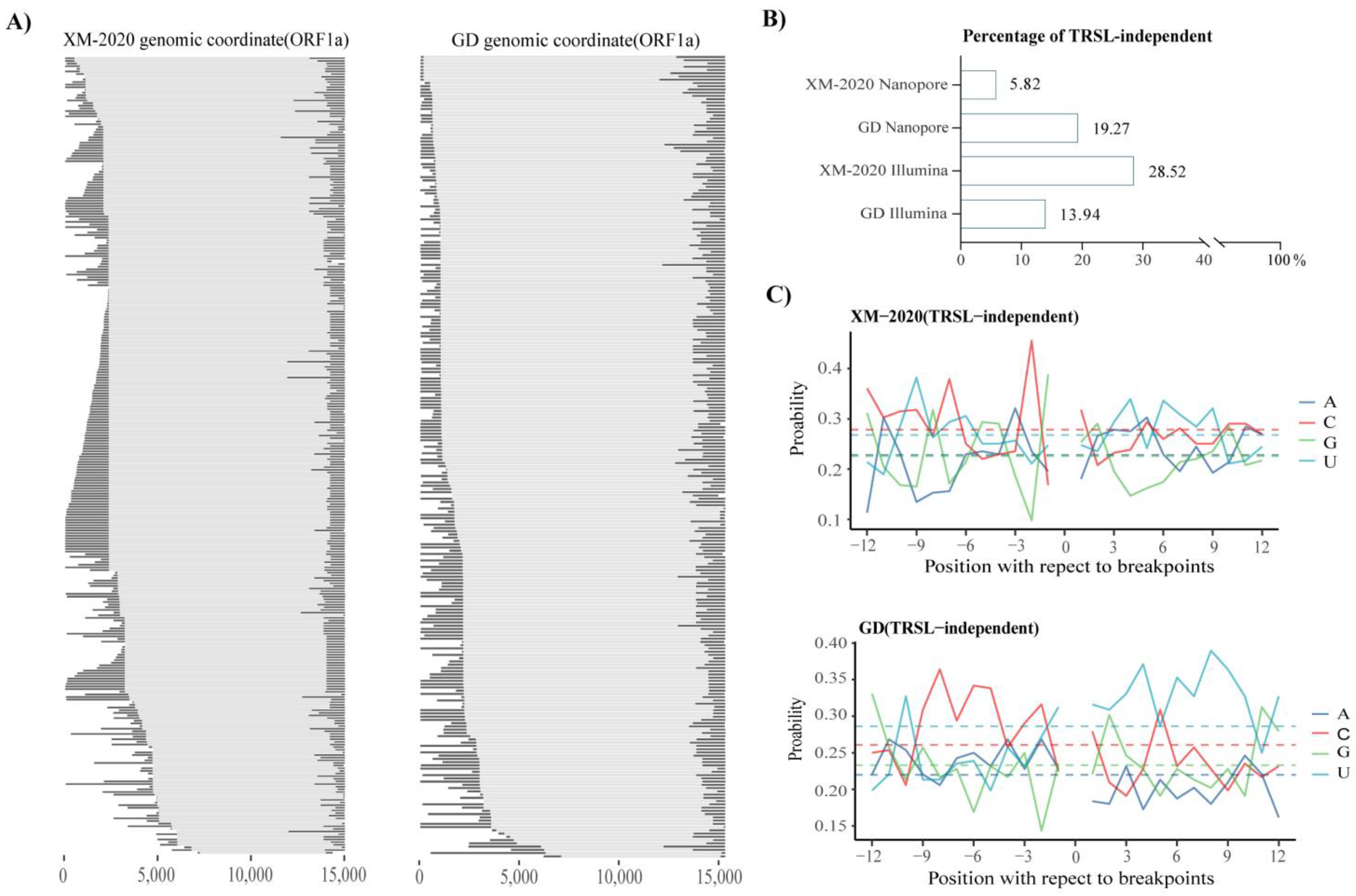
3.3. Analysis of Alternative Splicing Events during Transcription
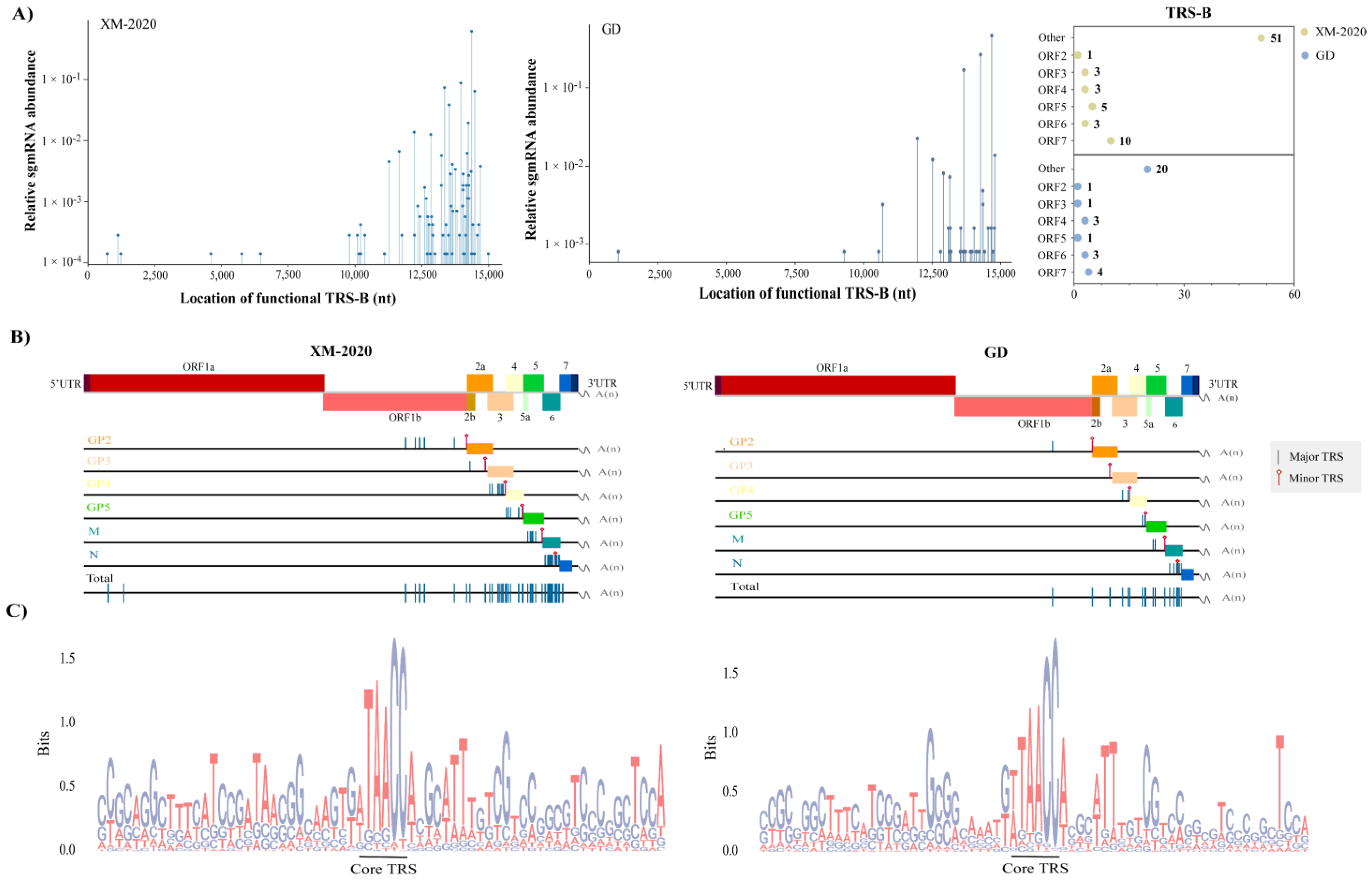
3.4. Identification of TRS-B Sites in PRRSV Genome
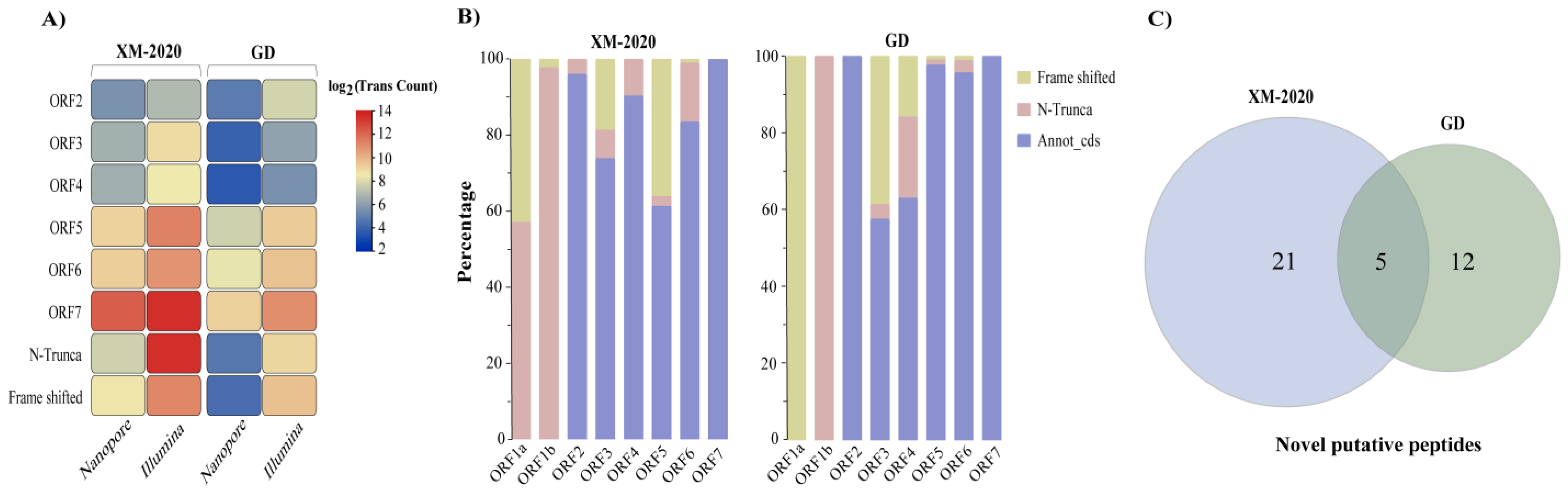
3.5. Gene Predictions and Annotations
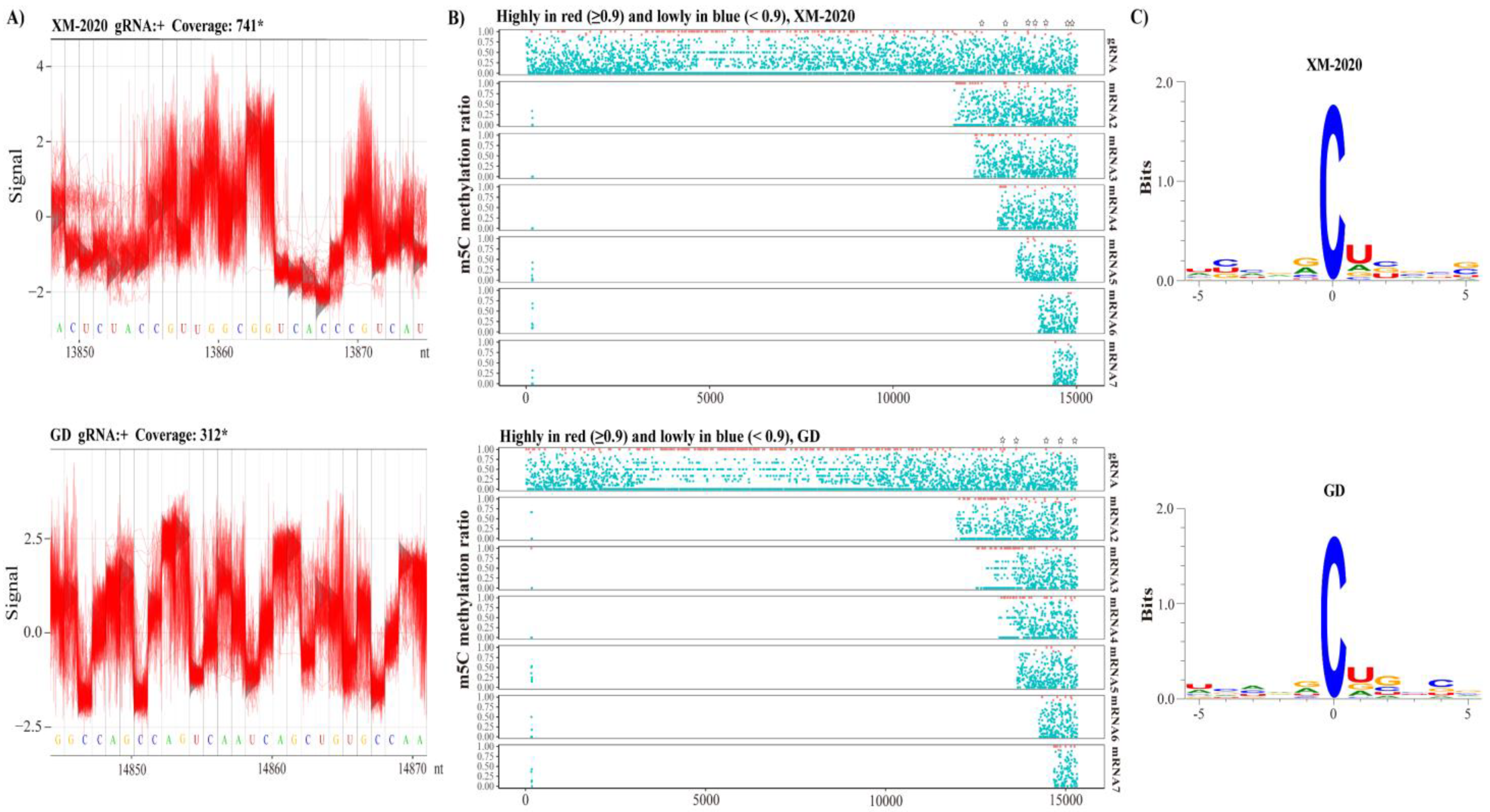
3.6. Revealing m5C Sites in gRNA and sg mRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lunney, J.K.; Fang, Y.; Ladinig, A.; Chen, N.; Li, Y.; Rowland, B.; Renukaradhya, G.J. Porcine Reproductive and Respiratory Syndrome Virus (PRRSV): Pathogenesis and Interaction with the Immune System. Annu Rev. Anim. Biosci. 2016, 4, 129–154. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Lauck, M.; Bailey, A.L.; Shchetinin, A.M.; Vishnevskaya, T.V.; Bào, Y.; Ng, T.F.; LeBreton, M.; Schneider, B.S.; Gillis, A.; et al. Reorganization and expansion of the nidoviral family Arteriviridae. Arch. Virol. 2016, 161, 755–768. [Google Scholar] [CrossRef]
- Brinton, M.A.; Gulyaeva, A.A.; Balasuriya, U.; Dunowska, M.; Faaberg, K.S.; Goldberg, T.; Leung, F.; Nauwynck, H.J.; Snijder, E.J.; Stadejek, T.; et al. ICTV Virus Taxonomy Profile: Arteriviridae 2021. J. Gen. Virol. 2021, 102, 001632. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shang, P.; Shyu, D.; Carrillo, C.; Naraghi-Arani, P.; Jaing, C.J.; Renukaradhya, G.J.; Firth, A.E.; Snijder, E.J.; Fang, Y. Nonstructural proteins nsp2TF and nsp2N of porcine reproductive and respiratory syndrome virus (PRRSV) play important roles in suppressing host innate immune responses. Virology 2018, 517, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.J.; Paul, P.S.; Morozov, I.; Halbur, P.G. A nested set of six or seven subgenomic mRNAs is formed in cells infected with different isolates of porcine reproductive and respiratory syndrome virus. J. Gen. Virol. 1996, 77, 1265–1270. [Google Scholar] [CrossRef] [PubMed]
