
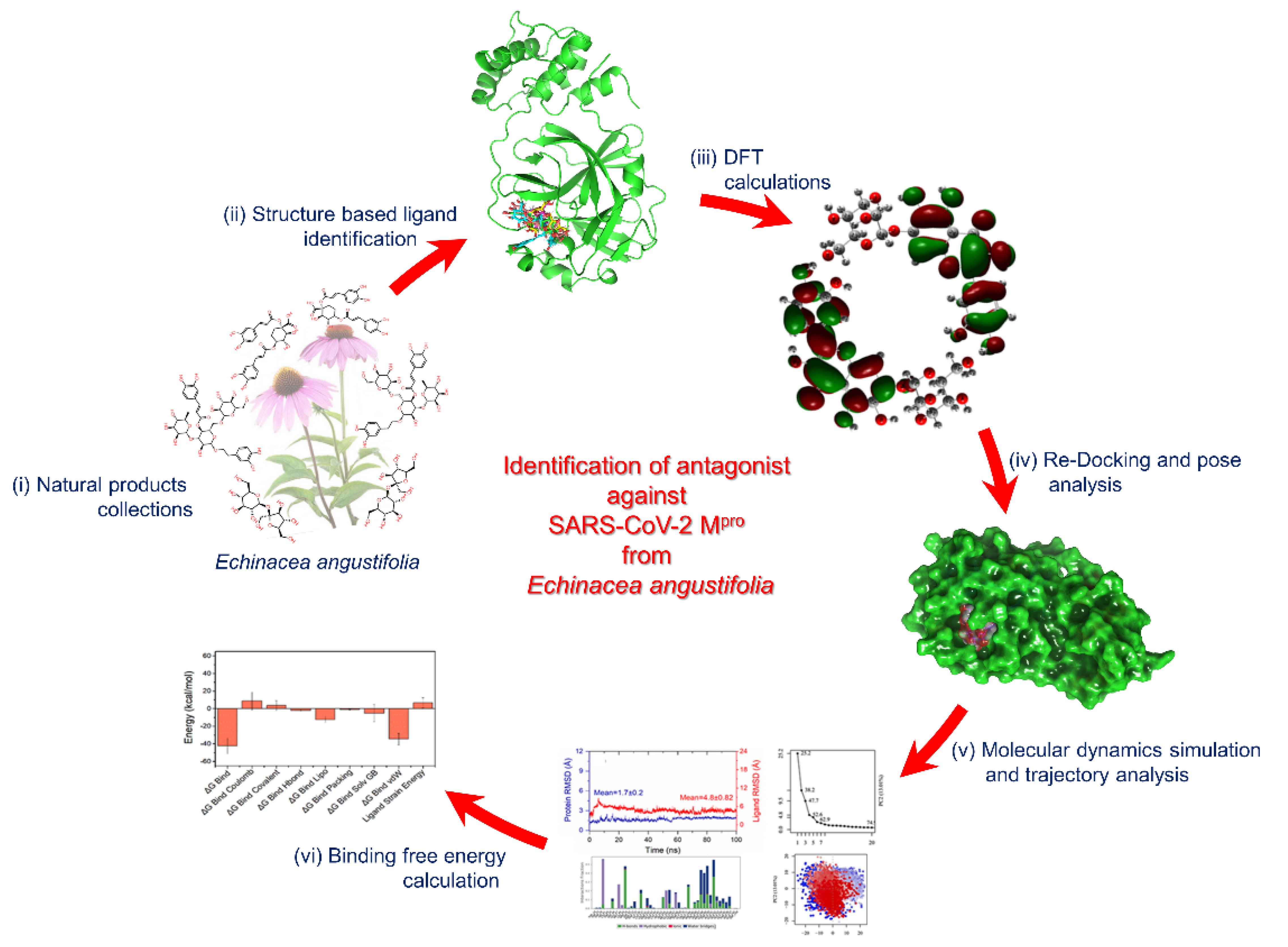
Structure-Based Identification of Natural Products as SARS-CoV-2 Mpro Antagonist from Echinacea angustifolia Using Computational Approaches

 , ,
, ,  , ,
, ,  ,
,  ,
,  , and
, and 
Abstract
:1. Introduction
2. Methodology
2.1. Receptor and Ligands Collection
2.2. Structure-Based Ligand Identification and Quantum Chemical Calculations
2.3. Re-Docking Simulation and Pose Profiling
2.4. Explicit Solvent Molecular Dynamics Simulations
2.5. Post Molecular Dynamics Simulation
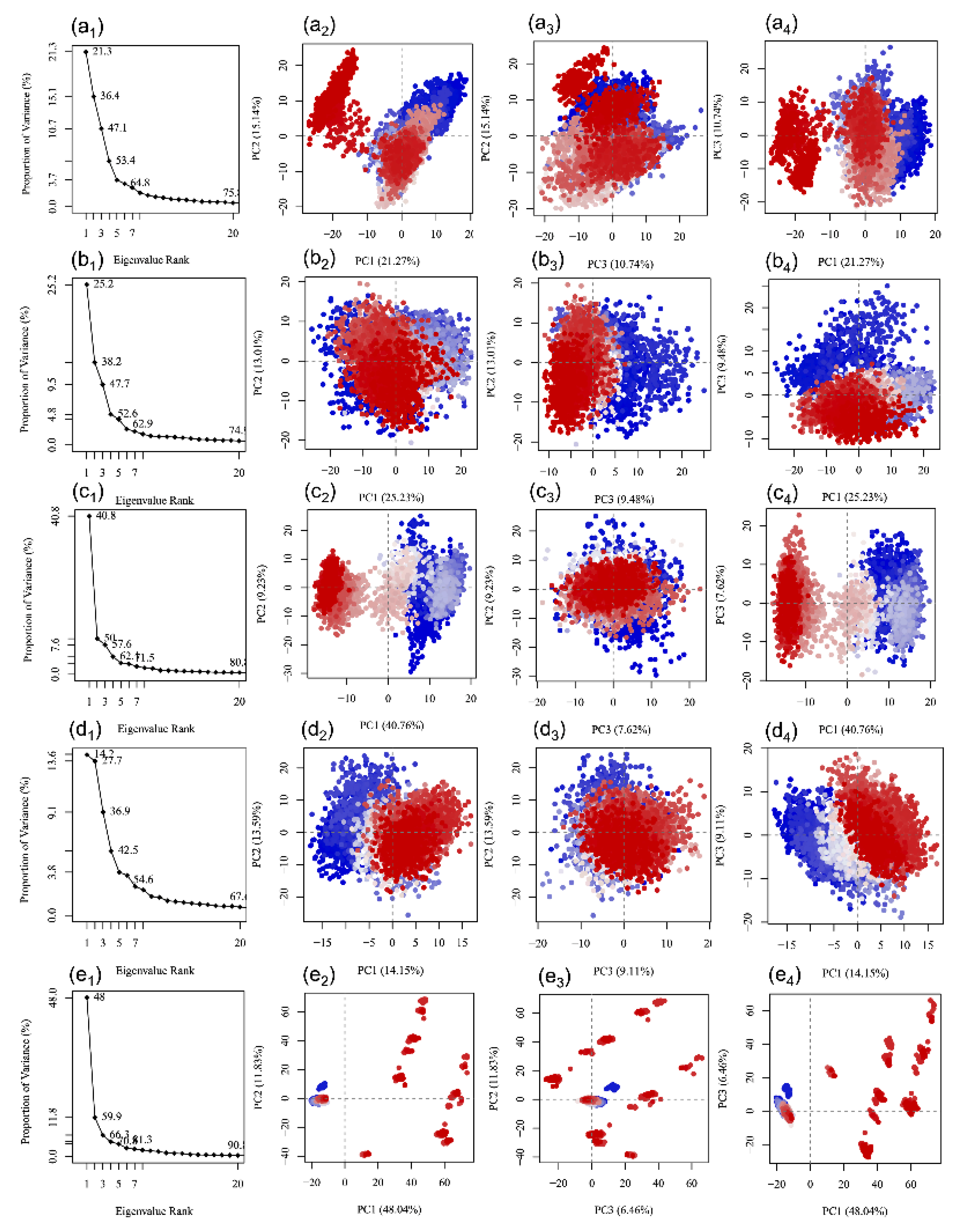
2.5.1. Essential Dynamics
2.5.2. Binding Free Energy Calculations
3. Results and Discussion
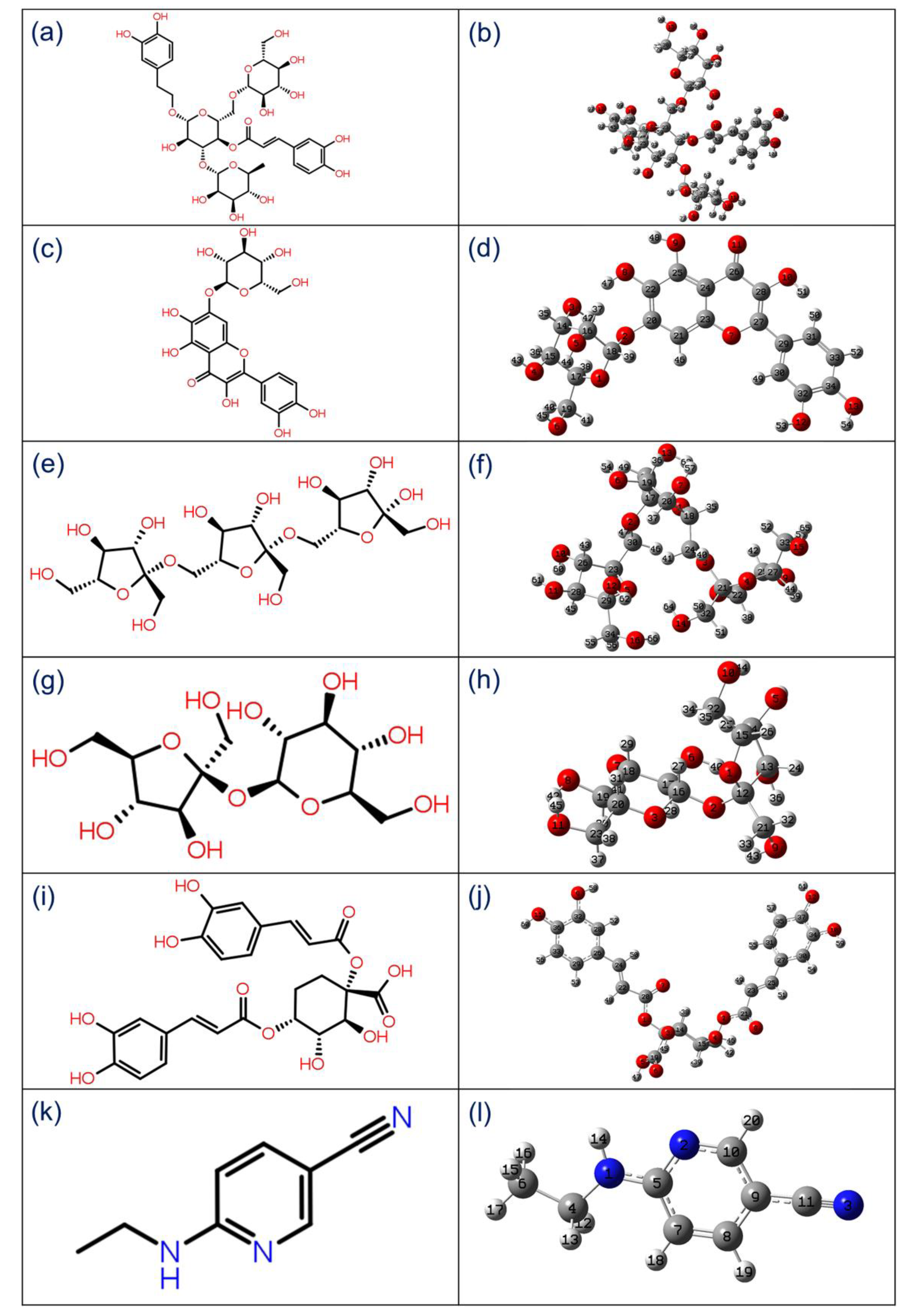
3.1. Structure-Based Ligand Identification
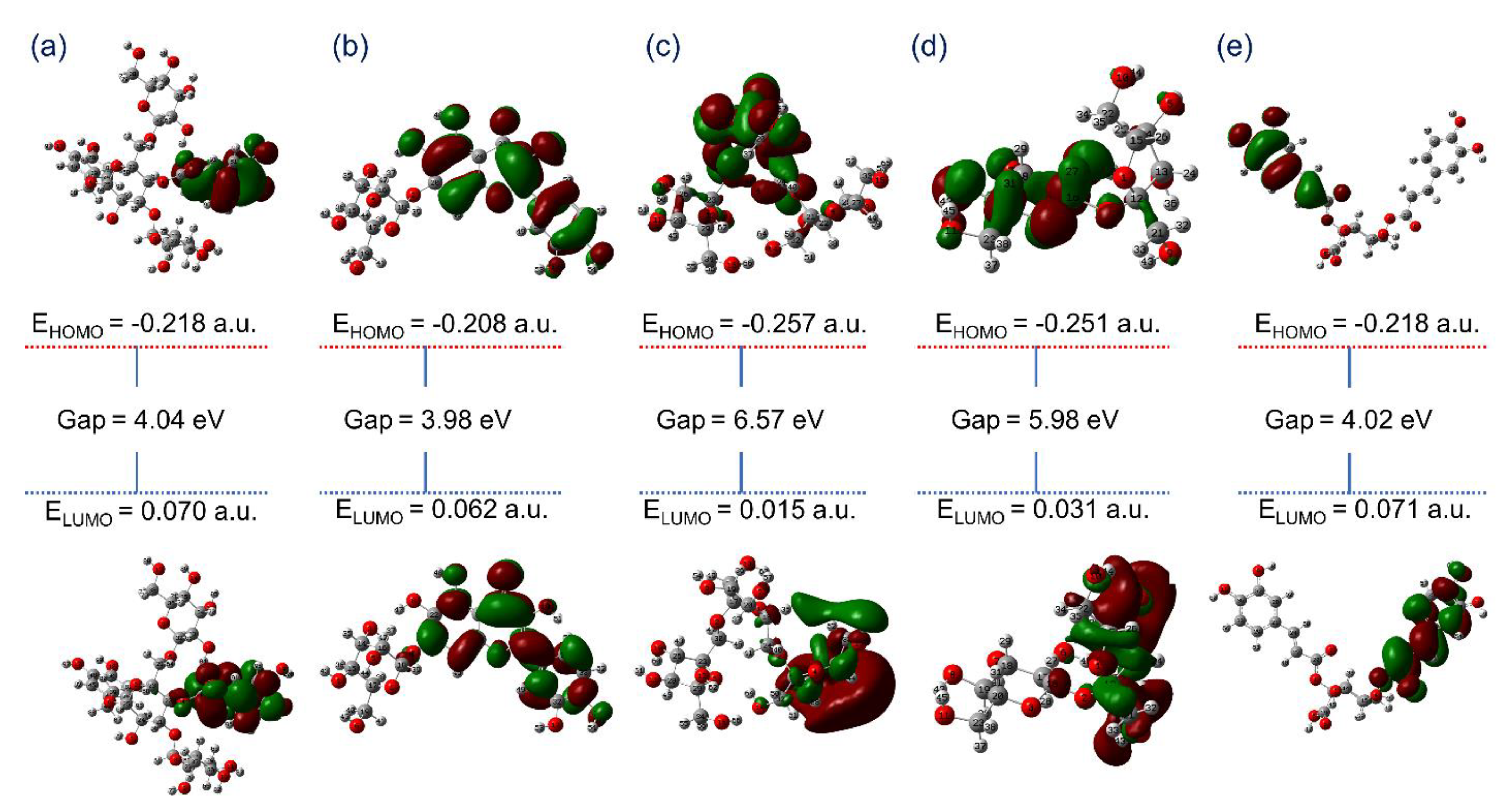
3.2. Quantum Chemical Calculations
3.2.1. Geometry Optimization
3.2.2. Frontier Molecular Orbitals Analysis
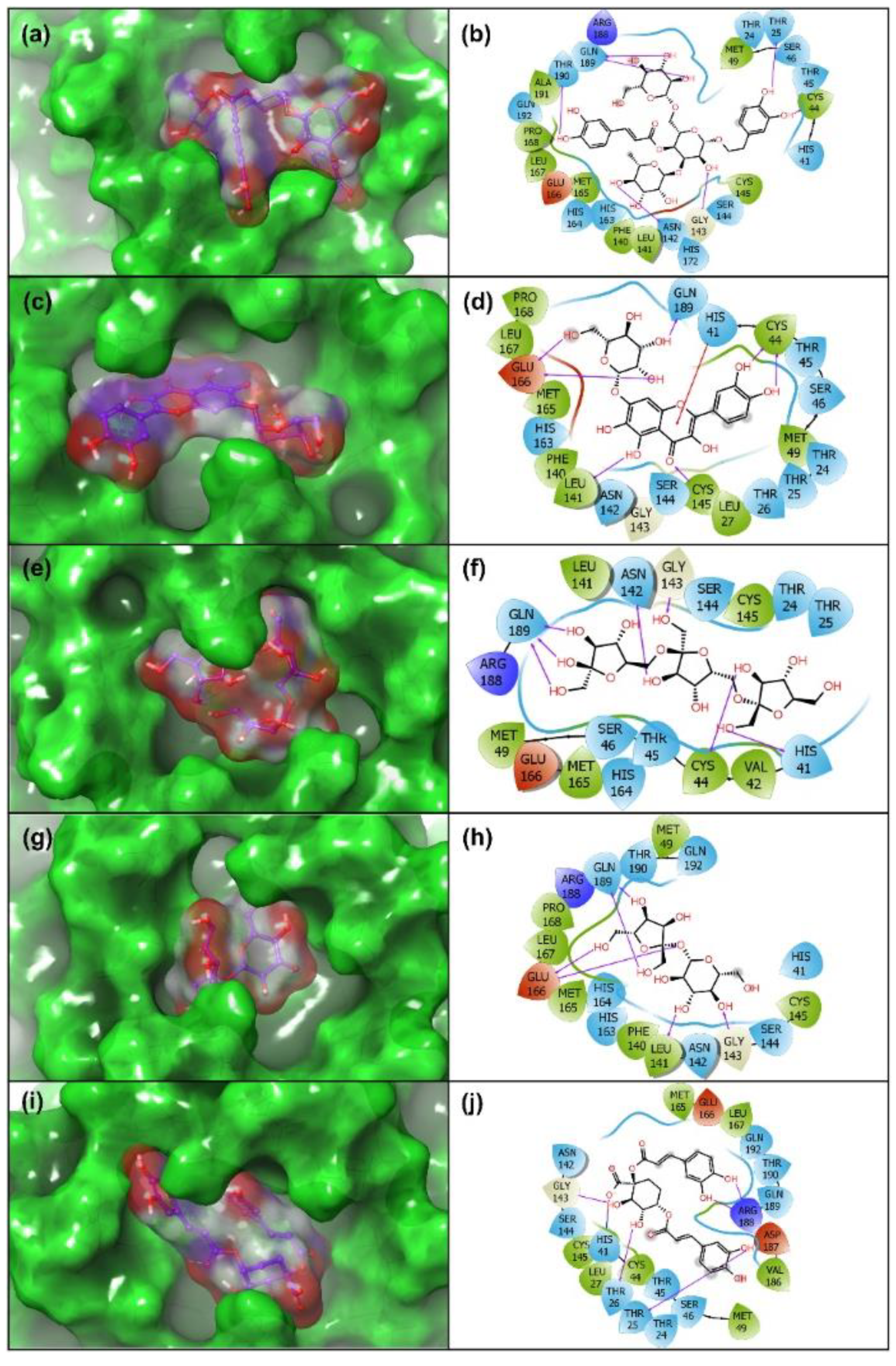
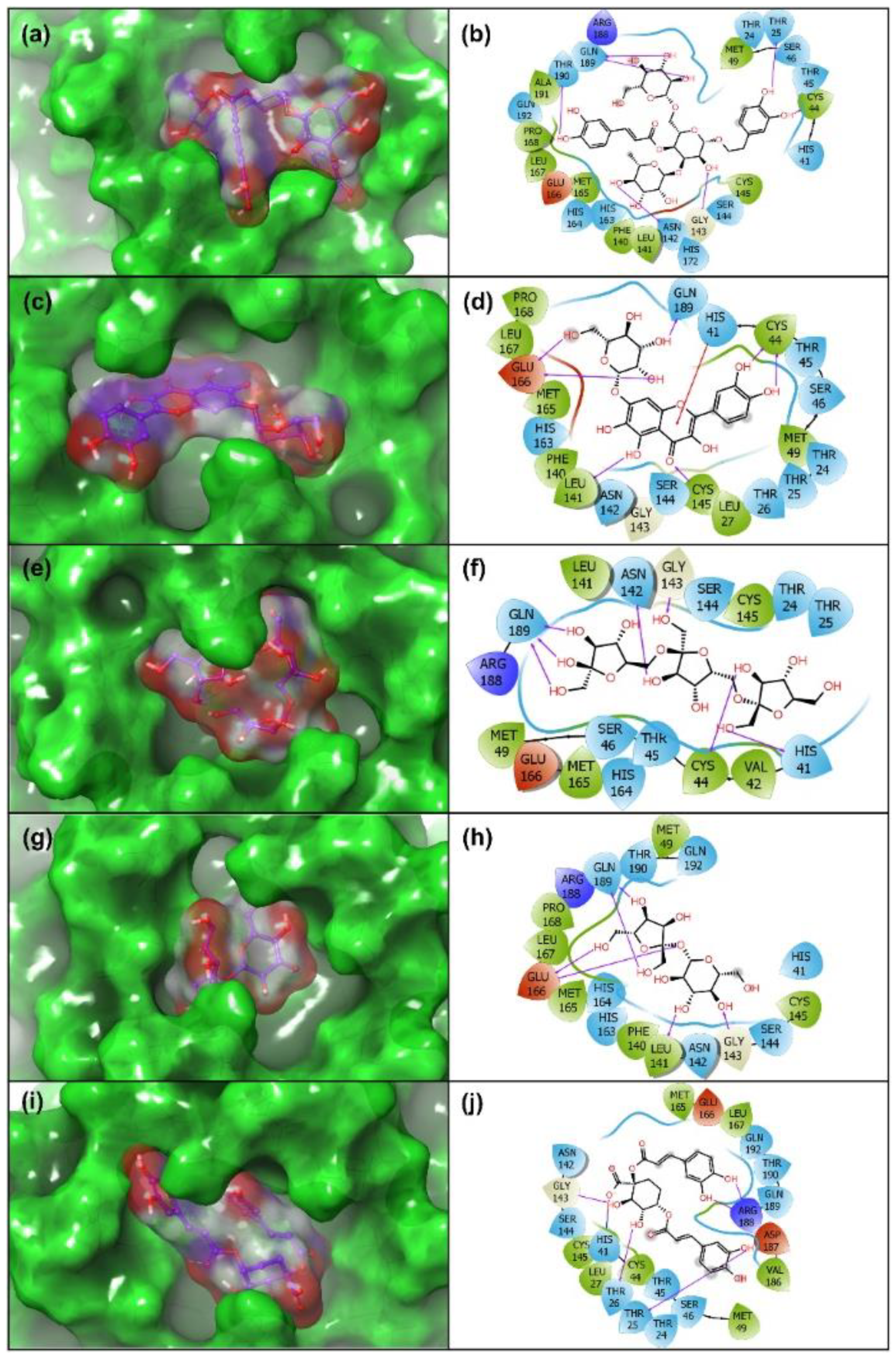
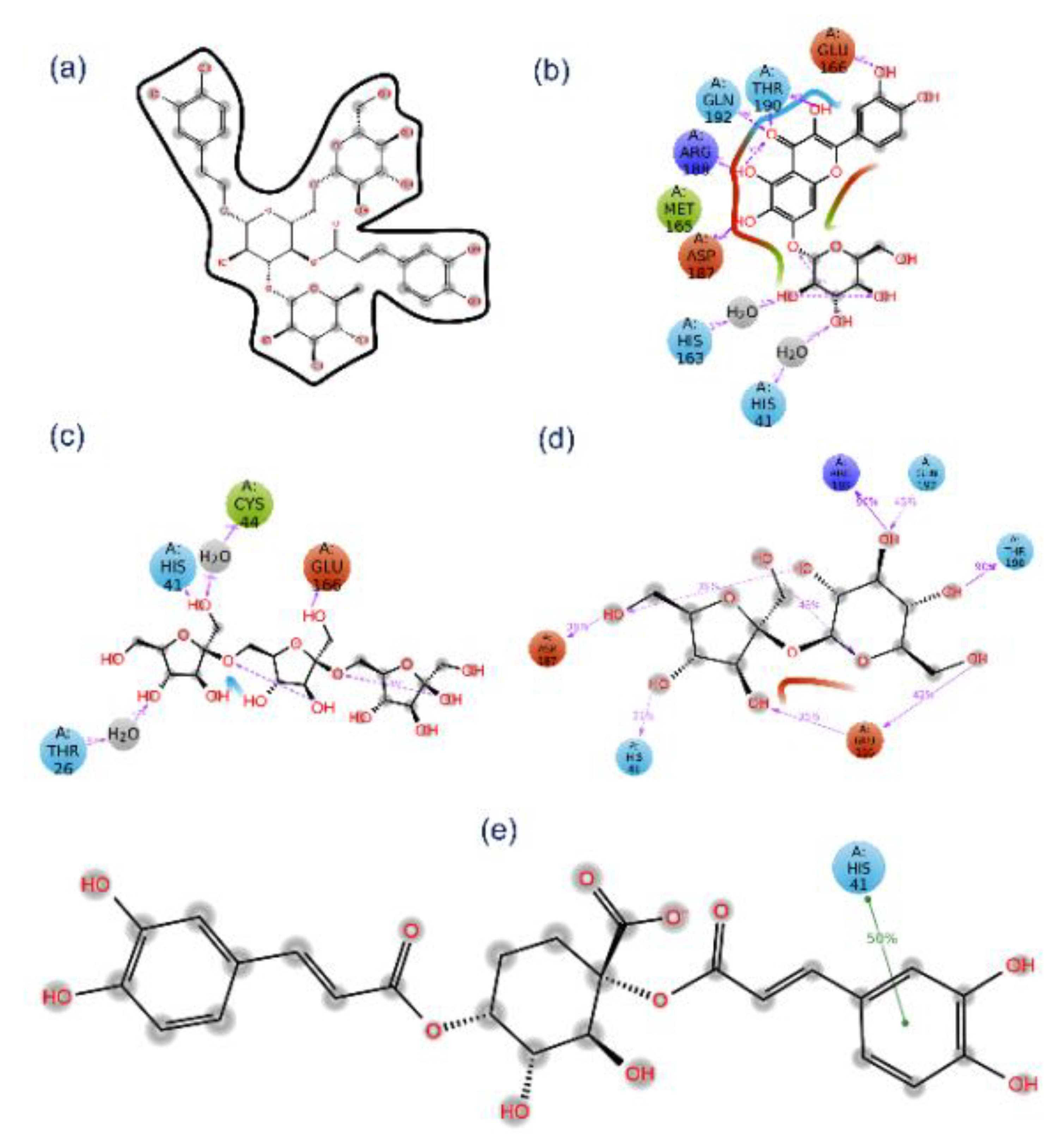
3.3. Re-Docking and Intermolecular Interaction Analysis
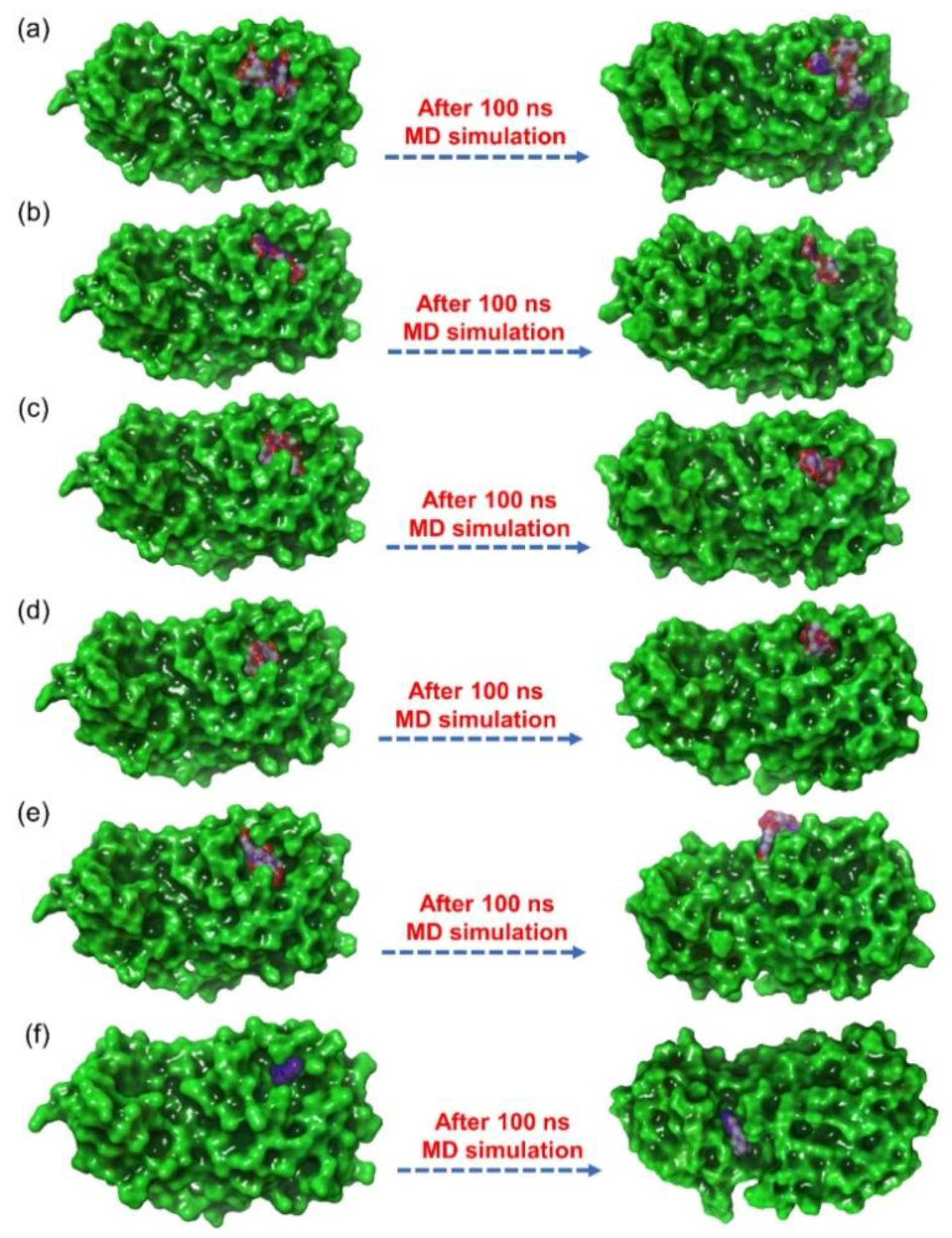
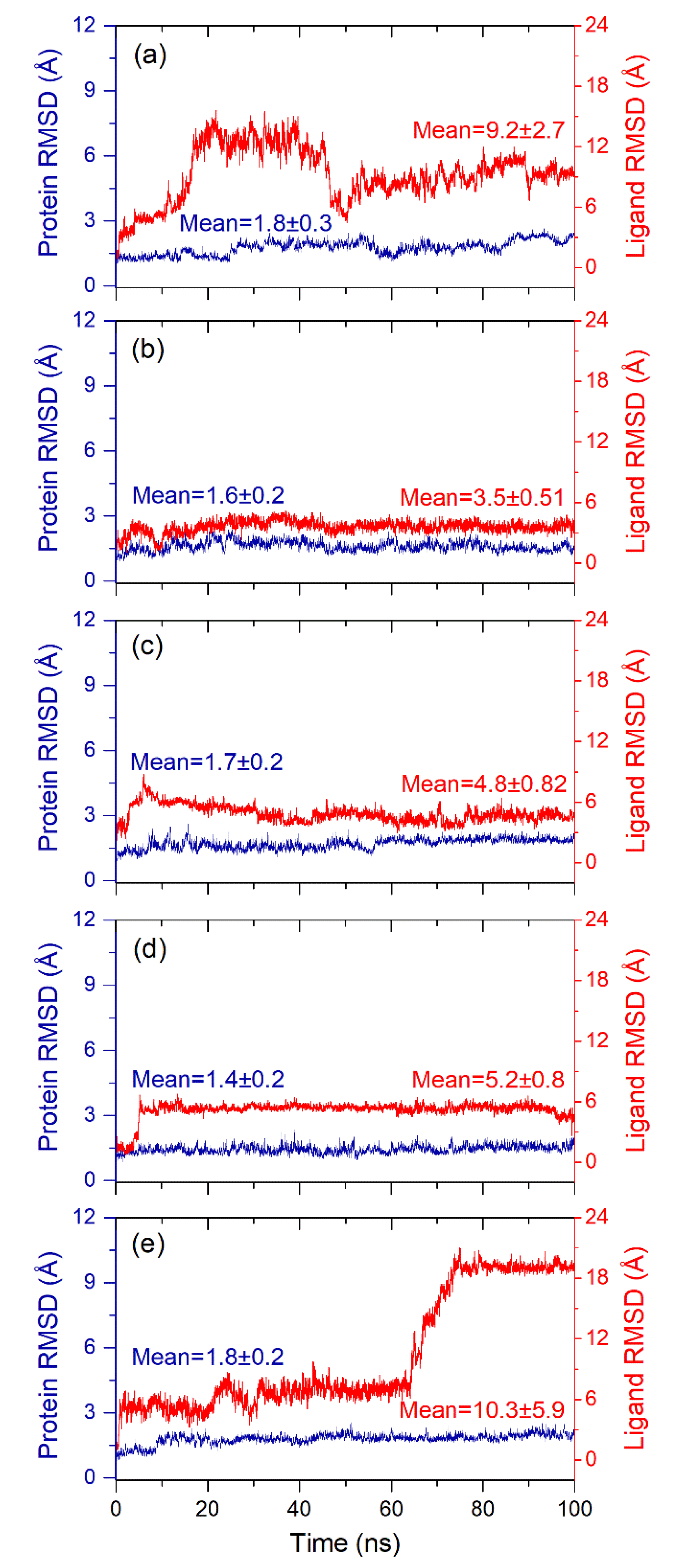
3.4. Explicit Solvent Molecular Dynamics Simulation Analysis
3.4.1. RMSD and RMSF Analysis
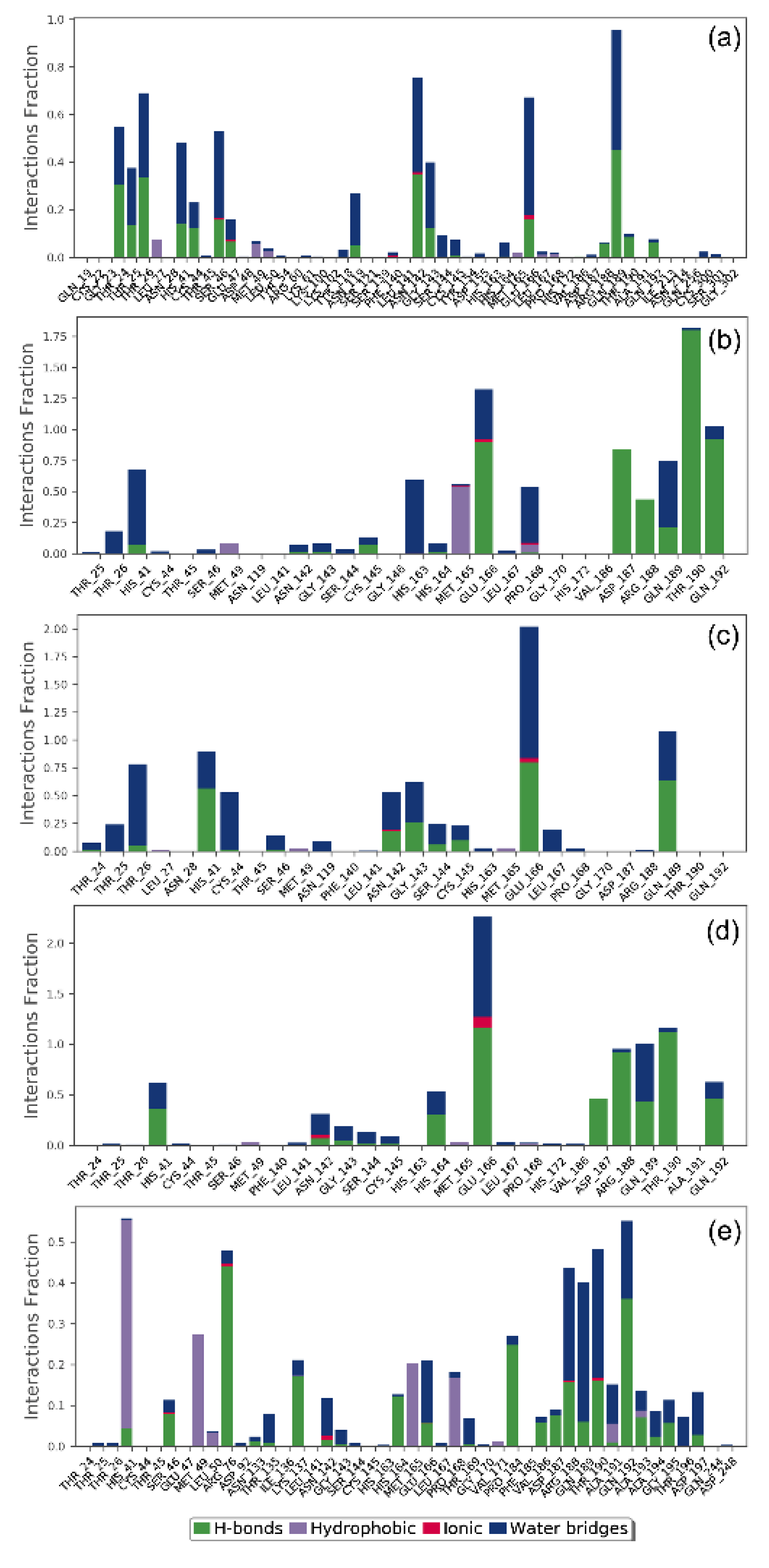
3.4.2. Protein-Ligand Contact Mapping
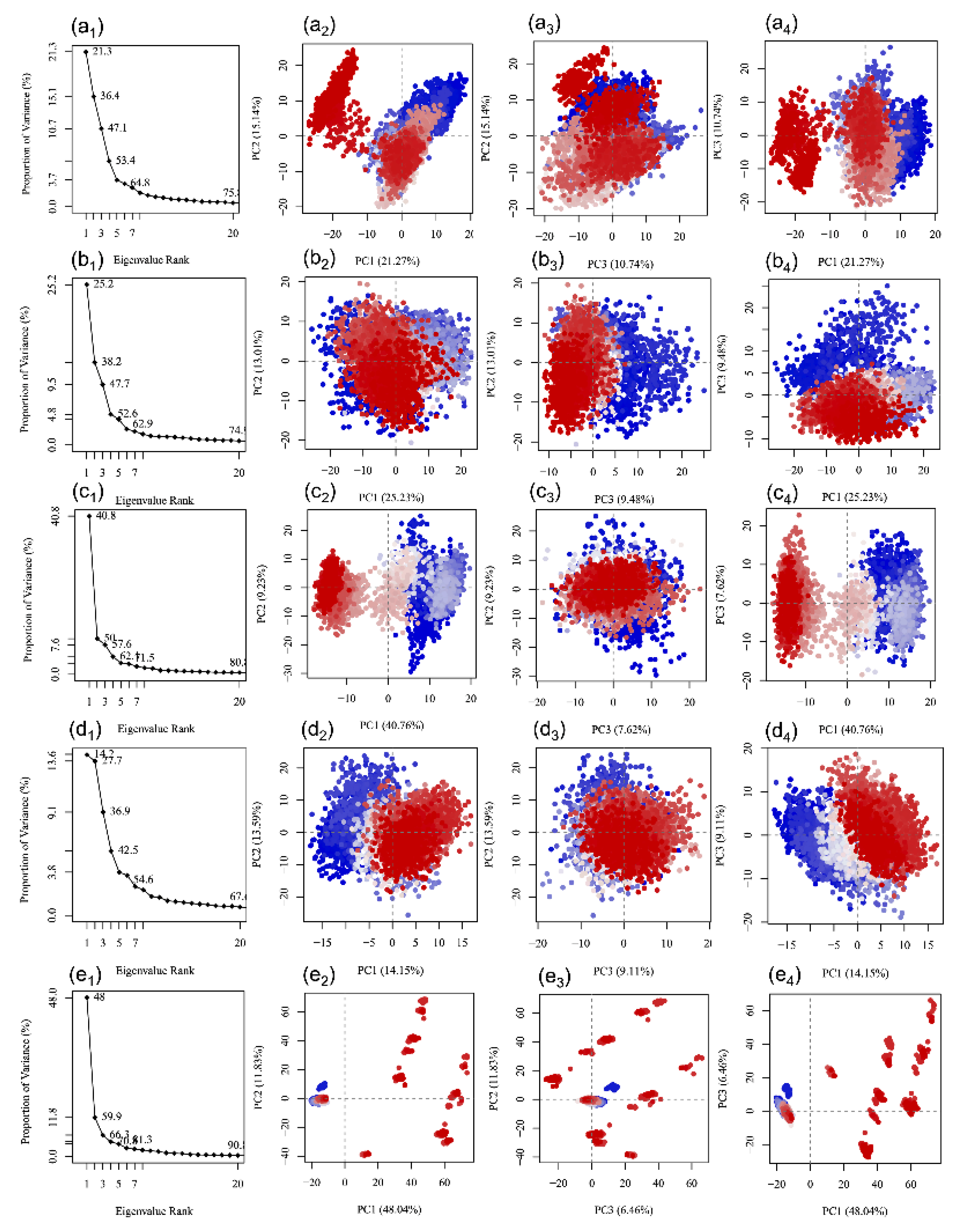
3.5. Essential Dynamics Analysis
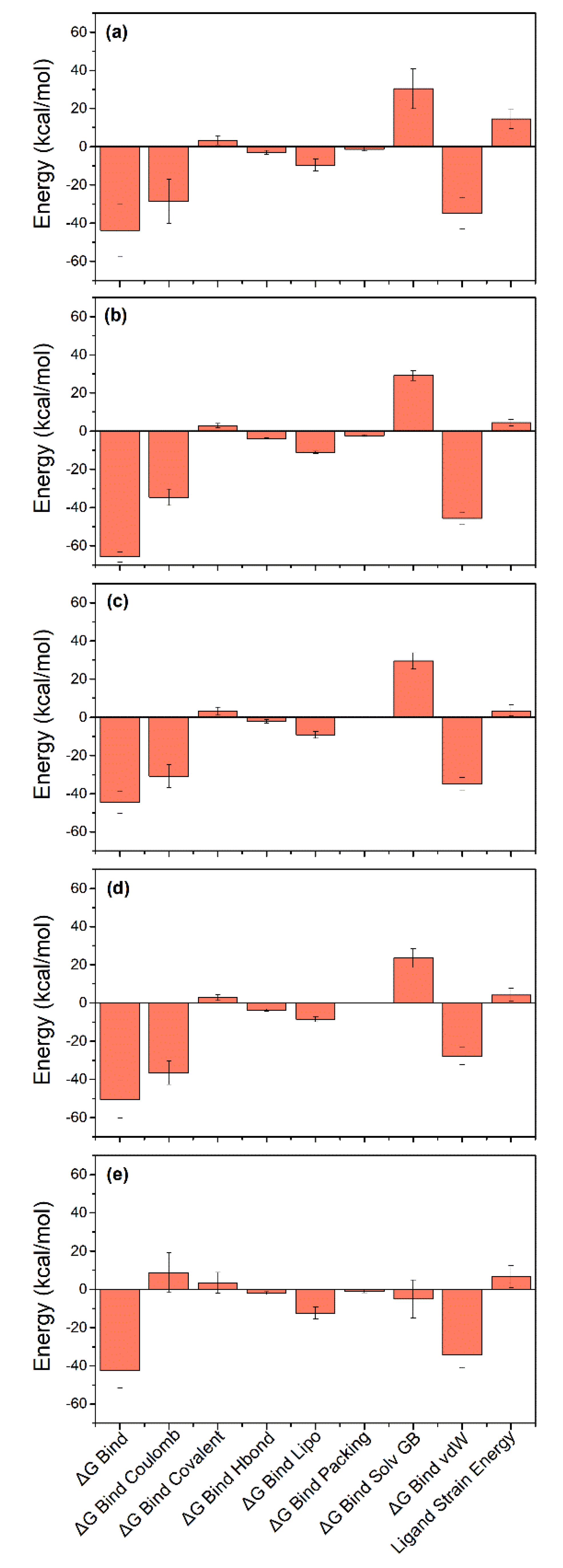
3.6. Binding Free Energy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kirtipal, N.; Bharadwaj, S.; Kang, S.G. From SARS to SARS-CoV-2, insights on structure, pathogenicity and immunity aspects of pandemic human coronaviruses. Infect. Genet. Evol. 2020, 85, 104502. [Google Scholar] [CrossRef]
- Coronavirus, N. Novel Coronavirus (2019-nCoV) Situation Reports—World Health Organization; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: Structure-based design, synthesis, and activity assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef]
- Ma, Y.; Zhao, Y.; Liu, J.; He, X.; Wang, B.; Fu, S.; Yan, J.; Niu, J.; Zhou, J.; Luo, B. Effects of temperature variation and humidity on the death of COVID-19 in Wuhan, China. Sci. Total. Environ. 2020, 724, 138226. [Google Scholar] [CrossRef] [PubMed]
