
Mammalian and Avian Host Cell Influenza A Restriction Factors

 , , , and
, , , and 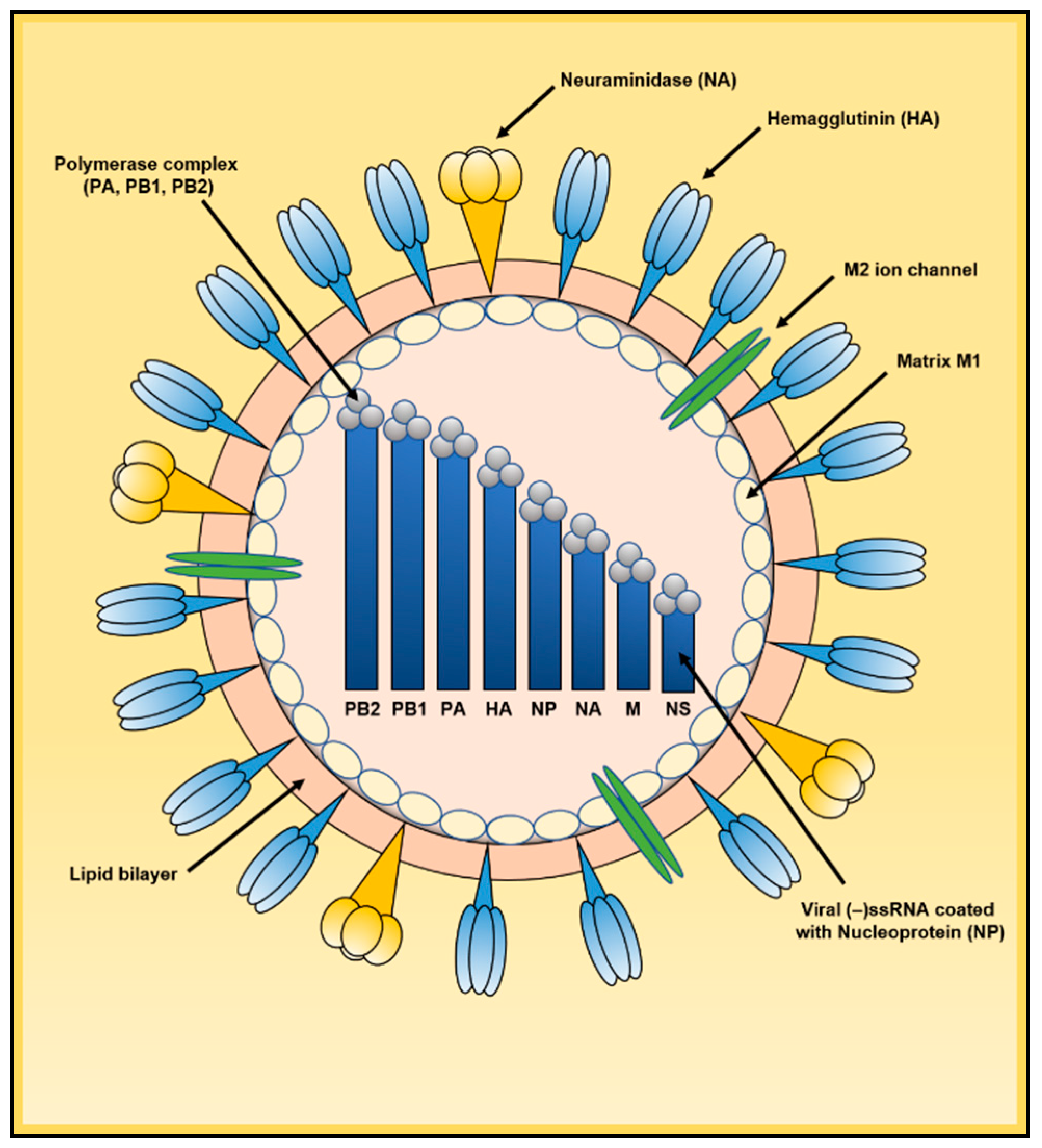{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
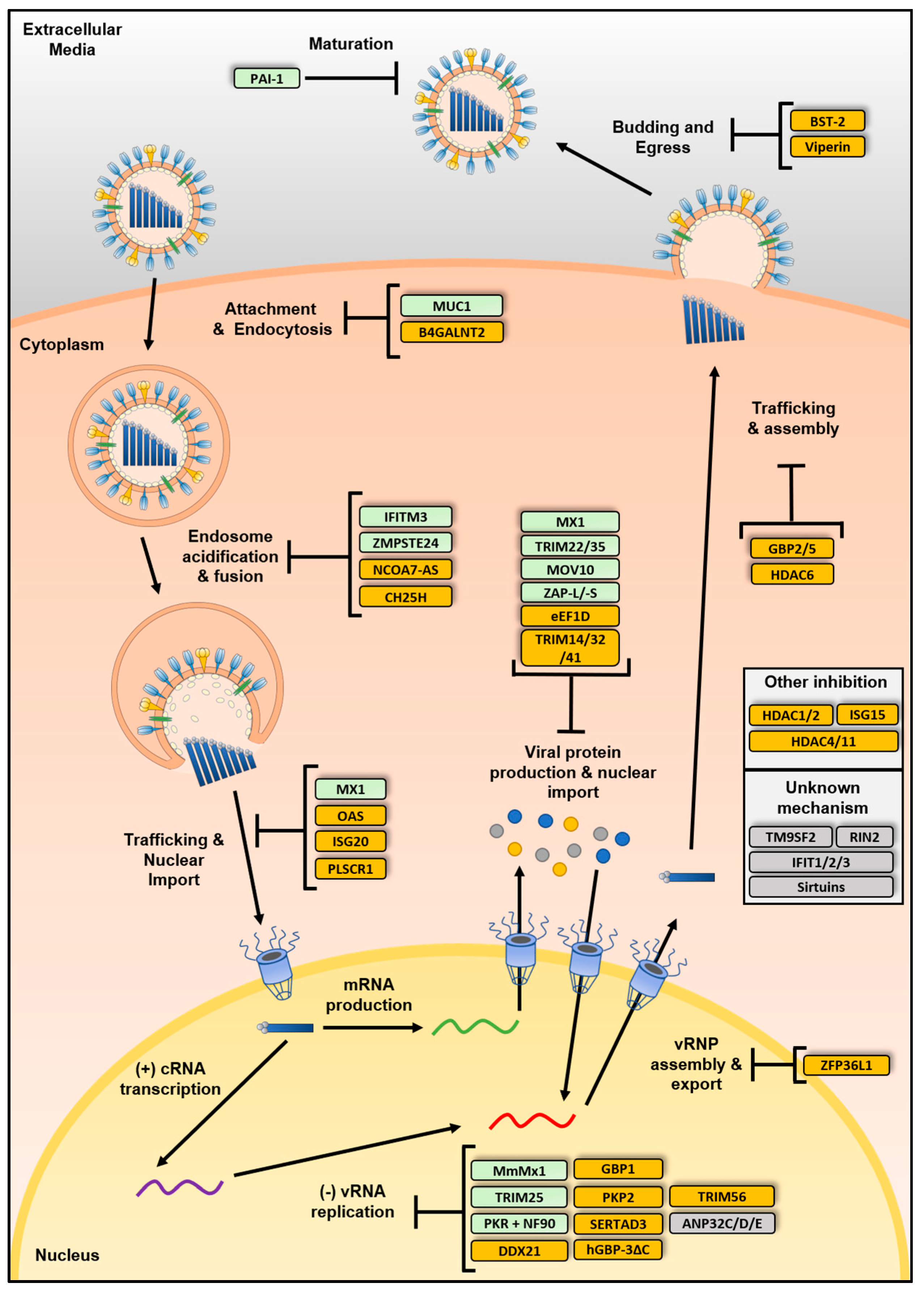
2. Influenza A Virus Life Cycle
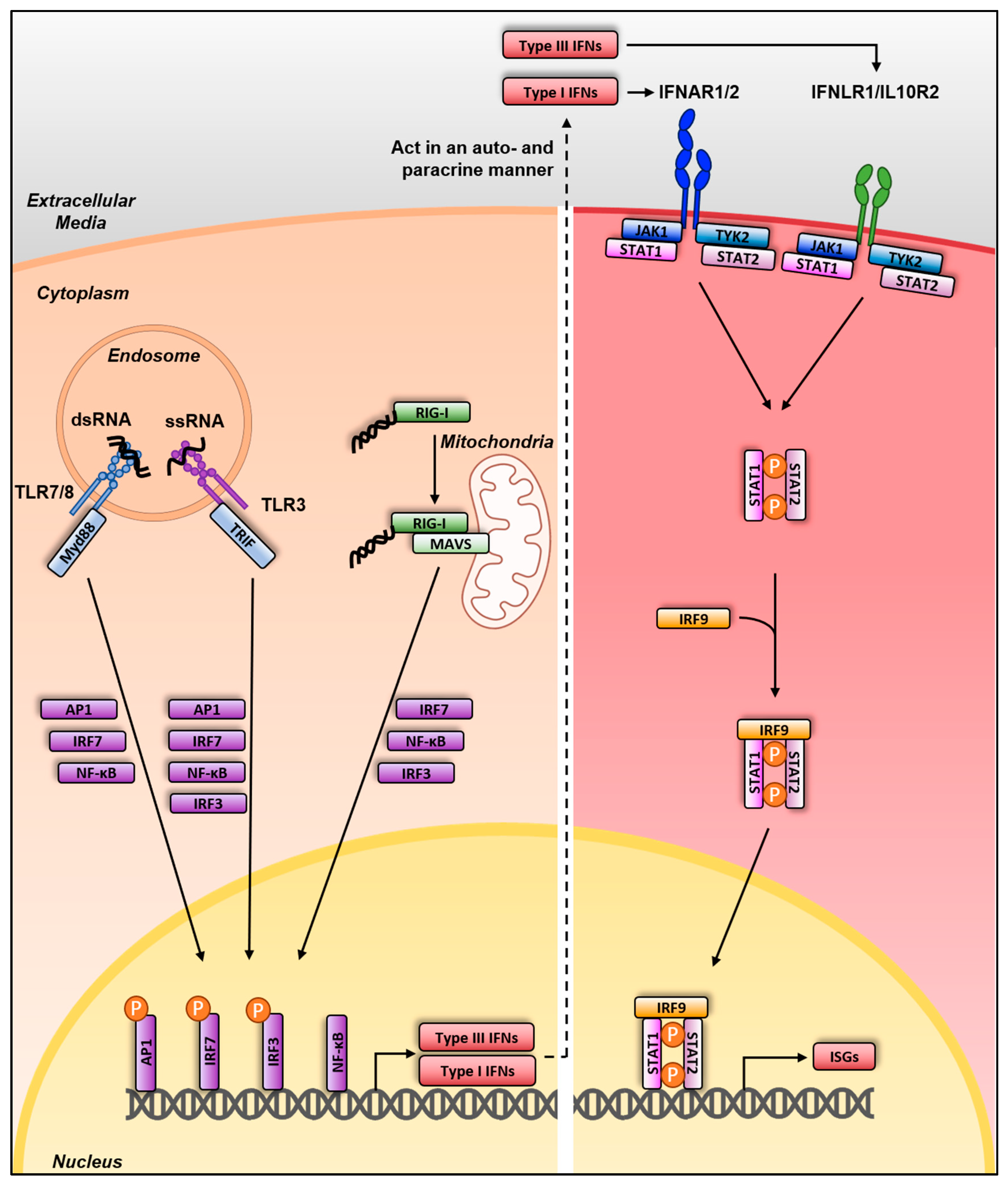
3. Sensing and Interferon Response
3.1. Sensing and Interferon Response in Mammalian Species
3.2. Sensing and Interferon Response in Avian Species
3.3. Viral Antagonism of Innate Immune Sensing by IAV
4. IAV Restriction Factors
4.1. Restriction Factors Inhibiting Viral Entry
4.1.1. IFITMs: Major, Pan-Viral Restriction Factors Preventing Cytosolic Entry
4.1.2. NCOA7-AS: A V-ATPase Regulator, Preventing Endocytosis-Mediated Viral Entry
4.1.3. ZMPSTE24: An IFITM Cofactor, Which Also Acts Independently
4.1.4. CH25H: A Restriction Factor In Vitro but an Enhancer of Inflammation In Vivo
4.1.5. B4GALNT2: A Factor with the Potential of Inhibiting Avian IAV Entry When Overexpressed
4.1.6. MUC1: A Decoy for IAV Viral Particles
4.2. Restriction Factors Inhibiting Viral Genome Replication
4.2.1. MX Dynamin-Like GTPases: Broad-Spectrum Antiviral Proteins with Possible Multiple Modes of Action
4.2.2. GBP Dynamin-Like GTPases: Indirect and Direct Inhibitors
4.2.3. TRIM Proteins: A Large Family of Antiviral Proteins Implicated in Innate Immune Signaling
- -
- Role of TRIM22, TRIM41 and TRIM14 in the degradation of viral components:
- -
- Role of TRIM25 and TRIM56 in the inhibition of viral genome replication:
4.2.4. ZAP and ZFP36L1: Antiviral Zinc Finger Proteins
4.2.5. IFITs: Friends or Foes?
4.2.6. OAS-Family Proteins: NS1-Counteracted dsRNA Binding Inhibitors
4.2.7. PKR and NF90: An Example of Host–Virus Coevolution
4.2.8. MOV10 and DDX21: Antiviral Cellular Helicases
4.2.9. HDACs: Indirect Inhibitors
4.2.10. ANP32s: Major Cofactors and Potential Minor Inhibitors
4.2.11. ISG15: ISGylation-Mediated Inhibition of NS1 Activities
4.2.12. ISG20: An Exonuclease Affecting IAV Replication When Overexpressed
4.3. Restriction Factors Affecting Viral Assembly, Egress or Maturation
4.3.1. Tetherin/BST-2: A General Inhibitor of Viral Release with a Controversial Activity on IAV
4.3.2. Viperin: An IAV Inhibitor When Overexpressed
4.3.3. PAI-1: An Inhibitory Protein Having an Effect in the Extracellular Media
4.4. Additional Factors Inhibiting IAV
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Asha, K.; Kumar, B. Emerging Influenza D Virus Threat: What We Know so Far! J. Clin. Med. 2019, 8, 192. [Google Scholar] [CrossRef] [Green Version]
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef]
- Parrish, C.R.; Murcia, P.R.; Holmes, E.C. Influenza Virus Reservoirs and Intermediate Hosts: Dogs, Horses, and New Possibilities for Influenza Virus Exposure of Humans. J. Virol. 2015, 89, 2990–2994. [Google Scholar] [CrossRef] [Green Version]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New World Bats Harbor Diverse Influenza A Viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciminski, K.; Schwemmle, M. Bat-Borne Influenza A Viruses: An Awakening. Cold Spring Harb. Perspect. Med. 2021, 11, a038612. [Google Scholar] [CrossRef] [Green Version]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Prim. 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Auladell, M.; Jia, X.; Hensen, L.; Chua, B.; Fox, A.; Nguyen, T.H.O.; Doherty, P.C.; Kedzierska, K. Recalling the Future: Immunological Memory Toward Unpredictable Influenza Viruses. Front. Immunol. 2019, 10, 1400. [Google Scholar] [CrossRef]
- Reid, A.H.; Taubenberger, J.K.; Fanning, T.G. Evidence of an absence: The genetic origins of the 1918 pandemic influenza virus. Nat. Rev. Genet. 2004, 2, 909–914. [Google Scholar] [CrossRef]
- Morens, D.M.; Taubenberger, J.K.; Fauci, A.S. The Persistent Legacy of the 1918 Influenza Virus. N. Engl. J. Med. 2009, 361, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Garten, R.J.; Davis, C.T.; Russell, C.A.; Shu, B.; Lindstrom, S.; Balish, A.; Sessions, W.M.; Xu, X.; Skepner, E.; Deyde, V.; et al. Antigenic and Genetic Characteristics of Swine-Origin 2009 A(H1N1) Influenza Viruses Circulating in Humans. Science 2009, 325, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, K.; Desselberger, U.; Palese, P. Recent human influenza A (H1N1) viruses are closely related genetically to strains isolated in 1950. Nat. Cell Biol. 1978, 274, 334–339. [Google Scholar] [CrossRef]
- Bloom, J.D.; Gong, L.I.; Baltimore, D. Permissive Secondary Mutations Enable the Evolution of Influenza Oseltamivir Resistance. Science 2010, 328, 1272–1275. [Google Scholar] [CrossRef] [Green Version]
- Bright, R.A.; Shay, D.K.; Shu, B.; Cox, N.J.; Klimov, A.I. Adamantane Resistance Among Influenza A Viruses Isolated Early During the 2005-2006 Influenza Season in the United States. JAMA 2006, 295, 891–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldhill, D.H.; te Velthuis, A.J.W.; Fletcher, R.A.; Langat, P.; Zambon, M.; Lackenby, A.; Barclay, W.S. The mechanism of resistance to favipiravir in influenza. Proc. Natl. Acad. Sci. USA 2018, 115, 11613–11618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omoto, S.; Speranzini, V.; Hashimoto, T.; Noshi, T.; Yamaguchi, H.; Kawai, M.; Kawaguchi, K.; Uehara, T.; Shishido, T.; Naito, A.; et al. Characterization of influenza virus variants induced by treatment with the endonuclease inhibitor baloxavir marboxil. Sci. Rep. 2018, 8, 9633. [Google Scholar] [CrossRef]
- Weis, W.; Brown, J.H.; Cusack, S.; Paulson, J.C.; Skehel, J.J.; Wiley, D.C. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nat. Cell Biol. 1988, 333, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, A. On the Mechanism Underlying Initiation of Influenza Virus Infection. Ergeb. Mikrobiol. Immun. Exp. Ther. 1959, 32, 1–22. [Google Scholar] [CrossRef]
- Matrosovich, M.; Gambaryan, A.; Teneberg, S.; Piskarev, V.; Yamnikova, S.; Lvov, D.; Robertson, J.; Karlsson, K.-A. Avian Influenza A Viruses Differ from Human Viruses by Recognition of Sialyloligosaccharides and Gangliosides and by a Higher Conservation of the HA Receptor-Binding Site. Virology 1997, 233, 224–234. [Google Scholar] [CrossRef] [Green Version]
- Gamblin, S.J.; Skehel, J.J. Influenza Hemagglutinin and Neuraminidase Membrane Glycoproteins. J. Biol. Chem. 2010, 285, 28403–28409. [Google Scholar] [CrossRef] [Green Version]
- Rust, M.J.; Lakadamyali, M.; Zhang, F.; Zhuang, X. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat. Struct. Mol. Biol. 2004, 11, 567–573. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhuang, X. Epsin 1 is a cargo-specific adaptor for the clathrin-mediated endocytosis of the influenza virus. Proc. Natl. Acad. Sci. USA 2008, 105, 11790–11795. [Google Scholar] [CrossRef] [Green Version]
- De Vries, E.; Tscherne, D.M.; Wienholts, M.J.; Cobos-Jiménez, V.; Scholte, F.; García-Sastre, A.; Rottier, P.J.M.; De Haan, C.A.M. Dissection of the Influenza A Virus Endocytic Routes Reveals Macropinocytosis as an Alternative Entry Pathway. PLoS Pathog. 2011, 7, e1001329. [Google Scholar] [CrossRef]
- Helenius, A. Unpacking the incoming influenza virus. Cell 1992, 69, 577–578. [Google Scholar] [CrossRef]
- Pinto, L.H.; Lamb, R.A. The M2 Proton Channels of Influenza A and B Viruses. J. Biol. Chem. 2006, 281, 8997–9000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Sieben, C.; Ludwig, K.; Höfer, C.T.; Chiantia, S.; Herrmann, A.; Eghiaian, F.; Schaap, I.A. pH-Controlled Two-Step Uncoating of Influenza Virus. Biophys. J. 2014, 106, 1447–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skehel, J.J.; Wiley, D.C. Receptor Binding and Membrane Fusion in Virus Entry: The Influenza Hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wilson, I.A. Structural Characterization of an Early Fusion Intermediate of Influenza Virus Hemagglutinin. J. Virol. 2011, 85, 5172–5182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cros, J.F.; García-Sastre, A.; Palese, P. An Unconventional NLS is Critical for the Nuclear Import of the Influenza A Virus Nucleoprotein and Ribonucleoprotein. Traffic 2005, 6, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Eisfeld, A.J.; Neumann, G.; Kawaoka, Y. At the centre: Influenza A virus ribonucleoproteins. Nat. Rev. Genet. 2015, 13, 28–41. [Google Scholar] [CrossRef] [Green Version]
- Plotch, S.J.; Bouloy, M.; Ulmanen, I.; Krug, R.M. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 1981, 23, 847–858. [Google Scholar] [CrossRef]
- Dias, A.; Bouvier, D.; Crépin, T.; McCarthy, A.A.; Hart, D.J.; Baudin, F.; Cusack, S.; Ruigrok, R.W.H. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nat. Cell Biol. 2009, 458, 914–918. [Google Scholar] [CrossRef]
- Velthuis, A.J.W.T.; Fodor, A.J.W.T.V.E. Influenza virus RNA polymerase: Insights into the mechanisms of viral RNA synthesis. Nat. Rev. Genet. 2016, 14, 479–493. [Google Scholar] [CrossRef] [Green Version]
- Pflug, A.; Lukarska, M.; Resa-Infante, P.; Reich, S.; Cusack, S. Structural insights into RNA synthesis by the influenza virus transcription-replication machine. Virus Res. 2017, 234, 103–117. [Google Scholar] [CrossRef]
- Peacock, T.P.; Sheppard, C.M.; Staller, E.; Barclay, W.S. Host Determinants of Influenza RNA Synthesis. Annu. Rev. Virol. 2019, 6, 215–233. [Google Scholar] [CrossRef]
- Huang, S.; Chen, J.; Chen, Q.; Wang, H.; Yao, Y.; Chen, Z.; Chen, J.; Chen, J. A Second CRM1-Dependent Nuclear Export Signal in the Influenza A Virus NS2 Protein Contributes to the Nuclear Export of Viral Ribonucleoproteins. J. Virol. 2012, 87, 767–778. [Google Scholar] [CrossRef] [Green Version]
- Cros, J.F.; Palese, P. Trafficking of viral genomic RNA into and out of the nucleus: Influenza, Thogoto and Borna disease viruses. Virus Res. 2003, 95, 3–12. [Google Scholar] [CrossRef]
