
The Mutation Profile of SARS-CoV-2 Is Primarily Shaped by the Host Antiviral Defense


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
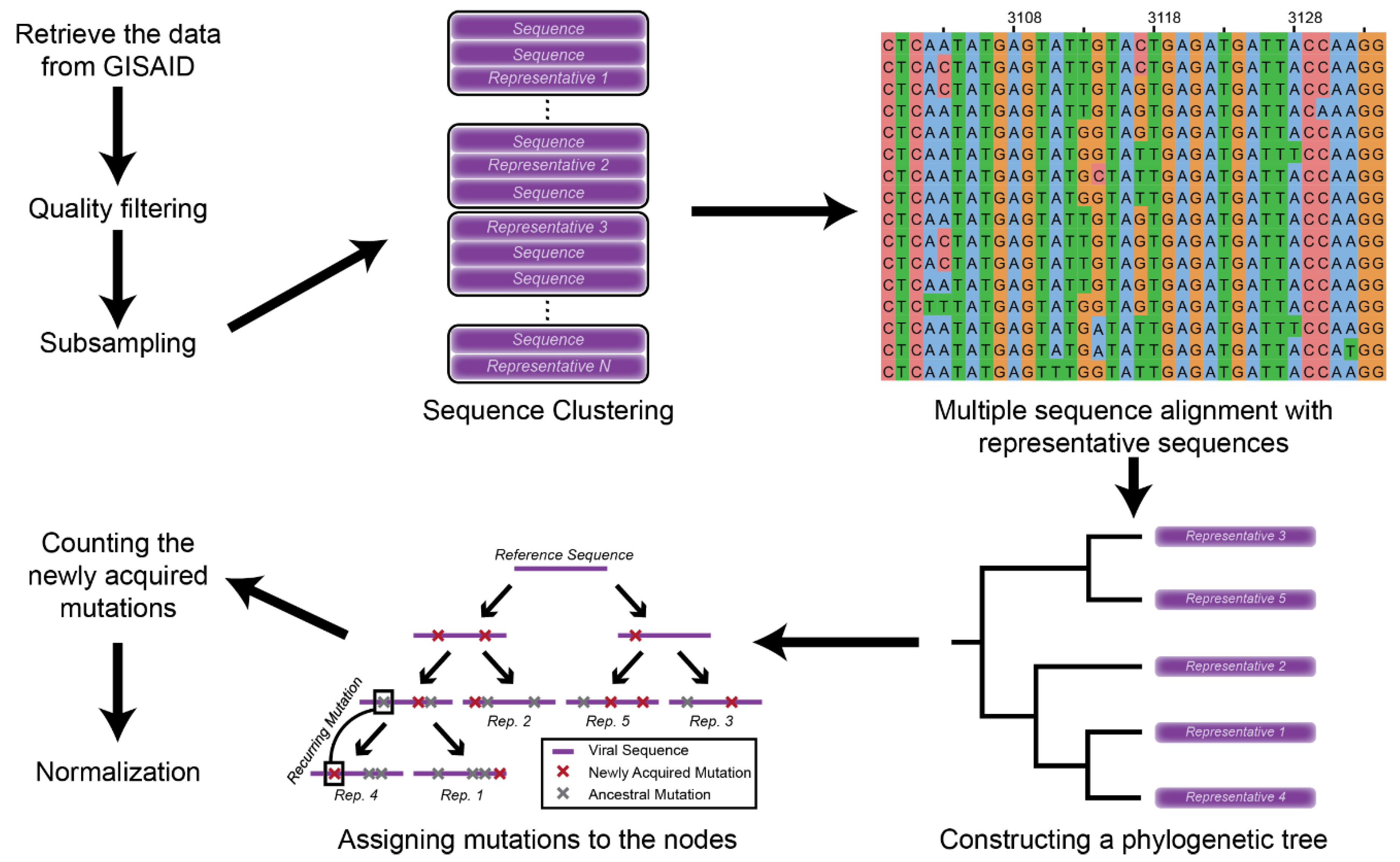
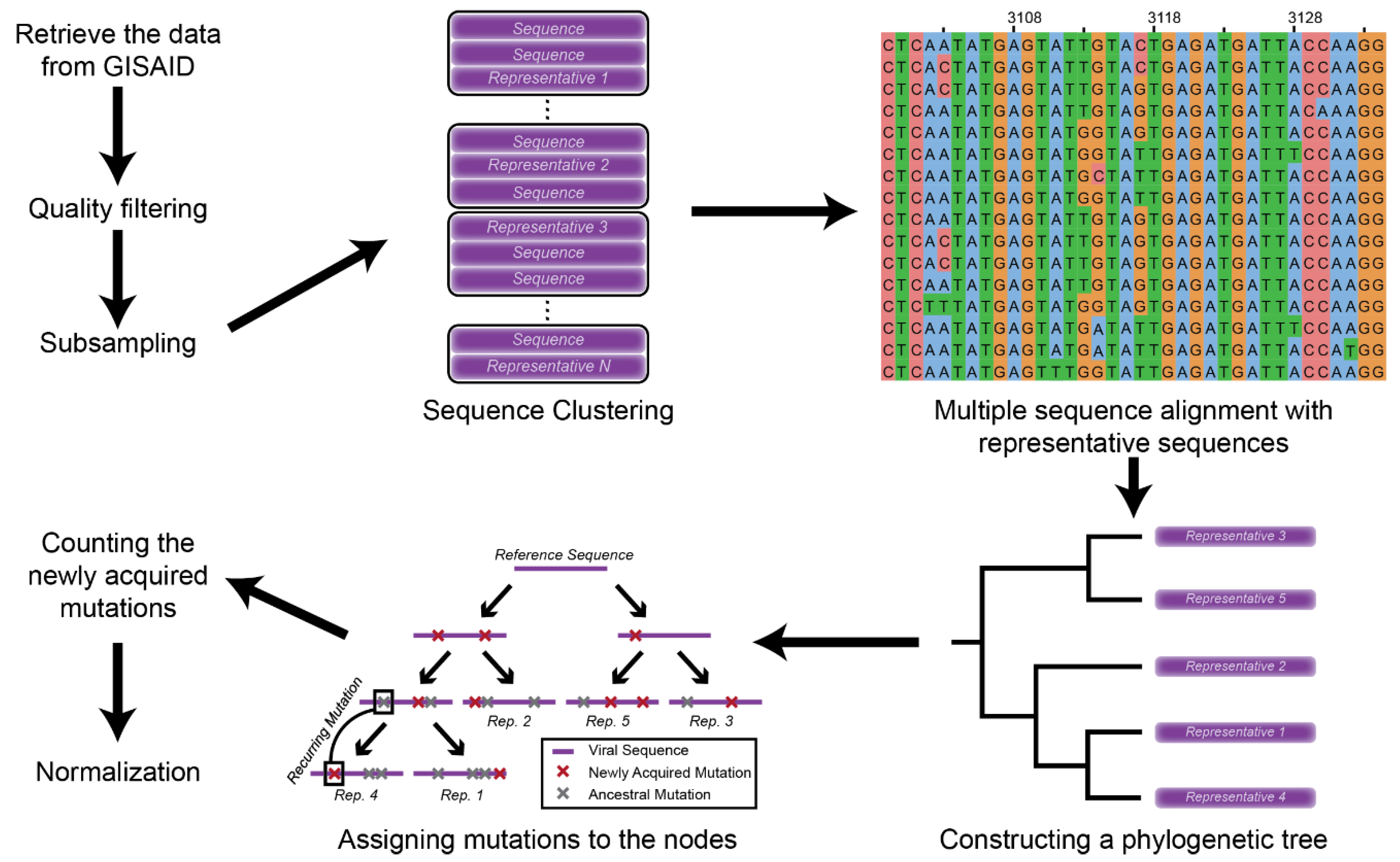
2.1. Data Retrieval and Mutation Assignment
2.2. Mutation Profile Analysis
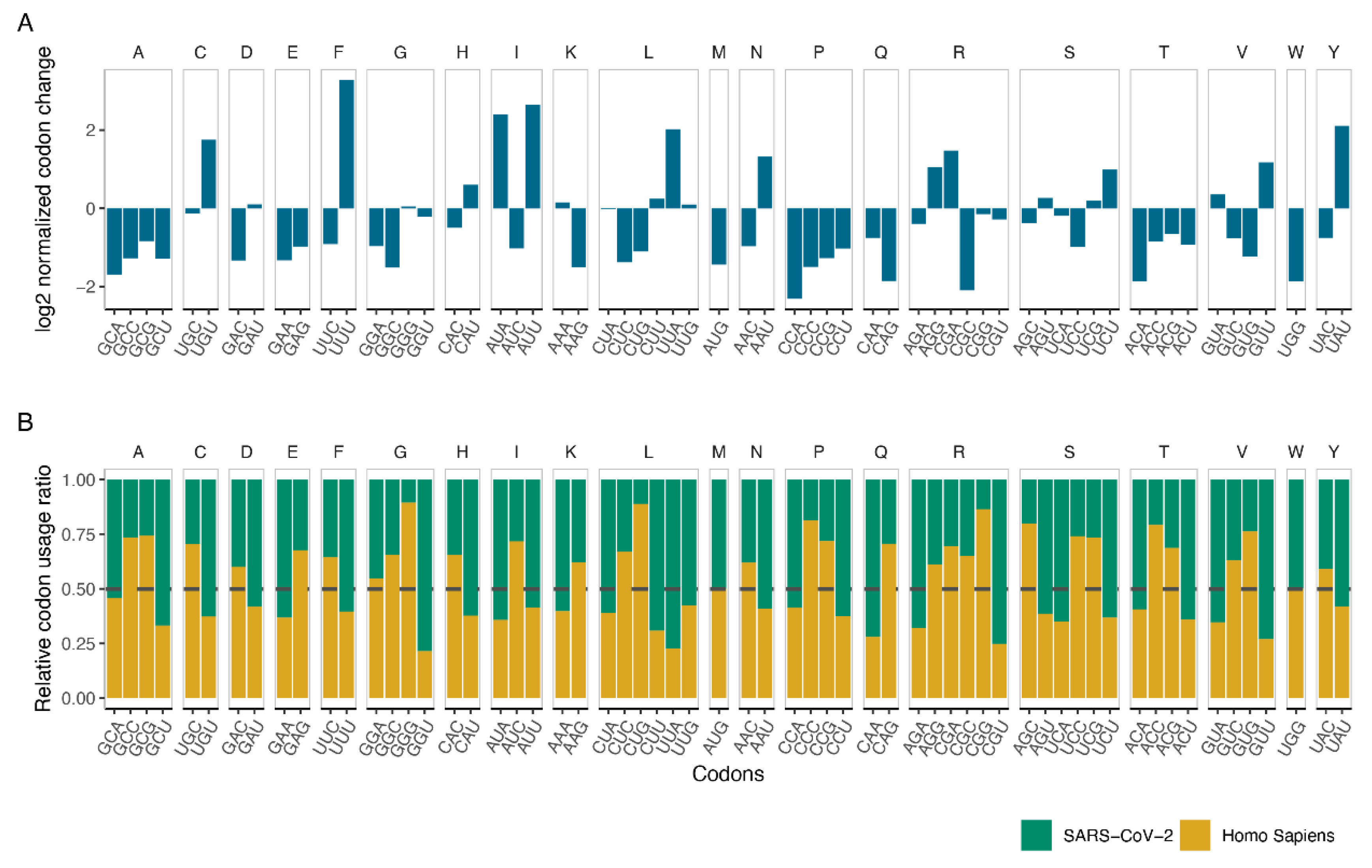
2.3. Measuring Codon Changes and Codon Usage
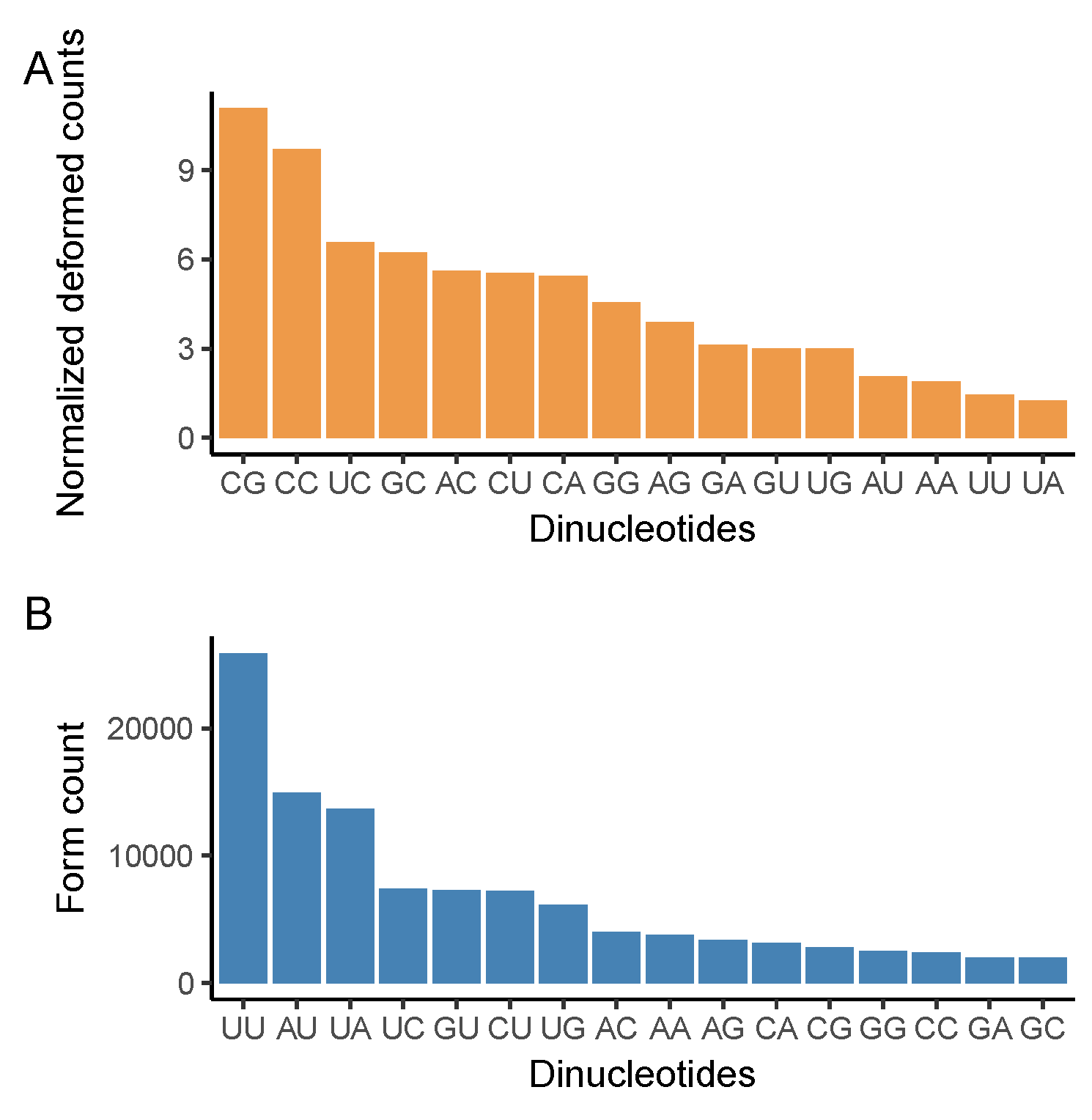
2.4. Dinucleotide Changes
3. Results
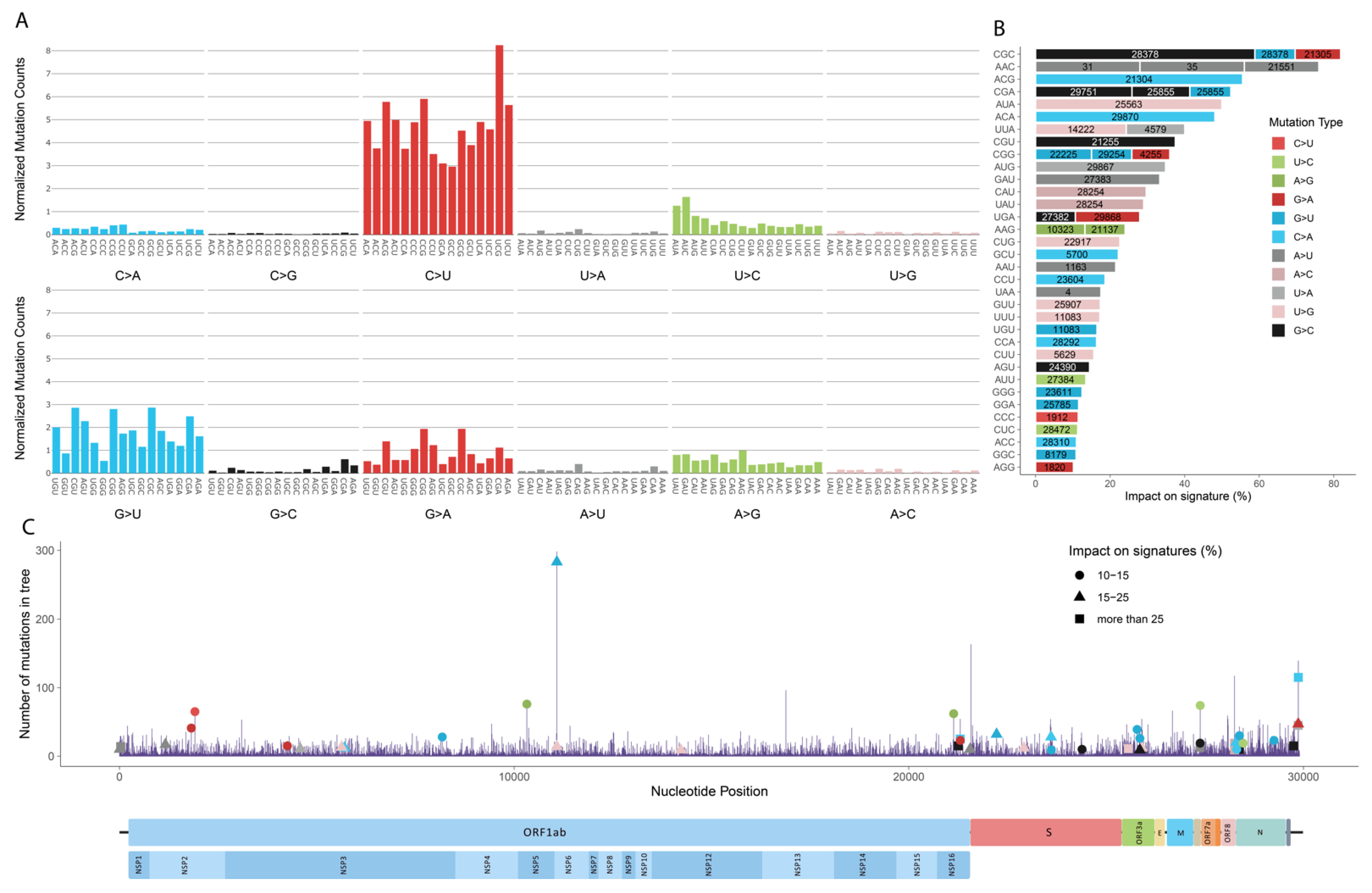
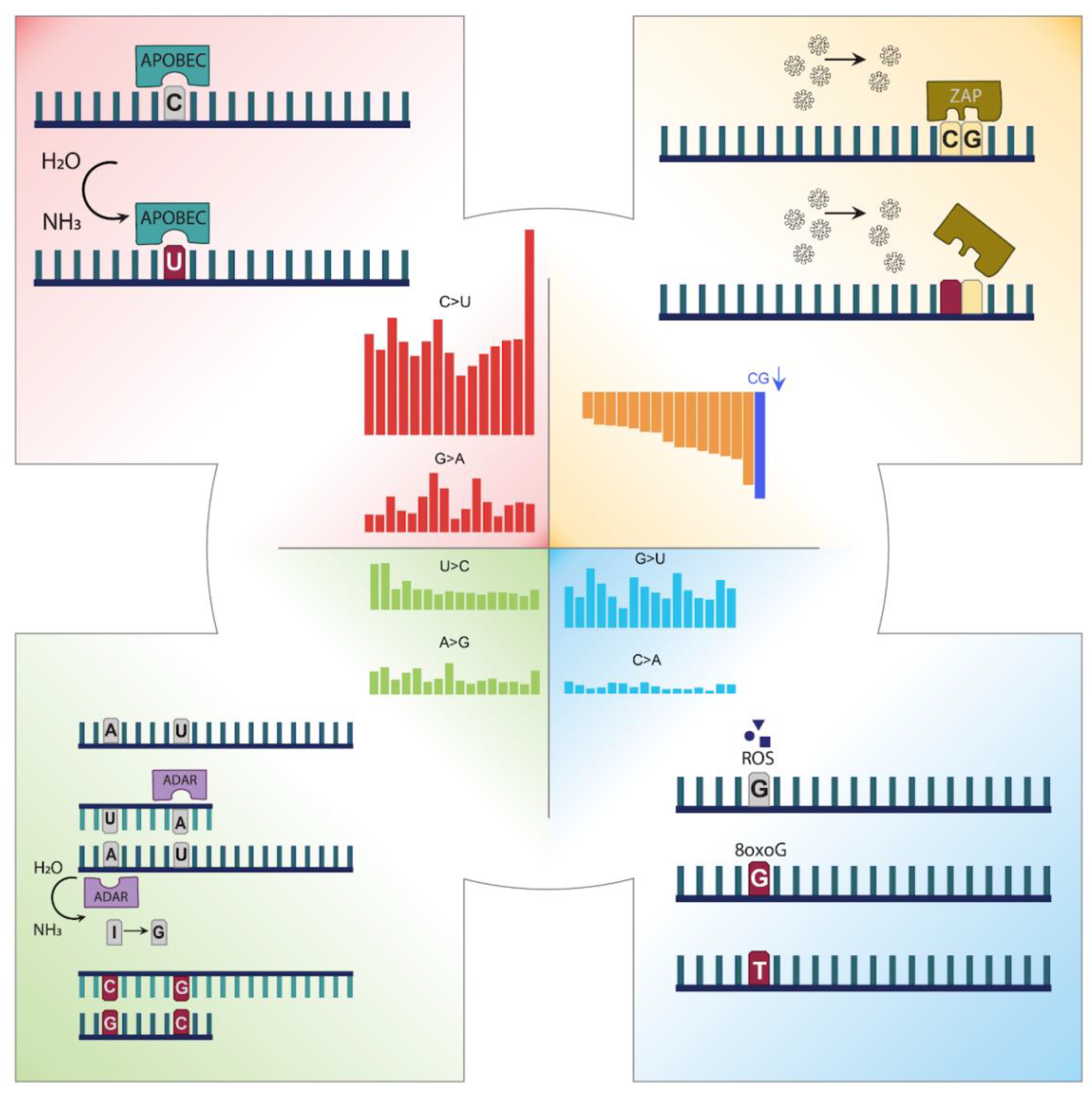
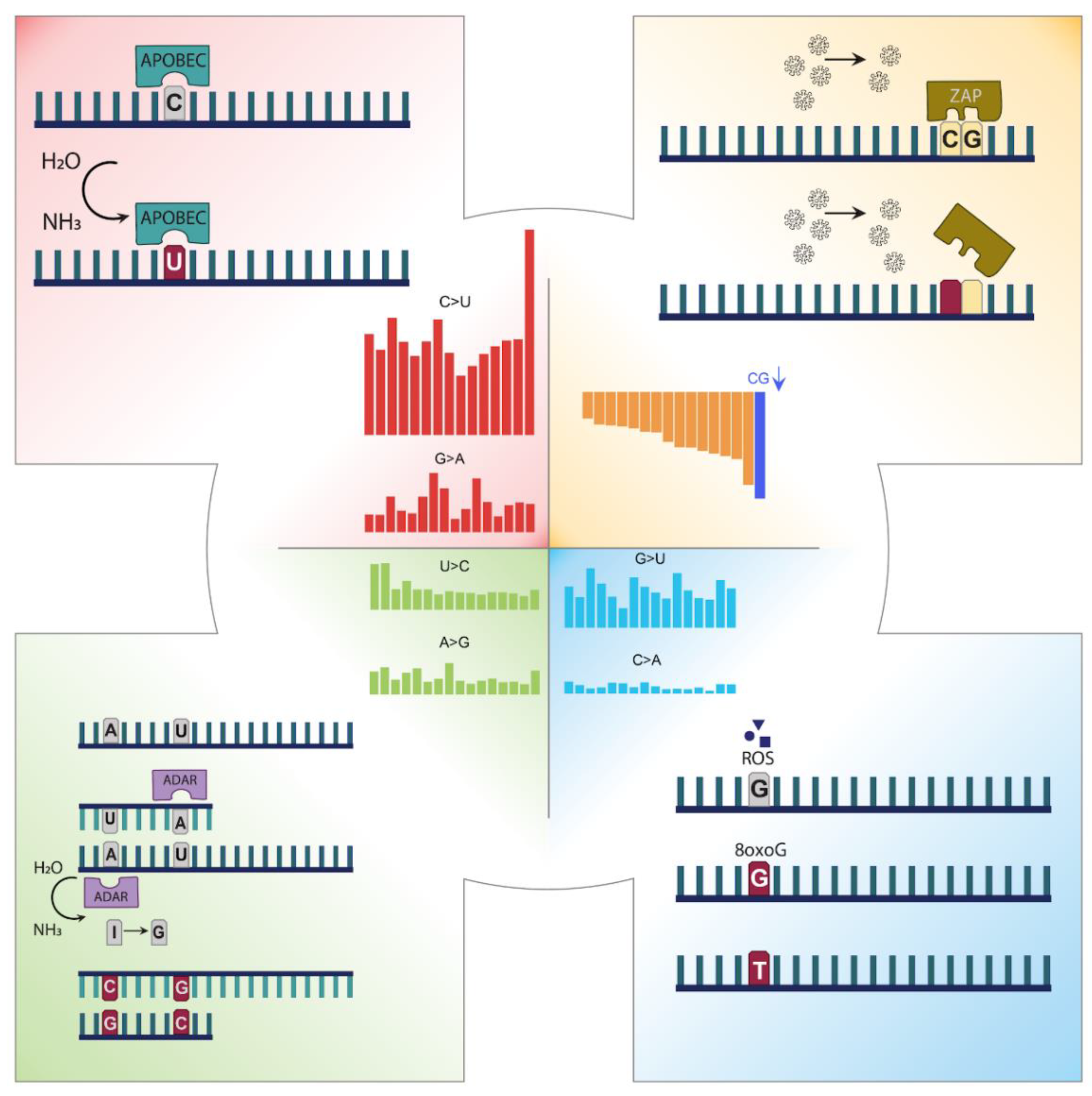
3.1. Mutation Profile of SARS-CoV-2 and Potentially Related Mechanisms
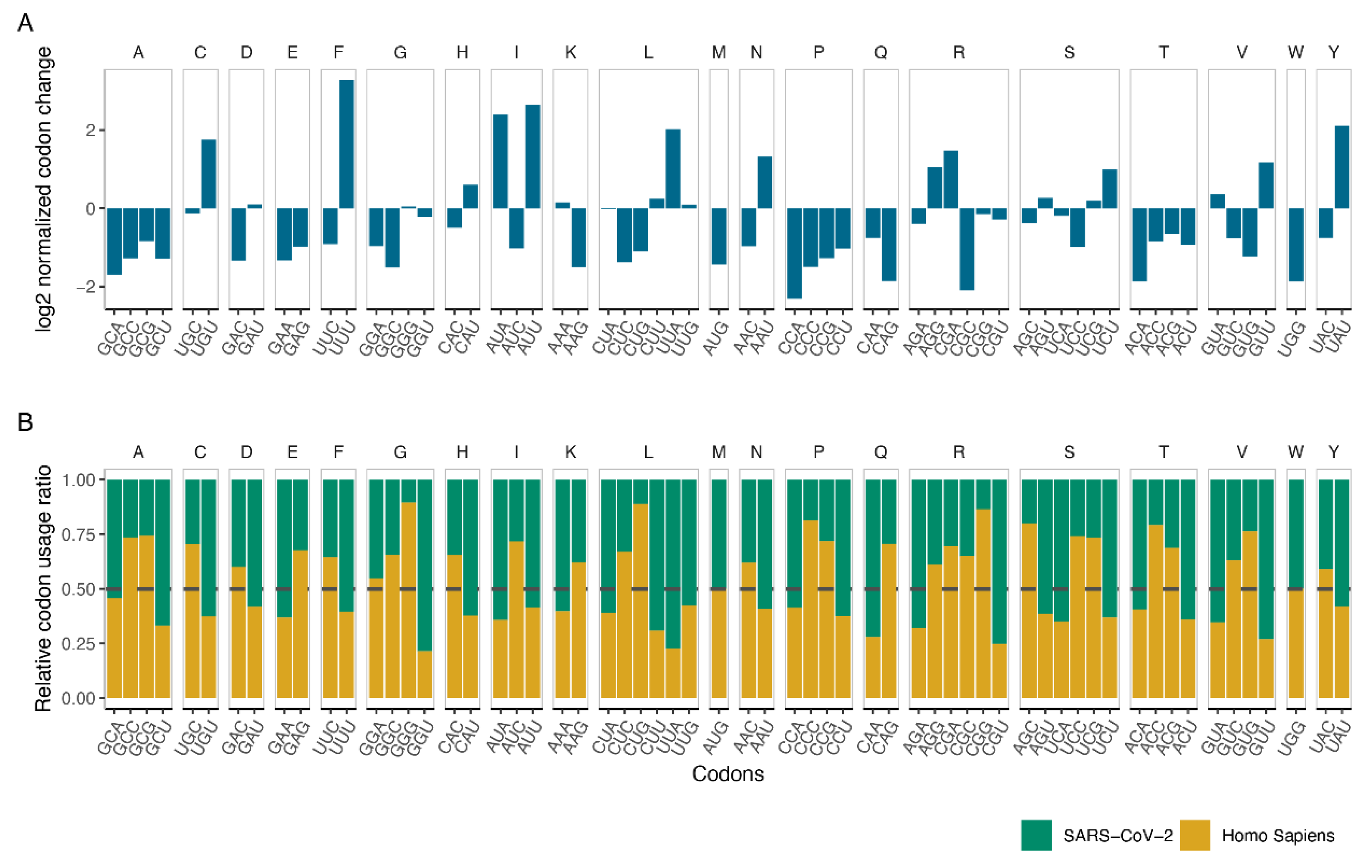
3.2. Codon Usage of SARS-CoV-2 Differentiates in Favor of A and U Containing Codons
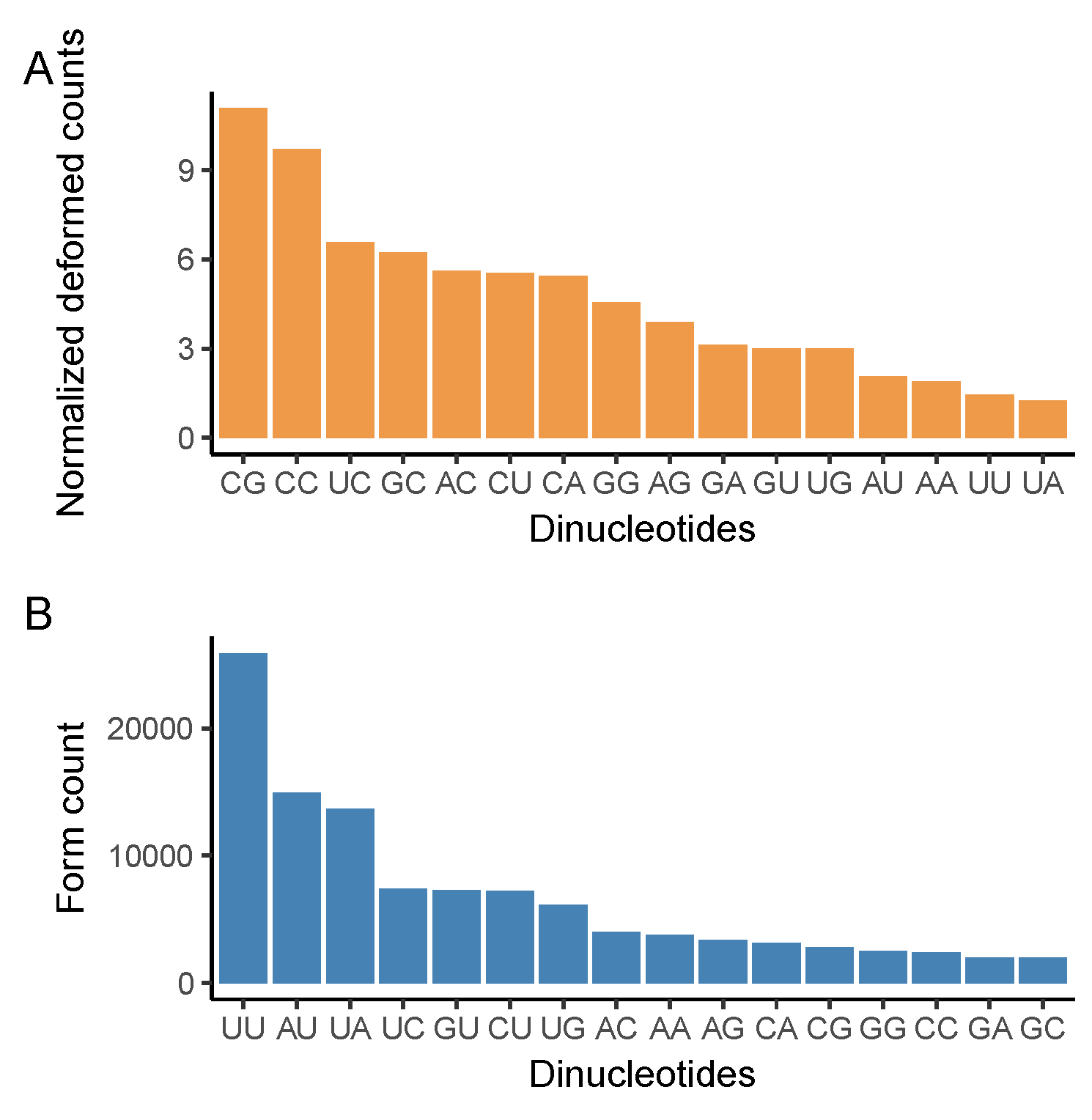
3.3. CG Nucleotide Deforms, While UU Nucleotide Forms
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2020, 1–14. [Google Scholar]