- Arya, A.; Dwivedi, V.D. Synergistic effect of vitamin D and Remdesivir can fight COVID-19. J. Biomol. Struct. Dyn. 2020, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, V.D.; Tripathi, I.P.; Mishra, S.K. In silico evaluation of inhibitory potential of triterpenoids from Azadirachta indica against therapeutic target of dengue virus, NS2B-NS3 protease. J. Vector Borne Dis. 2016, 53, 156–161. [Google Scholar]
- Benarba, B.; Pandiella, A. Medicinal plants as sources of active molecules against COVID-19. Front. Pharmacol. 2020, 11, 1189. [Google Scholar] [CrossRef]
- Joseph, J.; Thankamani, K.; Ajay, A.; Das, V.R.A.; Raj, V.S. Green tea and Spirulina extracts inhibit SARS, MERS, and SARS-2 spike pseudotyped virus entry in vitro. BioRxiv 2020. [Google Scholar] [CrossRef]
- Signer, J.; Jonsdottir, H.R.; Albrich, W.C.; Strasser, M.; Zust, R.; Ryter, S.; Ackermann-Gaumann, R.; Lenz, N.; Siegrist, D.; Suter, A.; et al. In vitro virucidal activity of Echinaforce®, an Echinacea purpurea preparation, against coronaviruses, including common cold coronavirus 229E and SARS-CoV-2. Virol. J. 2020, 17, 136. [Google Scholar] [CrossRef]
- Aucoin, M.; Cooley, K.; Saunders, P.R.; Carè, J.; Anheyer, D.; Medina, D.N.; Cardozo, V.; Remy, D.; Hannan, N.; Garber, A. The effect of Echinacea spp. on the prevention or treatment of COVID-19 and other respiratory tract infections in humans: A rapid review. Adv. Integr. Med. 2020, 7, 203–217. [Google Scholar] [CrossRef]
- Kembuan, G.; Lie, W.; Tumimomor, A. Potential usage of immune modulating supplements of the Echinacea genus for COVID-19 infection. Int. J. Med. Rev. Case Rep. 2020, 4. [Google Scholar] [CrossRef]
- Hudson, J.; Vimalanathan, S. Echinacea—A source of potent antivirals for respiratory virus infections. Pharmaceuticals 2011, 4, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Barnes, J.; Anderson, L.A.; Gibbons, S.; Phillipson, J.D. Echinacea species (Echinacea angustifolia (DC.) Hell., Echinacea pallida (Nutt.) Nutt., Echinacea purpurea (L.) Moench): A review of their chemistry, pharmacology and clinical properties. J. Pharm. Pharmacol. 2005, 57, 929–954. [Google Scholar] [CrossRef] [Green Version]