- Brunotte, L.; Flies, J.; Bolte, H.; Reuther, P.; Vreede, F.; Schwemmle, M. The Nuclear Export Protein of H5N1 Influenza A Viruses Recruits Matrix 1 (M1) Protein to the Viral Ribonucleoprotein to Mediate Nuclear Export. J. Biol. Chem. 2014, 289, 20067–20077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, E.A.; Digard, P.; Stuart, A.D. The Rab11 Pathway Is Required for Influenza A Virus Budding and Filament Formation. J. Virol. 2010, 84, 5848–5859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, I.F.D.C.; Fournier, G.; Sachse, M.; Pizarro-Cerda, J.; Risco, C.; Naffakh, N. Influenza virus genome reaches the plasma membrane via a modified endoplasmic reticulum and Rab11-dependent vesicles. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lakdawala, S.S.; Fodor, E.; Subbarao, K. Moving On Out: Transport and Packaging of Influenza Viral RNA into Virions. Annu. Rev. Virol. 2016, 3, 411–427. [Google Scholar] [CrossRef]
- Haralampiev, I.; Prisner, S.; Nitzan, M.; Schade, M.; Jolmes, F.; Schreiber, M.; Loidolt-Krüger, M.; Jongen, K.; Chamiolo, J.; Nilson, N.; et al. Selective flexible packaging pathways of the segmented genome of influenza A virus. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Malik, G.; Zhou, Y. Innate Immune Sensing of Influenza a Virus. Viruses 2020, 12, 755. [Google Scholar] [CrossRef]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H. Recent Advances and Contradictions in the Study of the Individual Roles of Ubiquitin Ligases That Regulate RIG-I-Like Receptor-Mediated Antiviral Innate Immune Responses. Front. Immunol. 2020, 11, 1296. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Tan, C.P.; Goubau, D.; Schulz, O.; Pichlmair, A.; Bier, K.; Robb, N.; Vreede, F.; Barclay, W.; Fodor, E.; et al. RIG-I Detects Viral Genomic RNA during Negative-Strand RNA Virus Infection. Cell 2010, 140, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Kandasamy, M.; Suryawanshi, A.; Tundup, S.; Perez, J.T.; Schmolke, M.; Manicassamy, S.; Manicassamy, B. RIG-I Signaling Is Critical for Efficient Polyfunctional T Cell Responses during Influenza Virus Infection. PLoS Pathog. 2016, 12, e1005754. [Google Scholar] [CrossRef]
- Le Goffic, R.; Pothlichet, J.; Vitour, D.; Fujita, T.; Meurs, E.; Chignard, M.; Si-Tahar, M. Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J. Immunol. 2007, 178, 3368–3372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Goffic, R.; Balloy, V.; Lagranderie, M.; Alexopoulou, L.; Escriou, N.; Flavell, R.; Chignard, M.; Si-Tahar, M. Detrimental Contribution of the Toll-Like Receptor (TLR)3 to Influenza A Virus–Induced Acute Pneumonia. PLoS Pathog. 2006, 2, e53. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.-U.; Kwon, H.-J.; Song, J.-H.; Byun, Y.-H.; Seong, B.L.; Kawai, T.; Akira, S.; Kweon, M.-N. MyD88 Signaling Is Indispensable for Primary Influenza A Virus Infection but Dispensable for Secondary Infection. J. Virol. 2010, 84, 12713–12722. [Google Scholar] [CrossRef] [Green Version]
- Wolf, A.I.; Buehler, D.; Hensley, S.E.; Cavanagh, L.L.; Wherry, E.J.; Kastner, P.; Chan, S.; Weninger, W. Plasmacytoid Dendritic Cells Are Dispensable during Primary Influenza Virus Infection. J. Immunol. 2009, 182, 871–879. [Google Scholar] [CrossRef] [Green Version]
- Wack, A.; Terczyńska-Dyla, E.; Hartmann, R. Guarding the frontiers: The biology of type III interferons. Nat. Immunol. 2015, 16, 802–809. [Google Scholar] [CrossRef]
- Wu, W.; Metcalf, J.P. The Role of Type I IFNs in Influenza: Antiviral Superheroes or Immunopathogenic Villains? J. Innate Immun. 2020, 12, 437–447. [Google Scholar] [CrossRef]
- Zhang, Q.; Zeng, L.-P.; Zhou, P.; Irving, A.T.; Li, S.; Shi, Z.-L.; Wang, L.-F. IFNAR2-dependent gene expression profile induced by IFN-α in Pteropus alecto bat cells and impact of IFNAR2 knockout on virus infection. PLoS ONE 2017, 12, e0182866. [Google Scholar] [CrossRef] [Green Version]
- Shepardson, K.M.; Larson, K.; Johns, L.L.; Stanek, K.; Cho, H.; Wellham, J.; Henderson, H.; Rynda-Apple, A. IFNAR2 Is Required for Anti-influenza Immunity and Alters Susceptibility to Post-influenza Bacterial Superinfections. Front. Immunol. 2018, 9, 2589. [Google Scholar] [CrossRef]
- Seo, S.-U.; Kwon, H.-J.; Ko, H.-J.; Byun, Y.-H.; Seong, B.L.; Uematsu, S.; Akira, S.; Kweon, M.-N. Type I Interferon Signaling Regulates Ly6Chi Monocytes and Neutrophils during Acute Viral Pneumonia in Mice. PLoS Pathog. 2011, 7, e1001304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koerner, I.; Kochs, G.; Kalinke, U.; Weiss, S.; Staeheli, P. Protective Role of Beta Interferon in Host Defense against Influenza A Virus. J. Virol. 2006, 81, 2025–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Sastre, A.; Durbin, R.K.; Zheng, H.; Palese, P.; Gertner, R.; Levy, D.E.; Durbin, J.E. The Role of Interferon in Influenza Virus Tissue Tropism. J. Virol. 1998, 72, 8550–8558. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.; Maini, M.K.; Wack, A. Disease-Promoting Effects of Type I Interferons in Viral, Bacterial, and Coinfections. J. Interf. Cytokine Res. 2015, 35, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Klinkhammer, J.; Schnepf, D.; Ye, L.; Schwaderlapp, M.; Gad, H.H.; Hartmann, R.; Garcin, D.; Mahlakõiv, T.; Staeheli, P. IFN-λ prevents influenza virus spread from the upper airways to the lungs and limits virus transmission. eLife 2018, 7, e33354. [Google Scholar] [CrossRef] [PubMed]
- Mordstein, M.; Kochs, G.; Dumoutier, L.; Renauld, J.-C.; Paludan, S.R.; Klucher, K.; Staeheli, P. Interferon-λ Contributes to Innate Immunity of Mice against Influenza A Virus but Not against Hepatotropic Viruses. PLoS Pathog. 2008, 4, e1000151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Schnepf, D.; Staeheli, P. Interferon-λ orchestrates innate and adaptive mucosal immune responses. Nat. Rev. Immunol. 2019, 19, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.-E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K.; et al. Interferon-λ Mediates Non-redundant Front-Line Antiviral Protection against Influenza Virus Infection without Compromising Host Fitness. Immunity 2017, 46, 875–890.e6. [Google Scholar] [CrossRef] [PubMed]
- Bennett, A.L.; Smith, D.W.; Cummins, M.J.; Jacoby, P.A.; Cummins, J.M.; Beilharz, M.W. Low-dose oral interferon alpha as prophylaxis against viral respiratory illness: A double-blind, parallel controlled trial during an influenza pandemic year. Influenza Other Respir. Viruses 2013, 7, 854–862. [Google Scholar] [CrossRef]
- Lim, H.K.; Huang, S.X.; Chen, J.; Kerner, G.; Gilliaux, O.; Bastard, P.; Dobbs, K.; Hernandez, N.; Goudin, N.; Hasek, M.L.; et al. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J. Exp. Med. 2019, 216, 2038–2056. [Google Scholar] [CrossRef] [PubMed]
- Ciancanelli, M.J.; Huang, S.X.L.; Luthra, P.; Garner, H.; Itan, Y.; Volpi, S.; Lafaille, F.G.; Trouillet, C.; Schmolke, M.; Albrecht, R.A.; et al. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 2015, 348, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, N.; Melki, I.; Jing, H.; Habib, T.; Huang, S.S.; Danielson, J.; Kula, T.; Drutman, S.; Belkaya, S.; Rattina, V.; et al. Life-threatening influenza pneumonitis in a child with inherited IRF9 deficiency. J. Exp. Med. 2018, 215, 2567–2585. [Google Scholar] [CrossRef] [PubMed]
- Hemann, E.A.; Green, R.; Turnbull, J.B.; Langlois, R.A.; Savan, R.; Gale, M. Interferon-λ modulates dendritic cells to facilitate T cell immunity during infection with influenza A virus. Nat. Immunol. 2019, 20, 1035–1045. [Google Scholar] [CrossRef]
- Ye, L.; Schnepf, D.; Becker, J.; Ebert, K.; Tanriver, Y.; Bernasconi, V.; Gad, H.H.; Hartmann, R.; Lycke, N.; Staeheli, P. Interferon-λ enhances adaptive mucosal immunity by boosting release of thymic stromal lymphopoietin. Nat. Immunol. 2019, 20, 593–601. [Google Scholar] [CrossRef]
- Kohlmeier, J.E.; Cookenham, T.; Roberts, A.D.; Miller, S.C.; Woodland, D.L. Type I Interferons Regulate Cytolytic Activity of Memory CD8+ T Cells in the Lung Airways during Respiratory Virus Challenge. Immunity 2010, 33, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Durbin, J.E.; Fernandez-Sesma, A.; Lee, C.-K.; Rao, T.D.; Frey, A.B.; Moran, T.M.; Vukmanovic, S.; García-Sastre, A.; Levy, D.E. Type I IFN Modulates Innate and Specific Antiviral Immunity. J. Immunol. 2000, 164, 4220–4228. [Google Scholar] [CrossRef] [Green Version]