- Shah, B.; Modi, P.; Sagar, S.R. In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci. 2020, 252, 117652. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yang, Y.; Ren, L. Genetic evolution analysis of 2019 novel coronavirus and coronavirus from other species. Infect. Genet. Evol. 2020, 82, 104285. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. Author Correction: A new coronavirus associated with human respiratory disease in China. Nature 2020, 580, E7. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2020, 184, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Adebali, O.; Bircan, A.; Circi, D.; İşlek, B.; Kilinc, Z.; Selcuk, B.; Turhan, B. Phylogenetic analysis of SARS-CoV-2 genomes in Turkey. Turk. J. Biol. 2020, 44, 146–156. [Google Scholar] [CrossRef]
- Popa, A.; Genger, J.W.; Nicholson, M.D.; Penz, T.; Schmid, D.; Aberle, S.W.; Agerer, B.; Lercher, A.; Endler, L.; Colaço, H.; et al. Genomic epidemiology of superspreading events in Austria reveals mutational dynamics and transmission properties of SARS-CoV-2. Sci. Transl. Med. 2020, 12, 573. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data-from vision to reality. Euro. Surveill. 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics 2016, 32, 1933–1942. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York City, NY, USA, 2016. [Google Scholar]
- Nakamura, Y.; Gojobori, T.; Ikemura, T. Codon usage tabulated from international DNA sequence databases: Status for the year 2000. Nucleic Acids Res. 2000, 28, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Dudley, J.P. APOBECs and virus restriction. Virology 2015, 479–480, 131–145. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Patnaik, S.K.; Taggart, R.T.; Kannisto, E.D.; Enriquez, S.M.; Gollnick, P.; Baysal, B.E. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat. Commun. 2015, 6, 6881. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Wong, B.H.; Huang, Y.; Lau, S.K.; Yuen, K.