- Barrett, B. Medicinal properties of Echinacea: A critical review. Phytomedicine 2003, 10, 66–86. [Google Scholar] [CrossRef]
- Rehman, F.; Sudhaker, M.; Roshan, S.; Khan, A. Antibacterial activity of Eachinacia angustfolia. Pharmacogn. J. 2012, 4, 67–70. [Google Scholar] [CrossRef]
- Hudson, J.; Vimalanathan, S.; Kang, L.; Amiguet, V.T.; Livesey, J.; Arnason, J.T. Characterization of antiviral activities in Echinacea. Root preparations. Pharm. Biol. 2005, 43, 790–796. [Google Scholar] [CrossRef]
- Fearon, D.; Powell, A.J.; Douangamath, A.; Owen, C.D.; Wild, C.; Krojer, T.; Lukacik, P.; Strain-Damerell, C.M.; Walsh, M.A.; von Delft, F. PanDDA analysis of COVID-19 main protease against the DSI-poised Fragment Library. Unpublished work. 2020. [Google Scholar]
- Erbel, P.; Schiering, N.; D’Arcy, A.; Renatus, M.; Kroemer, M.; Lim, S.P.; Yin, Z.; Keller, T.H.; Vasudevan, S.G.; Hommel, U. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat. Struct. Mol. Biol. 2006, 13, 372–373. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem substance and compound databases. Nucleic Acids Res. 2015, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E. Gaussian 03, Revision, B. 04; Gaussian, Inc.: Wallingford, CT, USA, 2003. [Google Scholar]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Lee, K.E.; Dwivedi, V.D.; Yadava, U.; Nees, M.; Kang, S.G. Density functional theory and molecular dynamics simulation support Ganoderma lucidum triterpenoids as broad range antagonist of matrix metalloproteinases. J. Mol. Liq. 2020, 311, 113322. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the ACM/IEEE SC 2006 Conference (SC’06), Tampa, FL, USA, 11–17 November 2006; IEEE: Piscataway, NJ, USA, 2006; p. 43. [Google Scholar] [CrossRef] [Green Version]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential dynamics of proteins. Proteins Struct. Funct. Bioinform. 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Daidone, I.; Amadei, A. Essential dynamics: Foundation and applications. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 762–770. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Mena-Ulecia, K.; Tiznado, W.; Caballero, J. Study of the differential activity of thrombin inhibitors using docking, QSAR, molecular dynamics, and MM-GBSA. PLoS ONE 2015, 10, e0142774. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, S.; Rao, A.K.; Dwivedi, V.D.; Mishra, S.K.; Yadava, U. Structure-based screening and validation of bioactive compounds as Zika virus methyltransferase (MTase) inhibitors through first-principle density functional theory, classical molecular simulation and QM/MM affinity estimation. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef]
- Genheden, S.; Kuhn, O.; Mikulskis, P.; Hoffmann, D.; Ryde, U. The normal-mode entropy in the MM/GBSA method: Effect of system truncation, buffer region, and dielectric constant. J. Chem. Inf. Model. 2012, 52, 2079–2088. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shalom, I.Y.; Pfeiffer-Marek, S.; Baringhaus, K.-H.; Gohlke, H. Efficient approximation of ligand rotational and translational entropy changes upon binding for use in MM-PBSA calculations. J. Chem. Inf. Model. 2017, 57, 170–189. [Google Scholar] [CrossRef]
- Duan, L.; Liu, X.; Zhang, J.Z. Interaction entropy: A new paradigm for highly efficient and reliable computation of protein–ligand binding free energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. [Google Scholar] [CrossRef]
- Genheden, S.; Akke, M.; Ryde, U. Conformational entropies and order parameters: Convergence, reproducibility, and transferability. J. Chem. Theory Comput. 2014, 10, 432–438. [Google Scholar] [CrossRef]
- Hikiri, S.; Yoshidome, T.; Ikeguchi, M. Computational methods for configurational entropy using internal and cartesian coordinates. J. Chem. Theory Comput. 2016, 12, 5990–6000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, K. Calculation of molecular entropies using temperature integration. J. Chem. Theory Comput. 2012, 9, 1164–1172. [Google Scholar] [CrossRef]