- Karpala, A.J.; Stewart, C.; McKay, J.; Lowenthal, J.W.; Bean, A.G.D. Characterization of Chicken Mda5 Activity: Regulation of IFN-β in the Absence of RIG-I Functionality. J. Immunol. 2011, 186, 5397–5405. [Google Scholar] [CrossRef]
- Barber, M.R.W.; Aldridge, J.R.; Webster, R.G.; Magor, K.E. Association of RIG-I with innate immunity of ducks to influenza. Proc. Natl. Acad. Sci. USA 2010, 107, 5913–5918. [Google Scholar] [CrossRef] [Green Version]
- Liniger, M.; Summerfield, A.; Zimmer, G.; McCullough, K.C.; Ruggli, N. Chicken Cells Sense Influenza A Virus Infection through MDA5 and CARDIF Signaling Involving LGP2. J. Virol. 2011, 86, 705–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Watanabe, C.; Suzuki, Y.; Tanikawa, T.; Uchida, Y.; Saito, T. Chicken MDA5 Senses Short Double-Stranded RNA with Implications for Antiviral Response against Avian Influenza Viruses in Chicken. J. Innate Immun. 2013, 6, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Uchikawa, E.; Lethier, M.; Malet, H.; Brunel, J.; Gerlier, D.; Cusack, S. Structural Analysis of dsRNA Binding to Anti-viral Pattern Recognition Receptors LGP2 and MDA5. Mol. Cell 2016, 62, 586–602. [Google Scholar] [CrossRef] [Green Version]
- Philbin, V.J.; Iqbal, M.; Boyd, Y.; Goodchild, M.J.; Beal, R.K.; Bumstead, N.; Young, J.; Smith, A.L. Identification and characterization of a functional, alternatively spliced Toll-like receptor 7 (TLR7) and genomic disruption of TLR8 in chickens. Immunology 2005, 114, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Liu, J.; Wu, Z.G.; Lin, C.Y.; Wang, M. The Interferon-α Genes from Three Chicken Lines and Its Effects on H9N2 Influenza Viruses. Anim. Biotechnol. 2004, 15, 77–88. [Google Scholar] [CrossRef]
- Penski, N.; Härtle, S.; Rubbenstroth, D.; Krohmann, C.; Ruggli, N.; Schusser, B.; Pfann, M.; Reuter, A.; Gohrbandt, S.; Hundt, J.; et al. Highly Pathogenic Avian Influenza Viruses Do Not Inhibit Interferon Synthesis in Infected Chickens but Can Override the Interferon-Induced Antiviral State. J. Virol. 2011, 85, 7730–7741. [Google Scholar] [CrossRef] [Green Version]
- Reuter, A.; Soubies, S.; Härtle, S.; Schusser, B.; Kaspers, B.; Staeheli, P.; Rubbenstroth, D.; Garcia-Sastre, A. Antiviral Activity of Lambda Interferon in Chickens. J. Virol. 2013, 88, 2835–2843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hale, B.G.; Randall, R.E.; Ortín, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef]
- Jureka, A.S.; Kleinpeter, A.B.; Tipper, J.L.; Harrod, K.S.; Petit, C.M. The influenza NS1 protein modulates RIG-I activation via a strain-specific direct interaction with the second CARD of RIG-I. J. Biol. Chem. 2020, 295, 1153–1164. [Google Scholar] [CrossRef]
- Mibayashi, M.; Martínez-Sobrido, L.; Loo, Y.-M.; Cárdenas, W.B.; Gale, M.; García-Sastre, A. Inhibition of Retinoic Acid-Inducible Gene I-Mediated Induction of Beta Interferon by the NS1 Protein of Influenza A Virus. J. Virol. 2006, 81, 514–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Chen, L.-M.; Zeng, H.; Gomez, J.A.; Plowden, J.; Fujita, T.; Katz, J.M.; Donis, R.O.; Sambhara, S. NS1 Protein of Influenza A Virus Inhibits the Function of Intracytoplasmic Pathogen Sensor, RIG-I. Am. J. Respir. Cell Mol. Biol. 2007, 36, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.-S.; Huang, I.-C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; García-Sastre, A. Influenza A Virus NS1 Targets the Ubiquitin Ligase TRIM25 to Evade Recognition by the Host Viral RNA Sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajsbaum, R.; Albrecht, R.A.; Wang, M.K.; Maharaj, N.P.; Versteeg, G.A.; Nistal-Villán, E.; García-Sastre, A.; Gack, M.U. Species-Specific Inhibition of RIG-I Ubiquitination and IFN Induction by the Influenza A Virus NS1 Protein. PLoS Pathog. 2012, 8, e1003059. [Google Scholar] [CrossRef] [PubMed]
- Koliopoulos, M.G.; Lethier, M.; Van Der Veen, A.G.; Haubrich, K.; Hennig, J.; Kowalinski, E.; Stevens, R.V.; Martin, S.R.; de Sousa, C.R.; Cusack, S.; et al. Molecular mechanism of influenza A NS1-mediated TRIM25 recognition and inhibition. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Mazur, I.; Anhlan, D.; Mitzner, D.; Wixler, L.; Schubert, U.; Ludwig, S. The proapoptotic influenza A virus protein PB1-F2 regulates viral polymerase activity by interaction with the PB1 protein. Cell. Microbiol. 2008, 10, 1140–1152. [Google Scholar] [CrossRef]
- Chen, W.; Calvo, P.A.; Malide, D.; Gibbs, J.; Schubert, U.; Bacik, I.; Basta, S.; O’Neill, R.; Schickli, J.; Palese, P.; et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 2001, 7, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Leymarie, O.; Jouvion, G.; Hervé, P.-L.; Chevalier, C.; Lorin, V.; Lecardonnel, J.; Da Costa, B.; Delmas, B.; Escriou, N.; Le Goffic, R. Kinetic Characterization of PB1-F2-Mediated Immunopathology during Highly Pathogenic Avian H5N1 Influenza Virus Infection. PLoS ONE 2013, 8, e57894. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.T.; Ramos, I.; Hai, R.; Schmolke, M.; García-Sastre, A.; Fernandez-Sesma, A.; Palese, P. The Influenza Virus Protein PB1-F2 Inhibits the Induction of Type I Interferon at the Level of the MAVS Adaptor Protein. PLoS Pathog. 2011, 7, e1002067. [Google Scholar] [CrossRef]
- Varga, Z.T.; Grant, A.; Manicassamy, B.; Palese, P. Influenza Virus Protein PB1-F2 Inhibits the Induction of Type I Interferon by Binding to MAVS and Decreasing Mitochondrial Membrane Potential. J. Virol. 2012, 86, 8359–8366. [Google Scholar] [CrossRef] [Green Version]
- Firth, A.E.; Jagger, B.W.; Wise, H.M.; Nelson, C.C.; Parsawar, K.; Wills, N.M.; Napthine, S.; Taubenberger, J.K.; Digard, P.; Atkins, J.F. Ribosomal frameshifting used in influenza A virus expression occurs within the sequence UCC_UUU_CGU and is in the +1 direction. Open Biol. 2012, 2, 120109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.-A.; Wills, N.M.; Xiao, Y.-L.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An Overlapping Protein-Coding Region in Influenza A Virus Segment 3 Modulates the Host Response. Science 2012, 337, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Khaperskyy, D.A.; Schmaling, S.; Larkins-Ford, J.; McCormick, C.; Gaglia, M.M. Selective Degradation of Host RNA Polymerase II Transcripts by Influenza A Virus PA-X Host Shutoff Protein. PLoS Pathog. 2016, 12, e1005427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Chaimayo, C.; McGuinness, J.; Takimoto, T. Critical Role of the PA-X C-Terminal Domain of Influenza A Virus in Its Subcellular Localization and Shutoff Activity. J. Virol. 2016, 90, 7131–7141. [Google Scholar] [CrossRef] [Green Version]
- Khaperskyy, D.A.; McCormick, C. Timing Is Everything: Coordinated Control of Host Shutoff by Influenza A Virus NS1 and PA-X Proteins. J. Virol. 2015, 89, 6528–6531. [Google Scholar] [CrossRef] [Green Version]
- Levene, R.E.; Gaglia, M.M. Host Shutoff in Influenza A Virus: Many Means to an End. Viruses 2018, 10, 475. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.C.; Zhong, G.; Huang, I.-C.; Farzan, M. IFITM-Family Proteins: The Cell’s First Line of Antiviral Defense. Annu. Rev. Virol. 2014, 1, 261–283. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.; Schwartz, O.; Compton, A.A. More than meets the I: The diverse antiviral and cellular functions of interferon-induced transmembrane proteins. Retrovirology 2017, 14, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Liu, J.; Li, M.; Yang, H.; Zhang, C. Evolutionary Dynamics of the Interferon-Induced Transmembrane Gene Family in Vertebrates. PLoS ONE 2012, 7, e49265. [Google Scholar] [CrossRef] [Green Version]
- Siegrist, F.; Ebeling, M.; Certa, U. The Small Interferon-Induced Transmembrane Genes and Proteins. J. Interf. Cytokine Res. 2011, 31, 183–197. [Google Scholar] [CrossRef]