-Y. Cytosine deamination and selection of CpG suppressed clones are the two major independent biological forces that shape codon usage bias in coronaviruses. Virology 2007, 369, 431–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giorgio, S.; Martignano, F.; Torcia, M.G.; Mattiuz, G.; Conticello, S.G. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci. Adv. 2020, 6, eabb5813. [Google Scholar] [CrossRef] [PubMed]
- Graudenzi, A.; Maspero, D.; Angaroni, F.; Piazza, R.; Ramazzotti, D. Mutational signatures and heterogeneous host response revealed via large-scale characterization of SARS-CoV-2 genomic diversity. BioRxiv 2020, 102116. [Google Scholar]
- Simmonds, P. Rampant C-->U Hypermutation in the Genomes of SARS-CoV-2 and Other Coronaviruses: Causes and Consequences for Their Short- and Long-Term Evolutionary Trajectories. mSphere 2020, 5, 3. [Google Scholar] [CrossRef]
- McDaniel, Y.Z.; Wang, D.; Love, R.P.; Adolph, M.B.; Mohammadzadeh, N.; Chelico, L.; Mansky, L.M. Deamination hotspots among APOBEC3 family members are defined by both target site sequence context and ssDNA secondary structure. Nucleic Acids Res. 2020, 48, 1353–1371. [Google Scholar] [CrossRef]