- Faaberg, K.S.; Elam, M.R.; Nelsen, C.J.; Murtaugh, M.P. Subgenomic RNA7 is transcribed with different leader-body junction sites in PRRSV (strain VR2332) infection of CL2621 cells. Adv. Exp. Med. Biol. 1998, 440, 275–279. [Google Scholar] [CrossRef]
- Van Marle, G.; Dobbe, J.C.; Gultyaev, A.P.; Luytjes, W.; Spaan, W.J.; Snijder, E.J. Arterivirus discontinuous mRNA transcription is guided by base pairing between sense and antisense transcription-regulating sequences. Proc. Natl. Acad. Sci. USA 1999, 96, 12056–12061. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Xu, Y.; Lin, Z.; Fan, J.; Dai, A.; Deng, X.; Mao, W.; Huang, X.; Yang, X.; Wei, C. Epidemiology investigation of PRRSV discharged by faecal and genetic variation of ORF5. Transbound. Emerg. Dis. 2021, 68, 2334–2344. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, G.; Yu, L.; Li, L.; Zhang, Y.; Zhou, Y.; Tong, W.; Liu, C.; Gao, F.; Tong, G. Genetic Diversity of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) From 1996 to 2017 in China. Front. Microbiol. 2020, 11, 618. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Yan, Y.; Shi, M.; Liu, H.Z.; Zhang, H.L.; Yang, Y.B.; Huang, X.Y.; Gauger, P.C.; Zhang, J.; Zhang, Y.H.; et al. Phylogenetics, Genomic Recombination, and NSP2 Polymorphic Patterns of Porcine Reproductive and Respiratory Syndrome Virus in China and the United States in 2014–2018. J. Virol. 2020, 94, e01813-19. [Google Scholar] [CrossRef]
- Paploski, I.; Corzo, C.; Rovira, A.; Murtaugh, M.P.; Sanhueza, J.M.; Vilalta, C.; Schroeder, D.C.; VanderWaal, K. Temporal Dynamics of Co-circulating Lineages of Porcine Reproductive and Respiratory Syndrome Virus. Front. Microbiol. 2019, 10, 2486. [Google Scholar] [CrossRef] [Green Version]