- Brass, A.L.; Huang, I.-C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; Van Der Weyden, L.; Fikrig, E.; et al. The IFITM Proteins Mediate Cellular Resistance to Influenza A H1N1 Virus, West Nile Virus, and Dengue Virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Li, J.; Winkler, C.A.; An, P.; Guo, J.-T. IFITM Genes, Variants, and Their Roles in the Control and Pathogenesis of Viral Infections. Front. Microbiol. 2019, 9, 3228. [Google Scholar] [CrossRef] [Green Version]
- Yount, J.S.; Karssemeijer, R.A.; Hang, H.C. S-Palmitoylation and Ubiquitination Differentially Regulate Interferon-induced Transmembrane Protein 3 (IFITM3)-mediated Resistance to Influenza Virus. J. Biol. Chem. 2012, 287, 19631–19641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, C.C.; Kondur, H.R.; Huang, I.-C.; Farzan, M. Interferon-induced Transmembrane Protein 3 Is a Type II Transmembrane Protein. J. Biol. Chem. 2013, 288, 32184–32193. [Google Scholar] [CrossRef] [Green Version]
- Weston, S.; Czieso, S.; White, I.J.; Smith, S.E.; Kellam, P.; Marsh, M. A Membrane Topology Model for Human Interferon Inducible Transmembrane Protein 1. PLoS ONE 2014, 9, e104341. [Google Scholar] [CrossRef]
- Ling, S.; Zhang, C.; Wang, W.; Cai, X.; Yu, L.; Wu, F.; Zhang, L.; Tian, C. Combined approaches of EPR and NMR illustrate only one transmembrane helix in the human IFITM3. Sci. Rep. 2016, 6, 24029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesarino, N.M.; McMichael, T.M.; Yount, J.S. Regulation of the trafficking and antiviral activity of IFITM3 by post-translational modifications. Future Microbiol. 2014, 9, 1151–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perreira, J.M.; Chin, C.R.; Feeley, E.M.; Brass, A.L. IFITMs Restrict the Replication of Multiple Pathogenic Viruses. J. Mol. Biol. 2013, 425, 4937–4955. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Hang, H.C. Site-Specific Bioorthogonal Labeling for Fluorescence Imaging of Intracellular Proteins in Living Cells. J. Am. Chem. Soc. 2016, 138, 14423–14433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spence, J.S.; He, R.; Hoffmann, H.-H.; Das, T.; Thinon, E.; Rice, C.M.; Peng, T.; Chandran, K.; Hang, H.C. IFITM3 directly engages and shuttles incoming virus particles to lysosomes. Nat. Chem. Biol. 2019, 15, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Compton, A.A.; Bruel, T.; Porrot, F.; Mallet, A.; Sachse, M.; Euvrard, M.; Liang, C.; Casartelli, N.; Schwartz, O. IFITM Proteins Incorporated into HIV-1 Virions Impair Viral Fusion and Spread. Cell Host Microbe 2014, 16, 736–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tartour, K.; Appourchaux, R.; Gaillard, J.; Nguyen, X.-N.; Durand, S.; Turpin, J.; Beaumont, E.; Roch, E.; Berger, G.; Mahieux, R.; et al. IFITM proteins are incorporated onto HIV-1 virion particles and negatively imprint their infectivity. Retrovirology 2014, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feeley, E.M.; Sims, J.S.; John, S.P.; Chin, C.R.; Pertel, T.; Chen, L.-M.; Gaiha, G.D.; Ryan, B.J.; Donis, R.O.; Elledge, S.J.; et al. IFITM3 Inhibits Influenza A Virus Infection by Preventing Cytosolic Entry. PLoS Pathog. 2011, 7, e1002337. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Markosyan, R.M.; Zheng, Y.-M.; Golfetto, O.; Bungart, B.; Li, M.; Ding, S.; Chen, L.; Liang, C.; Lee, J.C.; et al. IFITM Proteins Restrict Viral Membrane Hemifusion. PLoS Pathog. 2013, 9, e1003124. [Google Scholar] [CrossRef]
- Desai, T.M.; Marin, M.; Chin, C.R.; Savidis, G.; Brass, A.L.; Melikyan, G.B. IFITM3 Restricts Influenza A Virus Entry by Blocking the Formation of Fusion Pores following Virus-Endosome Hemifusion. PLoS Pathog. 2014, 10, e1004048. [Google Scholar] [CrossRef] [Green Version]
- Suddala, K.C.; Lee, C.C.; Meraner, P.; Marin, M.; Markosyan, R.M.; Desai, T.M.; Cohen, F.S.; Brass, A.L.; Melikyan, G.B. Interferon-induced transmembrane protein 3 blocks fusion of sensitive but not resistant viruses by partitioning into virus-carrying endosomes. PLoS Pathog. 2019, 15, e1007532. [Google Scholar] [CrossRef] [Green Version]
- John, S.P.; Chin, C.R.; Perreira, J.M.; Feeley, E.M.; Aker, A.M.; Savidis, G.; Smith, S.E.; Elia, A.E.H.; Everitt, A.R.; Vora, M.; et al. The CD225 Domain of IFITM3 Is Required for both IFITM Protein Association and Inhibition of Influenza A Virus and Dengue Virus Replication. J. Virol. 2013, 87, 7837–7852. [Google Scholar] [CrossRef] [Green Version]
- Jia, R.; Xu, F.; Qian, J.; Yao, Y.; Miao, C.; Zheng, Y.-M.; Liu, S.-L.; Guo, F.; Geng, Y.; Qiao, W.; et al. Identification of an endocytic signal essential for the antiviral action of IFITM3. Cell. Microbiol. 2014, 16, 1080–1093. [Google Scholar] [CrossRef]
- Wrensch, F.; Winkler, M.; Pöhlmann, S. IFITM Proteins Inhibit Entry Driven by the MERS-Coronavirus Spike Protein: Evidence for Cholesterol-Independent Mechanisms. Viruses 2014, 6, 3683–3698. [Google Scholar] [CrossRef] [Green Version]
- Yount, J.S.; Moltedo, B.; Yang, Y.-Y.; Charron, G.; Moran, T.M.; López, C.B.; Hang, H.C. Palmitoylome profiling reveals S-palmitoylation–dependent antiviral activity of IFITM3. Nat. Chem. Biol. 2010, 6, 610–614. [Google Scholar] [CrossRef]
- Chesarino, N.M.; McMichael, T.M.; Hach, J.C.; Yount, J.S. Phosphorylation of the Antiviral Protein Interferon-inducible Transmembrane Protein 3 (IFITM3) Dually Regulates Its Endocytosis and Ubiquitination. J. Biol. Chem. 2014, 289, 11986–11992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, R.; Pan, Q.; Ding, S.; Rong, L.; Liu, S.-L.; Geng, Y.; Qiao, W.; Liang, C. The N-Terminal Region of IFITM3 Modulates Its Antiviral Activity by Regulating IFITM3 Cellular Localization. J. Virol. 2012, 86, 13697–13707. [Google Scholar] [CrossRef] [Green Version]
- Everitt, A.R.; The GenISIS Investigators; Clare, S.; Pertel, T.; John, S.P.; Wash, R.S.; Smith, S.E.; Chin, C.R.; Feeley, E.M.; Sims, J.S.; et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nat. Cell Biol. 2012, 484, 519–523. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.C.; Huang, I.-C.; Kam, C.; Farzan, M. Ifitm3 Limits the Severity of Acute Influenza in Mice. PLoS Pathog. 2012, 8, e1002909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenney, A.D.; McMichael, T.M.; Imas, A.; Chesarino, N.M.; Zhang, L.; Dorn, L.E.; Wu, Q.; Alfaour, O.; Amari, F.; Chen, M.; et al. IFITM3 protects the heart during influenza virus infection. Proc. Natl. Acad. Sci. USA 2019, 116, 18607–18612. [Google Scholar] [CrossRef] [Green Version]
- Infusini, G.; Smith, J.M.; Yuan, H.; Pizzolla, A.; Ng, W.C.; Londrigan, S.L.; Haque, A.; Reading, P.C.; Villadangos, J.A.; Wakim, L.M. Respiratory DC Use IFITM3 to Avoid Direct Viral Infection and Safeguard Virus-Specific CD8+ T Cell Priming. PLoS ONE 2015, 10, e0143539. [Google Scholar] [CrossRef] [PubMed]
- Wakim, L.M.; Gupta, N.; Mintern, J.D.; Villadangos, A.J. Enhanced survival of lung tissue-resident memory CD8+ T cells during infection with influenza virus due to selective expression of IFITM3. Nat. Immunol. 2013, 14, 238–245. [Google Scholar] [CrossRef]
- Wellington, D.; Laurenson-Schafer, H.; Abdel-Haq, A.; Dong, T. IFITM3: How genetics influence influenza infection demographically. Biomed. J. 2019, 42, 19–26. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Zhao, Y.; Liu, N.; Peng, Y.-C.; Giannoulatou, E.; Jin, R.-H.; Yan, H.-P.; Wu, H.; Liu, J.-H.; Wang, D.-Y.; et al. Interferon-induced transmembrane protein-3 genetic variant rs12252-C is associated with severe influenza in Chinese individuals. Nat. Commun. 2013, 4, 1418. [Google Scholar] [CrossRef] [Green Version]
- Mehrbod, P.; Eybpoosh, S.; Fotouhi, F.; Targhi, H.S.; Mazaheri, V.; Farahmand, B. Association of IFITM3 rs12252 polymorphisms, BMI, diabetes, and hypercholesterolemia with mild flu in an Iranian population. Virol. J. 2017, 14, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Yang, P.; Dong, T.; Zhang, Y.; Shi, W.; Peng, X.; Cui, S.; Zhang, D.; Lu, G.; Liu, Y.; et al. IFITM3 Rs12252-C Variant Increases Potential Risk for Severe Influenza Virus Infection in Chinese Population. Front. Cell Infect. Microbiol. 2017, 7, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhang, A.; Wan, Y.; Liu, X.; Qiu, C.; Xi, X.; Ren, Y.; Wang, J.; Dong, Y.; Bao, M.; et al. Early hypercytokinemia is associated with interferon-induced transmembrane protein-3 dysfunction and predictive of fatal H7N9 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 769–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Tan, B.; Zhou, X.; Xue, J.; Zhang, X.; Wang, P.; Shao, C.; Li, Y.; Li, C.; Xia, H.; et al. Interferon-Inducible Transmembrane Protein 3 Genetic Variant rs12252 and Influenza Susceptibility and Severity: A Meta-Analysis. PLoS ONE 2015, 10, e0124985. [Google Scholar] [CrossRef]