- Choi, H.; Kang, H.; Park, H. Computational Prediction of molecular hydration entropy with hybrid scaled particle theory and free-energy perturbation method. J. Chem. Theory Comput. 2015, 11, 4933–4942. [Google Scholar] [CrossRef]
- Gyimesi, G.; Závodszky, P.; Szilágyi, A. Calculation of Configurational entropy differences from conformational ensembles using gaussian mixtures. J. Chem. Theory Comput. 2016, 13, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. II. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2010, 51, 69–82. [Google Scholar] [CrossRef]
- Cheminat, A.; Zawatzky, R.; Becker, H.; Brouillard, R. Caffeoyl conjugates from Echinacea species: Structures and biological activity. Phytochemistry 1988, 27, 2787–2794. [Google Scholar] [CrossRef]
- Vohra, S.; Adams, D.; Hudson, J.; Moore, J.; Vimalanathan, S.; Sharma, M.; Burt, A.; Lamont, E.; Lacaze, N.; Arnason, J.; et al. Selection of natural health products for clinical trials: A preclinical template. Can. J. Physiol. Pharmacol. 2009, 87, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D. Chapter Two—Health benefits of prebiotic fibers. In Advances in Food and Nutrition Research; Henry, J., Ed.; Academic Press: Cambridge, MA, USA, 2015; Volume 74, pp. 47–91. [Google Scholar]
- Srikanth, R.; Reddy, C.H.; Siddartha, G.; Ramaiah, M.J.; Uppuluri, K.B. Review on production, characterization and applications of microbial levan. Carbohydr. Polym. 2015, 120, 102–114. [Google Scholar] [CrossRef]
- Esawy, M.A.; Ahmed, E.F.; Helmy, W.A.; Mansour, N.M.; El-Senousy, W.M.; El-Safty, M.M. Production of levansucrase from novel honey Bacillus subtilis isolates capable of producing antiviral levans. Carbohydr. Polym. 2011, 86, 823–830. [Google Scholar] [CrossRef]
- Peiris, M. Pathogenesis of Avian flu H5N1 and SARS. Novartis Found. Symp. 2006, 279, 56–65. [Google Scholar]
- Wan, X.; Guo, H.; Liang, Y.; Zhou, C.; Liu, Z.; Li, K.; Niu, F.; Zhai, X.; Wang, L. The physiological functions and pharmaceutical applications of inulin: A review. Carbohydr. Polym. 2020, 246, 116589. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-B.; Miyake, S.; Umetsu, R.; Hayashi, K.; Chijimatsu, T.; Hayashi, T. Anti-influenza A virus effects of fructan from Welsh onion (Allium fistulosum L.). Food Chem. 2012, 134, 2164–2168. [Google Scholar] [CrossRef] [PubMed]