- Sawicki, S.G.; Sawicki, D.L. Coronaviruses use discontinuous extension for synthesis of subgenome-length negative strands. Adv. Exp. Med. Biol. 1995, 380, 499–506. [Google Scholar] [CrossRef] [Green Version]
- Sola, I.; Almazán, F.; Zúñiga, S.; Enjuanes, L. Continuous and Discontinuous RNA Synthesis in Coronaviruses. Annu. Rev. Virol. 2015, 2, 265–288. [Google Scholar] [CrossRef] [Green Version]
- Van den Born, E.; Posthuma, C.C.; Gultyaev, A.P.; Snijder, E.J. Discontinuous subgenomic RNA synthesis in arteriviruses is guided by an RNA hairpin structure located in the genomic leader region. J. Virol. 2005, 79, 6312–6324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasternak, A.O.; Spaan, W.J.; Snijder, E.J. Regulation of relative abundance of arterivirus subgenomic mRNAs. J. Virol. 2004, 78, 8102–8113. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.H.; Ngan, C.Y.; Goldfeder, R.L.; Idol, J.; Kuhlberg, C.; Maurya, R.; Kelly, K.; Omerza, G.; Renzette, N.; De-Abreu, F.; et al. Reduced subgenomic RNA expression is a molecular indicator of asymptomatic SARS-CoV-2 infection. Commun. Med. 2021, 1, 33. [Google Scholar] [CrossRef]
- Di, H.; Madden, J.C., Jr.; Morantz, E.K.; Tang, H.Y.; Graham, R.L.; Baric, R.S.; Brinton, M.A. Expanded subgenomic mRNA transcriptome and coding capacity of a nidovirus. Proc. Natl. Acad. Sci. USA 2017, 114, 8895–8904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, H.; McIntyre, A.A.; Brinton, M.A. New insights about the regulation of Nidovirus subgenomic mRNA synthesis. Virology 2018, 517, 38–43. [Google Scholar] [CrossRef]