- Xuan, Y.; Wang, L.N.; Li, W.; Zi, H.R.; Guo, Y.; Yan, W.J.; Chen, X.B.; Wei, P.M. IFITM3 rs12252 T>C polymorphism is associated with the risk of severe influenza: A meta-analysis. Epidemiol. Infect. 2015, 143, 2975–2984. [Google Scholar] [CrossRef]
- Kim, Y.-C.; Jeong, B.-H. No Correlation of the Disease Severity of Influenza A Virus Infection with the rs12252 Polymorphism of the Interferon-Induced Transmembrane Protein 3 Gene. Intervirology 2017, 60, 69–74. [Google Scholar] [CrossRef]
- David, S.; Correia, V.; Antunes, L.; Faria, R.; Ferrão, J.; Faustino, P.; Nunes, B.; Maltez, F.; Lavinha, J.; De Andrade, H.R. Population genetics of IFITM3 in Portugal and Central Africa reveals a potential modifier of influenza severity. Immunogenetics 2018, 70, 169–177. [Google Scholar] [CrossRef]
- Martins, J.S.C.; Oliveira, M.L.A.; Garcia, C.C.; Siqueira, M.M.; Matos, A.R. Investigation of Human IFITM3 Polymorphisms rs34481144A and rs12252C and Risk for Influenza A(H1N1)pdm09 Severity in a Brazilian Cohort. Front. Cell. Infect. Microbiol. 2020, 10, 352. [Google Scholar] [CrossRef]
- Williams, D.E.J.; Wu, W.-L.; Grotefend, C.R.; Radic, V.; Chung, C.; Chung, Y.-H.; Farzan, M.; Huang, I.-C. IFITM3 Polymorphism rs12252-C Restricts Influenza A Viruses. PLoS ONE 2014, 9, e110096. [Google Scholar] [CrossRef] [Green Version]
- Randolph, A.G.; Yip, W.-K.; Allen, E.K.; Rosenberger, C.M.; Agan, A.A.; Ash, S.A.; Zhang, Y.; Bhangale, T.R.; Finkelstein, D.; Cvijanovich, N.Z.; et al. Evaluation of IFITM3 rs12252 Association With Severe Pediatric Influenza Infection. J. Infect. Dis. 2017, 216, 14–21. [Google Scholar] [CrossRef]
- Makvandi-Nejad, S.; Laurenson-Schafer, H.; Wang, L.; Wellington, D.; Zhao, Y.; Jin, B.; Qin, L.; Kite, K.; Moghadam, H.K.; Song, C.; et al. Lack of Truncated IFITM3 Transcripts in Cells Homozygous for the rs12252-C Variant That is Associated With Severe Influenza Infection. J. Infect. Dis. 2018, 217, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Smith, N.; Yu, L.; Paton, I.R.; Gutowska, M.W.; Forrest, H.L.; Danner, A.F.; Seiler, J.P.; Digard, P.; Webster, R.G.; et al. A comparative analysis of host responses to avian influenza infection in ducks and chickens highlights a role for the interferon-induced transmembrane proteins in viral resistance. BMC Genom. 2015, 16, 574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanz, C.; Yángüez, E.; Andenmatten, D.; Stertz, S. Swine Interferon-Inducible Transmembrane Proteins Potently Inhibit Influenza A Virus Replication. J. Virol. 2014, 89, 863–869. [Google Scholar] [CrossRef] [Green Version]
- Benfield, C.T.O.; Smith, S.E.; Wright, E.; Wash, R.S.; Ferrara, F.; Temperton, N.J.; Kellam, P. Bat and pig IFN-induced transmembrane protein 3 restrict cell entry by influenza virus and lyssaviruses. J. Gen. Virol. 2015, 96, 991–1005. [Google Scholar] [CrossRef] [Green Version]
- Benfield, C.T.; MacKenzie, F.; Ritzefeld, M.; Mazzon, M.; Weston, S.; Tate, E.W.; Teo, B.H.; E Smith, S.; Kellam, P.; Holmes, E.C.; et al. Bat IFITM3 restriction depends on S-palmitoylation and a polymorphic site within the CD225 domain. Life Sci. Alliance 2020, 3, e201900542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.E.; Gibson, M.S.; Wash, R.S.; Ferrara, F.; Wright, E.; Temperton, N.; Kellam, P.; Fife, M. Chicken Interferon-Inducible Transmembrane Protein 3 Restricts Influenza Viruses and LyssavirusesIn Vitro. J. Virol. 2013, 87, 12957–12966. [Google Scholar] [CrossRef] [Green Version]
- Blyth, G.A.D.; Chan, W.F.; Webster, R.G.; Magor, K.E. Duck Interferon-Inducible Transmembrane Protein 3 Mediates Restriction of Influenza Viruses. J. Virol. 2015, 90, 103–116. [Google Scholar] [CrossRef] [Green Version]
- Finelli, M.J.; Oliver, P.L. TLDc proteins: New players in the oxidative stress response and neurological disease. Mamm. Genome 2017, 28, 395–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finelli, M.J.; Sanchez-Pulido, L.; Liu, K.X.; Davies, K.E.; Oliver, P.L. The Evolutionarily Conserved Tre2/Bub2/Cdc16 (TBC), Lysin Motif (LysM), Domain Catalytic (TLDc) Domain Is Neuroprotective against Oxidative Stress. J. Biol. Chem. 2016, 291, 2751–2763. [Google Scholar] [CrossRef] [Green Version]
- Shao, W.; Halachmi, S.; Brown, M. ERAP140, a Conserved Tissue-Specific Nuclear Receptor Coactivator. Mol. Cell. Biol. 2002, 22, 3358–3372. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Croze, E.; Yamaguchi, K.D.; Tran, T.; Reder, A.T.; Litvak, V.; Volkert, M.R. Induction of a Unique Isoform of theNCOA7Oxidation Resistance Gene by Interferon β-1b. J. Interf. Cytokine Res. 2015, 35, 186–199. [Google Scholar] [CrossRef] [Green Version]
- Doyle, T.; Moncorgé, O.; Bonaventure, B.; Pollpeter, D.; Lussignol, M.; Tauziet, M.; Apolonia, L.; Catanese, M.-T.; Goujon, C.; Malim, M.H. The interferon-inducible isoform of NCOA7 inhibits endosome-mediated viral entry. Nat. Microbiol. 2018, 3, 1369–1376. [Google Scholar] [CrossRef]
- Merkulova, M.; Păunescu, T.G.; Azroyan, A.; Marshansky, V.; Breton, S.; Brown, D. Mapping the H+ (V)-ATPase interactome: Identification of proteins involved in trafficking, folding, assembly and phosphorylation. Sci. Rep. 2015, 5, 14827. [Google Scholar] [CrossRef] [PubMed]
- Castroflorio, E.; Hoed, J.D.; Svistunova, D.; Finelli, M.J.; Cebrian-Serrano, A.; Corrochano, S.; Bassett, A.R.; Davies, B.; Oliver, P.L. The Ncoa7 locus regulates V-ATPase formation and function, neurodevelopment and behaviour. Cell. Mol. Life Sci. 2020, 1–22. [Google Scholar] [CrossRef]
- Fu, B.; Wang, L.; Li, S.; Dorf, M.E. ZMPSTE24 defends against influenza and other pathogenic viruses. J. Exp. Med. 2017, 214, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fu, B.; Li, W.; Patil, G.; Liu, L.; Dorf, M.E.; Li, S. Comparative influenza protein interactomes identify the role of plakophilin 2 in virus restriction. Nat. Commun. 2017, 8, 13876. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, J.; Li, M.; Chen, M.; Sun, C. Multifaceted Functions of CH25H and 25HC to Modulate the Lipid Metabolism, Immune Responses, and Broadly Antiviral Activities. Viruses 2020, 12, 727. [Google Scholar] [CrossRef]
- Blanc, M.; Hsieh, W.Y.; Robertson, K.A.; Kropp, K.A.; Forster, T.; Shui, G.; Lacaze, P.; Watterson, S.; Griffiths, S.J.; Spann, N.J.; et al. The Transcription Factor STAT-1 Couples Macrophage Synthesis of 25-Hydroxycholesterol to the Interferon Antiviral Response. Immunity 2013, 38, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Gold, E.S.; Diercks, A.H.; Podolsky, I.; Podyminogin, R.L.; Askovich, P.S.; Treuting, P.M.; Aderem, A. 25-Hydroxycholesterol acts as an amplifier of inflammatory signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 10666–10671. [Google Scholar] [CrossRef] [Green Version]
- Heaton, B.E.; Kennedy, E.M.; Dumm, R.E.; Harding, A.T.; Sacco, M.T.; Sachs, D.; Heaton, N.S. A CRISPR Activation Screen Identifies a Pan-avian Influenza Virus Inhibitory Host Factor. Cell Rep. 2017, 20, 1503–1512. [Google Scholar] [CrossRef] [Green Version]
- Meerzaman, D.; Shapiro, P.S.; Kim, K.C. Involvement of the MAP kinase ERK2 in MUC1 mucin signaling. Am. J. Physiol. Cell. Mol. Physiol. 2001, 281, L86–L91. [Google Scholar] [CrossRef]
- Mukherjee, P.; Tinder, T.L.; Basu, G.D.; Gendler, S.J. MUC1 (CD227) interacts with lck tyrosine kinase in Jurkat lymphoma cells and normal T cells. J. Leukoc. Biol. 2004, 77, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Rahn, J.J.; Shen, Q.; Mah, B.K.; Hugh, J.C. MUC1 Initiates a Calcium Signal after Ligation by Intercellular Adhesion Molecule-1. J. Biol. Chem. 2004, 279, 29386–29390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Lillehoj, E.P.; Kim, K.C. MUC1 tyrosine phosphorylation activates the extracellular signal-regulated kinase. Biochem. Biophys. Res. Commun. 2004, 321, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Lillehoj, E.P.; Kim, H.; Chun, E.Y.; Kim, K.C. Pseudomonas aeruginosastimulates phosphorylation of the airway epithelial membrane glycoprotein Muc1 and activates MAP kinase. Am. J. Physiol. Cell. Mol. Physiol. 2004, 287, L809–L815. [Google Scholar] [CrossRef] [Green Version]