- Danino, O.; Gottlieb, H.E.; Grossman, S.; Bergman, M. Antioxidant activity of 1,3-dicaffeoylquinic acid isolated from Inula visosa. Food Res. Int. 2009, 42, 1273–1280. [Google Scholar] [CrossRef]
- Slanina, J.; Táborská, E.; Bochořáková, H.; Slaninová, I.; Humpa, O.; Robinson, W.; Schram, K.H. New and facile method of preparation of the anti-HIV-1 agent, 1,3-dicaffeoylquinic acid. Tetrahedron Lett. 2001, 42, 3383–3385. [Google Scholar] [CrossRef]
- Li, L.; Chang, S.; Xiang, J.; Li, Q.; Liang, H.; Tang, Y.; Liu, Y. Screen anti-influenza lead compounds that target the PA C subunit of H5N1 viral RNA polymerase. PLoS ONE 2012, 7, e35234. [Google Scholar]
- Fukui, K. The role of frontier orbitals in chemical reactions (Nobel Lecture). Angew. Chem. Int. Ed. 1982, 21, 801–809. [Google Scholar] [CrossRef]
- Pearson, R.G. Electronic spectra and chemical reactivity. J. Am. Chem. Soc. 1988, 110, 2092–2097. [Google Scholar] [CrossRef]
- Thanthiriwatte, K.S.; De Silva, K.N. Non-linear optical properties of novel fluorenyl derivatives—Ab initio quantum chemical calculations. J. Mol. Struct. THEOCHEM 2002, 617, 169–175. [Google Scholar] [CrossRef]
- Gohlke, H.; Klebe, G. Approaches to the description and prediction of the binding affinity of small-molecule ligands to mac-romolecular receptors. Angew. Chem. Int. Ed. Engl. 2002, 41, 2644–2676. [Google Scholar] [CrossRef]
- Thangavel, M.; Chandramohan, V.; Shankaraiah, L.H.; Jayaraj, R.L.; Poomani, K.; Magudeeswaran, S.; Govindasamy, H.; Vijayakumar, R.; Rangasamy, B.; Dharmar, M. Design and molecular dynamic Investigations of 7, 8-Dihydroxyflavone derivatives as potential neuroprotective agents against alpha-synuclein. Sci. Rep. 2020, 10, 1–10. [Google Scholar]