- Zhang, S.; Li, R.; Zhang, L.; Chen, S.; Xie, M.; Yang, L.; Xia, Y.; Foyer, C.H.; Zhao, Z.; Lam, H.M. New insights into Arabidopsis transcriptome complexity revealed by direct sequencing of native RNAs. Nucleic Acids Res. 2020, 48, 7700–7711. [Google Scholar] [CrossRef] [PubMed]
- Viehweger, A.; Krautwurst, S.; Lamkiewicz, K.; Madhugiri, R.; Ziebuhr, J.; Hölzer, M.; Marz, M. Direct RNA nanopore sequencing of full-length coronavirus genomes provides novel insights into structural variants and enables modification analysis. Genome Res. 2019, 29, 1545–1554. [Google Scholar] [CrossRef] [Green Version]
- Wahyuningtyas, R.; Lai, Y.S.; Wu, M.L.; Chen, H.W.; Chung, W.B.; Chaung, H.C.; Chang, K.T. Recombinant Antigen of Type 2 Porcine Reproductive and Respiratory Syndrome Virus (PRRSV-2) Promotes M1 Repolarization of Porcine Alveolar Macrophages and Th1 Type Response. Vaccines 2021, 9, 1009. [Google Scholar] [CrossRef]
- Roach, N.P.; Sadowski, N.; Alessi, A.F.; Timp, W.; Taylor, J.; Kim, J.K. The full-length transcriptome of C. elegans using direct RNA sequencing. Genome Res. 2020, 30, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Knyazev, A.; Glushkevich, A.; Fesenko, I. Direct RNA sequencing dataset of SMG1 KO mutant Physcomitrella (Physcomitrium patens). Data Brief. 2020, 33, 106602. [Google Scholar] [CrossRef]
- Zimin, A.V.; Puiu, D.; Luo, M.C.; Zhu, T.; Koren, S.; Marçais, G.; Yorke, J.A.; Dvořák, J.; Salzberg, S.L. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 2017, 27, 787–792. [Google Scholar] [CrossRef] [Green Version]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [Green Version]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Wang, J.R.; Holt, J.; McMillan, L.; Jones, C.D. FMLRC: Hybrid long read error correction using an FM-index. BMC Bioinform. 