- Rusinova, I.; Forster, S.; Yu, S.; Kannan, A.; Masse, M.; Cumming, H.; Chapman, R.; Hertzog, P.J. INTERFEROME v2.0: An updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2012, 41, D1040–D1046. [Google Scholar] [CrossRef]
- Ng, G.Z.; Menheniott, T.R.; Every, A.L.; Stent, A.; Judd, L.M.; Chionh, Y.T.; Dhar, P.; Komen, J.C.; Giraud, A.S.; Wang, T.C.; et al. The MUC1 mucin protects againstHelicobacter pyloripathogenesis in mice by regulation of the NLRP3 inflammasome. Gut 2016, 65, 1087–1099. [Google Scholar] [CrossRef] [PubMed]
- McAuley, J.L.; Corcilius, L.; Tan, H.-X.; Payne, R.J.; A McGuckin, M.; E Brown, L. The cell surface mucin MUC1 limits the severity of influenza A virus infection. Mucosal Immunol. 2017, 10, 1581–1593. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, R.; Schmid, S.L. The dynamin superfamily. Curr. Biol. 2018, 28, R411–R416. [Google Scholar] [CrossRef] [Green Version]
- Haller, O.; Staeheli, P.; Schwemmle, M.; Kochs, G. Mx GTPases: Dynamin-like antiviral machines of innate immunity. Trends Microbiol. 2015, 23, 154–163. [Google Scholar] [CrossRef]
- Goujon, C.; Moncorgé, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hué, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nat. Cell Biol. 2013, 502, 559–562. [Google Scholar] [CrossRef]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.J.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T.; et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nat. Cell Biol. 2013, 502, 563–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Pan, Q.; Ding, S.; Qian, J.; Xu, F.; Zhou, J.; Cen, S.; Guo, F.; Liang, C. The Interferon-Inducible MxB Protein Inhibits HIV-1 Infection. Cell Host Microbe 2013, 14, 398–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schilling, M.; Bulli, L.; Weigang, S.; Graf, L.; Naumann, S.; Patzina, C.; Wagner, V.; Bauersfeld, L.; Goujon, C.; Hengel, H.; et al. Human MxB Protein Is a Pan-herpesvirus Restriction Factor. J. Virol. 2018, 92, e01056-18. [Google Scholar] [CrossRef] [Green Version]
- Crameri, M.; Bauer, M.; Caduff, N.; Walker, R.; Steiner, F.; Franzoso, F.D.; Gujer, C.; Boucke, K.; Kucera, T.; Zbinden, A.; et al. MxB is an interferon-induced restriction factor of human herpesviruses. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, D.-R.; An, N.; Liu, Z.-L.; Xu, F.-W.; Raniga, K.; Li, Q.-J.; Zhou, R.; Wang, J.; Zhang, Y.-X.; Zhou, J.-M.; et al. Human MxB Inhibits the Replication of Hepatitis C Virus. J. Virol. 2018, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-X.; Niklasch, M.; Liu, T.; Wang, Y.; Shi, B.; Yuan, W.; Baumert, T.F.; Yuan, Z.; Tong, S.; Nassal, M.; et al. Interferon-inducible MX2 is a host restriction factor of hepatitis B virus replication. J. Hepatol. 2020, 72, 865–876. [Google Scholar] [CrossRef]
- Haller, O.; Arnheiter, H.; Pavlovic, J.; Staeheli, P. The Discovery of the Antiviral Resistance GeneMx: A Story of Great Ideas, Great Failures, and Some Success. Annu. Rev. Virol. 2018, 5, 33–51. [Google Scholar] [CrossRef]
- Kolb, E.; Laine, E.; Strehler, D.; Staeheli, P. Resistance to influenza virus infection of Mx transgenic mice expressing Mx protein under the control of two constitutive promoters. J. Virol. 1992, 66, 1709–1716. [Google Scholar] [CrossRef] [Green Version]
- Deeg, C.M.; Hassan, E.; Mutz, P.; Rheinemann, L.; Götz, V.; Magar, L.; Schilling, M.; Kallfass, C.; Nürnberger, C.; Soubies, S.; et al. In vivo evasion of MxA by avian influenza viruses requires human signature in the viral nucleoprotein. J. Exp. Med. 2017, 214, 1239–1248. [Google Scholar] [CrossRef]
- Holzinger, D.; Jorns, C.; Stertz, S.; Boisson-Dupuis, S.; Thimme, R.; Weidmann, M.; Casanova, J.-L.; Haller, O.; Kochs, G. Induction of MxA Gene Expression by Influenza A Virus Requires Type I or Type III Interferon Signaling. J. Virol. 2007, 81, 7776–7785. [Google Scholar] [CrossRef] [Green Version]
- Pitossi, F.; Blank, A.; Schröder, A.; Schwarz, A.; Hüssi, P.; Schwemmle, M.; Pavlovic, J.; Staeheli, P. A functional GTP-binding motif is necessary for antiviral activity of Mx proteins. J. Virol. 1993, 67, 6726–6732. [Google Scholar] [CrossRef] [Green Version]
- Ponten, A.; Sick, C.; Weeber, M.; Haller, O.; Kochs, G. Dominant-negative mutants of human MxA protein: Domains in the carboxy-terminal moiety are important for oligomerization and antiviral activity. J. Virol. 1997, 71, 2591–2599. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Von Der Malsburg, A.; Paeschke, S.; Behlke, J.; Haller, O.; Kochs, G.; Daumke, O. Structural basis of oligomerization in the stalk region of dynamin-like MxA. Nat. Cell Biol. 2010, 465, 502–506. [Google Scholar] [CrossRef] [Green Version]
- Haller, O.; Gao, S.; von der Malsburg, A.; Daumke, O.; Kochs, G. Dynamin-like MxA GTPase: Structural Insights into Oligomerization and Implications for Antiviral Activity. J. Biol. Chem. 2010, 285, 28419–28424. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P.S.; Patzina, C.; Emerman, M.; Haller, O.; Malik, H.S.; Kochs, G. Evolution-Guided Identification of Antiviral Specificity Determinants in the Broadly Acting Interferon-Induced Innate Immunity Factor MxA. Cell Host Microbe 2012, 12, 598–604. [Google Scholar] [CrossRef] [Green Version]
- Patzina, C.; Haller, O.; Kochs, G. Structural Requirements for the Antiviral Activity of the Human MxA Protein against Thogoto and Influenza A Virus. J. Biol. Chem. 2014, 289, 6020–6027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goujon, C.; Moncorgé, O.; Bauby, H.; Doyle, T.; Barclay, W.S.; Malim, M.H. Transfer of the Amino-Terminal Nuclear Envelope Targeting Domain of Human MX2 Converts MX1 into an HIV-1 Resistance Factor. J. Virol. 2014, 88, 9017–9026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goujon, C.; Greenbury, R.A.; Papaioannou, S.; Doyle, T.; Malim, M.H. A Triple-Arginine Motif in the Amino-Terminal Domain and Oligomerization Are Required for HIV-1 Inhibition by Human MX2. J. Virol. 2015, 89, 4676–4680. [Google Scholar] [CrossRef] [Green Version]
- Dicks, M.D.J.; Goujon, C.; Pollpeter, D.; Betancor, G.; Apolonia, L.F.S.; Bergeron, J.R.C.; Malim, M.H. Oligomerization Requirements for MX2-Mediated Suppression of HIV-1 Infection. J. Virol. 2015, 90, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, P.; Mänz, B.; Haller, O.; Schwemmle, M.; Kochs, G. The Viral Nucleoprotein Determines Mx Sensitivity of Influenza A Viruses. J. Virol. 2011, 85, 8133–8140. [Google Scholar] [CrossRef] [Green Version]
- Mänz, B.; Dornfeld, D.; Götz, H.; Zell, R.; Zimmermann, P.; Haller, O.; Kochs, G.; Schwemmle, M. Pandemic Influenza A Viruses Escape from Restriction by Human MxA through Adaptive Mutations in the Nucleoprotein. PLoS Pathog. 2013, 9, e1003279. [Google Scholar] [CrossRef]
- Riegger, D.; Hai, R.; Dornfeld, D.; Mänz, B.; Leyva-Grado, V.; Sánchez-Aparicio, M.T.; Albrecht, R.A.; Palese, P.; Haller, O.; Schwemmle, M.; et al. The Nucleoprotein of Newly Emerged H7N9 Influenza A Virus Harbors a Unique Motif Conferring Resistance to Antiviral Human MxA. J. Virol. 2014, 89, 2241–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashenberg, O.; Padmakumar, J.; Doud, M.B.; Bloom, J.D. Deep mutational scanning identifies sites in influenza nucleoprotein that affect viral inhibition by MxA. PLoS Pathog. 2017, 13, e1006288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmann, J.; Stertz, S.; Grimm, D.; Steel, J.; García-Sastre, A.; Haller, O.; Kochs, G. Influenza A Virus Strains Differ in Sensitivity to the Antiviral Action of Mx-GTPase. J. Virol. 2008, 82, 3624–3631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhelst, J.; Parthoens, E.; Schepens, B.; Fiers, W.; Saelens, X. Interferon-Inducible Protein Mx1 Inhibits Influenza Virus by Interfering with Functional Viral Ribonucleoprotein Complex Assembly. J. Virol. 2012, 86, 13445–13455. [Google Scholar] [CrossRef] [Green Version]