- Connelly, P.R.; Snyder, P.W.; Zhang, Y.; McClain, B.; Quinn, B.P.; Johnston, S.; Medek, A.; Tanoury, J.; Griffith, J.; Walters, W.P.; et al. The potency–insolubility conundrum in pharmaceuticals: Mechanism and solution for hepatitis C protease inhibitors. Biophys. Chem. 2015, 196, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Sanphui, P.; Rajput, L.; Gopi, S.P.; Desiraju, G.R. New multi-component solid forms of anti-cancer drug Erlotinib: Role of auxiliary interactions in determining a preferred conformation. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 291–300. [Google Scholar] [CrossRef]
- Brigo, A.; Lee, K.W.; Mustata, G.I.; Briggs, J.M. Comparison of multiple molecular dynamics trajectories calculated for the drug-resistant HIV-1 integrase T66I/M154I catalytic domain. Biophys. J. 2005, 88, 3072–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsui, V.; Case, D.A. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers 2000, 56, 275–291. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Azhar, E.I.; Kamal, M.A.; Bajrai, L.H.; Dubey, A.; Jha, K.; Yadava, U.; Kang, S.G.; Dwivedi, V.D. SARS-CoV-2 M(pro) inhibitors: Identification of anti-SARS-CoV-2 M(pro) compounds from FDA approved drugs. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. no. | Compound | Docking Score (kcal/mol) | H-Bond | π-π/ * π- Cation Stacking | Hydrophobic | Polar | Negative | Positive | Glycine/ * Salt Bridge |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Echinacoside | −14.17 | Thr25, Cys44, Asn142, Gly143, Gln189, Thr190 | - | Leu27, Val42, Cys44, Met49, Phe140, Leu141, Cys145, Met165, Leu167, Pro168, Ala191 | Thr24, Thr25, His41, Thr45, Ser46, Asn142, Ser144, His163, His164, His172, Gln189, Thr190, Gln192 | Glu166 | Arg188 | Gly143 |
| 2 | Quercetagetin 7-Glucoside | −15.20 | Cys44(2), Leu141, Cys145, Glu166(2), Gln189 | * His41 | Leu27, Cys44, Met49, Phe140, Leu141, Cys145, Met165, Leu167, Pro168 | Thr24, Thr25, Thr26, His41, Thr45, Ser46, Asn142, Ser144, His163, Gln189 | Glu166 | - | Gly143 |
| 3 | Levan N | −12.92 | His41, Cys44 Asn142, Gly143, Gln189(3) | - | Val42, Cys44, Met49, Leu141, Cys145, Met165 | Thr24, Thr25, His41, Thr45, Ser46, Asn142, Ser144, His164, Gln189 | Glu166 | Arg188 | Gly143 |