2018, 19, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Lorenz, D.A.; Sathe, S.; Einstein, J.M.; Yeo, G.W. Direct RNA sequencing enables m6A detection in endogenous transcript isoforms at base-specific resolution. RNA 2020, 26, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasternak, A.O.; Gultyaev, A.P.; Spaan, W.J.; Snijder, E.J. Genetic manipulation of arterivirus alternative mRNA leader-body junction sites reveals tight regulation of structural protein expression. J. Virol. 2000, 74, 11642–11653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Zhang, K.; Zhu, X.Q.; Liu, C.; Lu, J.; Gao, F.; Zhou, Y.; Zheng, H.; Lin, T.; Li, L.; et al. Genetic manipulation of a transcription-regulating sequence of porcine reproductive and respiratory syndrome virus reveals key nucleotides determining its activity. Arch. Virol. 2014, 159, 1927–1940. [Google Scholar] [CrossRef] [PubMed]
- Olasz, F.; Dénes, B.; Bálint, Á.; Magyar, T.; Belák, S.; Zádori, Z. Characterisation of the nucleic acid binding features of the PRRSV 7ap and its ability to induce antinuclear antibodies. Acta Vet. Hung. 2017, 65, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Olasz, F.; Dénes, B.; Bálint, Á.; Magyar, T.; Belák, S.; Zádori, Z. Immunological and biochemical characterisation of 7ap, a short protein translated from an alternative frame of ORF7 of PRRSV. Acta Vet. Hung. 2016, 64, 273–287. [Google Scholar] [CrossRef] [Green Version]
- Tijms, M.A.; van Dinten, L.C.; Gorbalenya, A.E.; Snijder, E.J. A zinc finger-containing papain-like protease couples subgenomic mRNA synthesis to genome translation in a positive-stranded RNA virus. Proc. Natl. Acad. Sci. USA 2001, 98, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Y.; Brian, D.A. Subgenomic messenger RNA amplification in coronaviruses. Proc. Natl. Acad. Sci. USA 2010, 107, 12257–12262. [Google Scholar] [CrossRef] [Green Version]