- Schmidt, N.; Lareau, C.A.; Keshishian, H.; Ganskih, S.; Schneider, C.; Hennig, T.; Melanson, R.; Werner, S.; Wei, Y.; Zimmer, M.; et al. The SARS-CoV-2 RNA-protein interactome in infected human cells. Nat. Microbiol. 2020, 6, 339–353. [Google Scholar] [CrossRef]
- Niocel, M.; Appourchaux, R.; Nguyen, X.-N.; Delpeuch, M.; Cimarelli, A. The DNA damage induced by the Cytosine Deaminase APOBEC3A Leads to the production of ROS. Sci. Rep. 2019, 9, 4714. [Google Scholar] [CrossRef] [PubMed]
- Molteni, C.G.; Principi, N.; Esposito, S. Reactive oxygen and nitrogen species during viral infections. Free Radic. Res. 2014, 48, 1163–1169. [Google Scholar] [CrossRef]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, Y.; Nemoto, K.; Matsumoto, S.; Nakamura, Y.; Kiyotani, K. SARS-CoV-2 genomic variations associated with mortality rate of COVID-19. J. Hum. Genet. 2020, 65, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Berrio, A.; Gartner, V.; Wray, G.A. Positive selection within the genomes of SARS-CoV-2 and other Coronaviruses independent of impact on protein function. PeerJ 2020, 8, e10234. [Google Scholar] [CrossRef]