| 4 | Inulin From Chicory | −11.72 | Leu141, Gly143, Glu166 (2), Gln189(2) | - | Met49, Phe140, Leu141, Cys145, Met165, Leu167, Pro168 | His41, Asn142, Ser144, His163, His164, Gln189, Thr190, Gln192 | Glu166 | Arg188 | Gly143 |
| 5 | 1,3-Dicaffeoylquinic Acid | −10.01 | Thr26,Thr25, Gly143, Arg188 (2) | - | Leu27, Cys44, Met49, Cys145, Met165, Leu167, Val186 | Thr24, Thr25, Thr26, His41, Thr45, Ser46, Asn142, Ser144, Gln189, Thr190, Gln192 | Glu166, Asp187 | Arg188 | Gly143/* His41 |
| 6 | 6-(ethylamino)pyridine-3-carbonitrile | −3.57 | Arg188 | - | Met49, Cys145, Met165, Leu167, Pro168 | His41, His164, Gln189, Thr190, Gln192 | Glu166, Asp187 | Arg188 | Gly143 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bharadwaj, S.; El-Kafrawy, S.A.; Alandijany, T.A.; Bajrai, L.H.; Shah, A.A.; Dubey, A.; Sahoo, A.K.; Yadava, U.; Kamal, M.A.; Azhar, E.I.; et al. Structure-Based Identification of Natural Products as SARS-CoV-2 Mpro Antagonist from Echinacea angustifolia Using Computational Approaches. Viruses 2021, 13, 305. https://doi.org/10.3390/v13020305
Bharadwaj S, El-Kafrawy SA, Alandijany TA, Bajrai LH, Shah AA, Dubey A, Sahoo AK, Yadava U, Kamal MA, Azhar EI, et al. Structure-Based Identification of Natural Products as SARS-CoV-2 Mpro Antagonist from Echinacea angustifolia Using Computational Approaches. Viruses. 2021; 13(2):305. https://doi.org/10.3390/v13020305
Chicago/Turabian StyleBharadwaj, Shiv, Sherif Aly El-Kafrawy, Thamir A. Alandijany, Leena Hussein Bajrai, Altaf Ahmad Shah, Amit Dubey, Amaresh Kumar Sahoo, Umesh Yadava, Mohammad Amjad Kamal, Esam Ibraheem Azhar, and et al. 2021. "Structure-Based Identification of Natural Products as SARS-CoV-2 Mpro Antagonist from Echinacea angustifolia Using Computational Approaches" Viruses 13, no. 2: 305. https://doi.org/10.3390/v13020305
APA StyleBharadwaj, S., El-Kafrawy, S. A., Alandijany, T. A., Bajrai, L. H., Shah, A. A., Dubey, A., Sahoo, A. K., Yadava, U., Kamal, M. A., Azhar, E. I., Kang, S. G., & Dwivedi, V. D. (2021). Structure-Based Identification of Natural Products as SARS-CoV-2 Mpro Antagonist from Echinacea angustifolia Using Computational Approaches. Viruses, 13(2), 305. https://doi.org/10.3390/v13020305