- Van Dinten, L.C.; den Boon, J.A.; Wassenaar, A.L.; Spaan, W.J.; Snijder, E.J. An infectious arterivirus cDNA clone: Identification of a replicase point mutation that abolishes discontinuous mRNA transcription. Proc. Natl. Acad. Sci. USA 1997, 94, 991–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921. [Google Scholar] [CrossRef]
- Chang, J.J.; Rawlinson, D.; Pitt, M.E.; Taiaroa, G.; Gleeson, J.; Zhou, C.; Mordant, F.L.; De Paoli-Iseppi, R.; Caly, L.; Purcell, D.; et al. Transcriptional and epi-transcriptional dynamics of SARS-CoV-2 during cellular infection. Cell Rep. 2021, 35, 109108. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Jiang, A.; Feng, J.; Li, G.; Guo, D.; Sajid, M.; Wu, K.; Zhang, Q.; Ponty, Y.; Will, S.; et al. The SARS-CoV-2 subgenome landscape and its novel regulatory features. Mol. Cell. 2021, 81, 2135–2147. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Yuan, S.; Murtaugh, M.P.; Faaberg, K.S. Heteroclite subgenomic RNAs are produced in porcine reproductive and respiratory syndrome virus infection. Virology 2000, 275, 158–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Murtaugh, M.P.; Faaberg, K.S. Packaged heteroclite subgenomic RNAs of PRRSV. Adv. Exp. Med. Biol. 2001, 494, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Faaberg, K.S.; Murtaugh, M.P.; Yuan, S. Predicted RNA folding suggests PRRSV major and heteroclite subgenomic transcripts result from polymerase switching at unpaired nucleotides. Adv. Exp. Med. Biol. 2001, 494, 37–42. [Google Scholar] [CrossRef]