- Van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.S.; Boshier, F.A.T.; et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Yin, C. Genotyping coronavirus SARS-CoV-2: Methods and implications. Genomics 2020, 112, 3588–3596. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2020, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, B.; van Hemert, F. On the biased nucleotide composition of the human coronavirus RNA genome. Virus Res. 2015, 202, 41–47. [Google Scholar] [CrossRef]
- Xia, X. Extreme Genomic CpG Deficiency in SARS-CoV-2 and Evasion of Host Antiviral Defense. Mol. Biol. Evol. 2020, 37, 2699–2705. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, X.; Wang, N.; Wang, H.; Yin, B.; Yang, X.; Jiang, W. GC usage of SARS-CoV-2 genes might adapt to the environment of human lung expressed genes. Mol. Genet. Genom. 2020, 295, 1537–1546. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mao, J.M.; Wang, G.D.; Luo, Z.P.; Yang, L.; Yao, Q.; Chen, K.P. Human SARS-CoV-2 has evolved to reduce CG dinucleotide in its open reading frames. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kosuge, M.; Furusawa-Nishii, E.; Ito, K.; Saito, Y.; Ogasawara, K. Point mutation bias in SARS-CoV-2 variants results in increased ability to stimulate inflammatory responses. Sci. Rep. 2020, 10, 17766. [Google Scholar] [CrossRef]
- Klimczak, L.J.; Randall, T.A.; Saini, N.; Li, J.L.; Gordenin, D.A. Similarity between mutation spectra in hypermutated genomes of rubella virus and in SARS-CoV-2 genomes accumulated during the COVID-19 pandemic. PLoS ONE 2020, 15, e0237689. [Google Scholar] [CrossRef]
- Caudill, V.R.; Qin, S.; Winstead, R.; Kaur, J.; Tisthammer, K.; Pineda, E.G.; Solis, C.; Cobey, S.; Bedford, T.; Carja, O.; et al. CpG-creating mutations are costly in many human viruses. Evol. Ecol. 2020, 34, 339–359. [Google Scholar] [CrossRef] [Green Version]