- Yuan, S.; Murtaugh, M.P.; Schumann, F.A.; Mickelson, D.; Faaberg, K.S. Characterization of heteroclite subgenomic RNAs associated with PRRSV infection. Virus Res. 2004, 105, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Ke, T.Y.; Liao, W.Y.; Wu, H.Y. A leaderless genome identified during persistent bovine coronavirus infection is associated with attenuation of gene expression. PLoS ONE 2013, 8, 82176. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, M.A.; Senanayake, S.D.; Brian, D.A. A translation-attenuating intraleader open reading frame is selected on coronavirus mRNAs during persistent infection. Proc. Natl. Acad. Sci. USA 1993, 90, 11733–11737. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.H.; Yang, C.Y.; Wang, M.; Ou, S.C.; Lo, C.Y.; Tsai, T.L.; Wu, H.Y. Effects of Coronavirus Persistence on the Genome Structure and Subsequent Gene Expression, Pathogenicity and Adaptation Capability. Cells 2020, 9, 2322. [Google Scholar] [CrossRef]
- Bentley, K.; Alnaji, F.G.; Woodford, L.; Jones, S.; Woodman, A.; Evans, D.J. Imprecise recombinant viruses evolve via a fitness-driven, iterative process of polymerase template-switching events. PLoS Pathog. 2021, 17, 1009676. [Google Scholar] [CrossRef]
- Boussier, J.; Munier, S.; Achouri, E.; Meyer, B.; Crescenzo-Chaigne, B.; Behillil, S.; Enouf, V.; Vignuzzi, M.; van der Werf, S.; Naffakh, N. RNA-seq accuracy and reproducibility for the mapping and quantification of influenza defective viral genomes. RNA 2020, 26, 1905–1918. [Google Scholar] [CrossRef]
- Vignuzzi, M.; López, C.B. Defective viral genomes are key drivers of the virus-host interaction. Nat. Microbiol. 2019, 4, 1075–1087. [Google Scholar] [CrossRef]
- Orr, M.W.; Mao, Y.; Storz, G.; Qian, S.B. Alternative ORFs and small ORFs: Shedding light on the dark proteome. Nucleic Acids Res. 2020, 48, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wright, M.; Gogol, M.M.; Bradford, W.D.; Zhang, N.; Bazzini, A.A. Translation of small downstream ORFs enhances translation of canonical main open reading frames. EMBO J. 2020, 39, 104763. [Google Scholar] [CrossRef] [PubMed]
- Trixl, L.; Lusser, A. The dynamic RNA modification 5-methylcytosine and its emerging role as an epitranscriptomic mark. Wiley Interdiscip. Rev. RNA 2019, 10, 1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.; Wang, P.; Ma, X.; Wu, Y.; Luo, C.; Qiu, L.; Zeshan, B.; Yang, Z.; Zhou, Y.; Wang, X. Nanopore-Based Direct RNA-Sequencing Reveals a High-Resolution Transcriptional Landscape of Porcine Reproductive and Respiratory Syndrome Virus. Viruses 2021, 13, 2531. https://doi.org/10.3390/v13122531
Zhang R, Wang P, Ma X, Wu Y, Luo C, Qiu L, Zeshan B, Yang Z, Zhou Y, Wang X. Nanopore-Based Direct RNA-Sequencing Reveals a High-Resolution Transcriptional Landscape of Porcine Reproductive and Respiratory Syndrome Virus. Viruses. 2021; 13(12):2531. https://doi.org/10.3390/v13122531
Chicago/Turabian StyleZhang, Riteng, Peixin Wang, Xin Ma, Yifan Wu, Chen Luo, Li Qiu, Basit Zeshan, Zengqi Yang, Yefei Zhou, and Xinglong Wang. 2021. "Nanopore-Based Direct RNA-Sequencing Reveals a High-Resolution Transcriptional Landscape of Porcine Reproductive and Respiratory Syndrome Virus" Viruses 13, no. 12: 2531. https://doi.org/10.3390/v13122531
APA StyleZhang, R., Wang, P., Ma, X., Wu, Y., Luo, C., Qiu, L., Zeshan, B., Yang, Z., Zhou, Y., & Wang, X. (2021). Nanopore-Based Direct RNA-Sequencing Reveals a High-Resolution Transcriptional Landscape of Porcine Reproductive and Respiratory Syndrome Virus. Viruses, 13(12), 2531. https://doi.org/10.3390/v13122531