- Chand, M. Investigation of Novel SARS-COV-2 Variant: Variant of Concern 202012/01; Public Health England: London, UK, 2020. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azgari, C.; Kilinc, Z.; Turhan, B.; Circi, D.; Adebali, O. The Mutation Profile of SARS-CoV-2 Is Primarily Shaped by the Host Antiviral Defense. Viruses 2021, 13, 394. https://doi.org/10.3390/v13030394
Azgari C, Kilinc Z, Turhan B, Circi D, Adebali O. The Mutation Profile of SARS-CoV-2 Is Primarily Shaped by the Host Antiviral Defense. Viruses. 2021; 13(3):394. https://doi.org/10.3390/v13030394
Chicago/Turabian StyleAzgari, Cem, Zeynep Kilinc, Berk Turhan, Defne Circi, and Ogun Adebali. 2021. "The Mutation Profile of SARS-CoV-2 Is Primarily Shaped by the Host Antiviral Defense" Viruses 13, no. 3: 394. https://doi.org/10.3390/v13030394
APA StyleAzgari, C., Kilinc, Z., Turhan, B., Circi, D., & Adebali, O. (2021). The Mutation Profile of SARS-CoV-2 Is Primarily Shaped by the Host Antiviral Defense. Viruses, 13(3), 394. https://doi.org/10.3390/v13030394