- Strandén, A.M.; Staeheli, P.; Pavlovic, J. Function of the Mouse Mx1 Protein Is Inhibited by Overexpression of the PB2 Protein of Influenza Virus. Virology 1993, 197, 642–651. [Google Scholar] [CrossRef]
- Gao, S.; Von Der Malsburg, A.; Dick, A.; Faelber, K.; Schröder, G.F.; Haller, O.; Kochs, G.; Daumke, O. Structure of Myxovirus Resistance Protein A Reveals Intra- and Intermolecular Domain Interactions Required for the Antiviral Function. Immunity 2011, 35, 514–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.; Killip, M.J.; Staeheli, P.; Randall, R.E.; Jackson, D. The Human Interferon-Induced MxA Protein Inhibits Early Stages of Influenza A Virus Infection by Retaining the Incoming Viral Genome in the Cytoplasm. J. Virol. 2013, 87, 13053–13058. [Google Scholar] [CrossRef] [Green Version]
- Götz, V.; Magar, L.; Dornfeld, D.; Giese, S.; Pohlmann, A.; Höper, D.; Kong, B.-W.; Jans, D.A.; Beer, M.; Haller, O.; et al. Influenza A viruses escape from MxA restriction at the expense of efficient nuclear vRNP import. Sci. Rep. 2016, 6, 23138. [Google Scholar] [CrossRef] [Green Version]
- Pavlovic, J.; Haller, O.; Staeheli, P. Human and mouse Mx proteins inhibit different steps of the influenza virus multiplication cycle. J. Virol. 1992, 66, 2564–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duc, T.T.T.; Farnir, F.; Michaux, C.; Desmecht, D.; Cornet, A. Detection of new biallelic polymorphisms in the human MxA gene. Mol. Biol. Rep. 2012, 39, 8533–8538. [Google Scholar] [CrossRef] [PubMed]
- Graf, L.; Dick, A.; Sendker, F.; Barth, E.; Marz, M.; Daumke, O.; Kochs, G. Effects of allelic variations in the human myxovirus resistance protein A on its antiviral activity. J. Biol. Chem. 2018, 293, 3056–3072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tazi-Ahnini, R.; di Giovine, F.S.; McDonagh, A.J.; Messenger, A.G.; Amadou, C.; Cox, A.; Duff, G.W.; Cork, M.J. Structure and polymorphism of the human gene for the interferon-induced p78 protein (MX1): Evidence of association with alopecia areata in the Down syndrome region. Qual. Life Res. 2000, 106, 639–645. [Google Scholar] [CrossRef]
- Haller, O.; Kochs, G. Mx genes: Host determinants controlling influenza virus infection and trans-species transmission. Qual. Life Res. 2019, 139, 695–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlovic, J.; Zürcher, T.; Haller, O.; Staeheli, P. Resistance to influenza virus and vesicular stomatitis virus conferred by expression of human MxA protein. J. Virol. 1990, 64, 3370–3375. [Google Scholar] [CrossRef] [Green Version]
- Verhelst, J.; Spitaels, J.; Nürnberger, C.; De Vlieger, D.; Ysenbaert, T.; Staeheli, P.; Fiers, W.; Saelens, X. Functional Comparison of Mx1 from Two Different Mouse Species Reveals the Involvement of Loop L4 in the Antiviral Activity against Influenza A Viruses. J. Virol. 2015, 89, 10879–10890. [Google Scholar] [CrossRef] [Green Version]
- Pavlovic, J.; Arzet, A.H.; Hefti, H.P.; Frese, M.; Rost, D.; Ernst, B.; Kolb, E.; Staeheli, P.; Haller, O. Enhanced virus resistance of transgenic mice expressing the human MxA protein. J. Virol. 1995, 69, 4506–4510. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Ishitsuka, A.; Noguchi, M.; Hirohama, M.; Fujiyasu, Y.; Petric, P.P.; Schwemmle, M.; Staeheli, P.; Nagata, K.; Kawaguchi, A. Influenza restriction factor MxA functions as inflammasome sensor in the respiratory epithelium. Sci. Immunol. 2019, 4, eaau4643. [Google Scholar] [CrossRef]
- Palm, M.; Garigliany, M.-M.; Cornet, F.; Desmecht, D. Interferon-inducedSus scrofaMx1 blocks endocytic traffic of incoming influenza A virus particles. Vet. Res. 2010, 41, 29. [Google Scholar] [CrossRef] [Green Version]
- Meier, E.; Kunz, G.; Haller, O.; Arnheiter, H. Activity of rat Mx proteins against a rhabdovirus. J. Virol. 1990, 64, 6263–6269. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, J.; Hölzer, M.; Schilling, M.; Patzina, C.; Schoen, A.; Hoenen, T.; Zimmer, G.; Marz, M.; Weber, F.; Müller, M.A.; et al. Evolution and Antiviral Specificities of Interferon-Induced Mx Proteins of Bats against Ebola, Influenza, and Other RNA Viruses. J. Virol. 2017, 91, e00361-17. [Google Scholar] [CrossRef] [Green Version]
- Benfield, C.T.O.; Lyall, J.W.; Kochs, G.; Tiley, L.S. Asparagine 631 Variants of the Chicken Mx Protein Do Not Inhibit Influenza Virus Replication in Primary Chicken Embryo Fibroblasts or In Vitro Surrogate Assays. J. Virol. 2008, 82, 7533–7539. [Google Scholar] [CrossRef] [Green Version]
- Bernasconi, D.; Schultz, U.; Staeheli, P. The Interferon-Induced Mx Protein of Chickens Lacks Antiviral Activity. J. Interf. Cytokine Res. 1995, 15, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Schusser, B.; Reuter, A.; Von Der Malsburg, A.; Penski, N.; Weigend, S.; Kaspers, B.; Staeheli, P.; Härtle, S. Mx Is Dispensable for Interferon-Mediated Resistance of Chicken Cells against Influenza A Virus. J. Virol. 2011, 85, 8307–8315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daviet, S.; Van Borm, S.; Habyarimana, A.; Ahanda, M.-L.E.; Morin, V.; Oudin, A.; Berg, T.V.D.; Zoorob, R. Induction of Mx and PKR Failed to Protect Chickens from H5N1 Infection. Viral Immunol. 2009, 22, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Bazzigher, L.; Schwarz, A.; Staeheli, P. No Enhanced Influenza Virus Resistance of Murine and Avian Cells Expressing Cloned Duck Mx Protein. Virology 1993, 195, 100–112. [Google Scholar] [CrossRef]
- Tretina, K.; Park, E.-S.; Maminska, A.; MacMicking, J.D. Interferon-induced guanylate-binding proteins: Guardians of host defense in health and disease. J. Exp. Med. 2019, 216, 482–500. [Google Scholar] [CrossRef] [Green Version]
- Ngo, C.C.; Man, S.M. Mechanisms and functions of guanylate-binding proteins and related interferon-inducible GTPases: Roles in intracellular lysis of pathogens. Cell. Microbiol. 2017, 19, e12791. [Google Scholar] [CrossRef] [Green Version]
- Nordmann, A.; Wixler, L.; Boergeling, Y.; Wixler, V.; Ludwig, S. A new splice variant of the human guanylate-binding protein 3 mediates anti-influenza activity through inhibition of viral transcription and replication. FASEB J. 2011, 26, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Shi, Z.; Yan, W.; Wei, J.; Shao, N.; Deng, X.; Wang, S.; Li, B.; Tong, G.; Ma, Z. Nonstructural Protein 1 of Influenza A Virus Interacts with Human Guanylate-Binding Protein 1 to Antagonize Antiviral Activity. PLoS ONE 2013, 8, e55920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Cao, Z.; Wang, L.; Wan, Y.; Peng, N.; Wang, Q.; Chen, X.; Zhou, Y.; Zhu, Y. Inducible GBP5 Mediates the Antiviral Response via Interferon-Related Pathways during Influenza A Virus Infection. J. Innate Immun. 2017, 9, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Braun, E.; Hotter, D.; Koepke, L.; Zech, F.; Groß, R.; Sparrer, K.M.; Müller, J.A.; Pfaller, C.K.; Heusinger, E.; Wombacher, R.; et al. Guanylate-Binding Proteins 2 and 5 Exert Broad Antiviral Activity by Inhibiting Furin-Mediated Processing of Viral Envelope Proteins. Cell Rep. 2019, 27, 2092–2104.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Xu, W.W.; Zhang, Z.; Liu, J.; Sun, L.; Sun, W.; Jiao, P.; Sang, X.; Ren, Z.; Yu, Z.; et al. The innate immunity of guinea pigs against highly pathogenic avian influenza virus infection. Oncotarget 2017, 8, 30422–30437. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Zhang, Q.; Wu, W.; Chen, L.; Gu, S.; Ye, Y.; Zhong, Y.; Huang, Q.; Liu, S. Inducible Guanylate-Binding Protein 7 Facilitates Influenza A Virus Replication by Suppressing Innate Immunity via NF-κB and JAK-STAT Signaling Pathways. J. Virol. 2021, 95. [Google Scholar] [CrossRef] [PubMed]
- King, J.K.; Yeh, S.; Lin, M.; Liu, C.; Lai, M.; Kao, J.; Chen, D.; Chen, P. Genetic polymorphisms in interferon pathway and response to interferon treatment in hepatitis B patients: A pilot study. Hepatology 2002, 36, 1416–1424. [Google Scholar] [CrossRef]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef]
- Di Pietro, A.; Kajaste-Rudnitski, A.; Oteiza, A.; Nicora, L.; Towers, G.J.; Mechti, N.; Vicenzi, E. TRIM22 Inhibits Influenza A Virus Infection by Targeting the Viral Nucleoprotein for Degradation. J. Virol. 2013, 87, 4523–4533. [Google Scholar] [CrossRef] [Green Version]
- Carthagena, L.; Bergamaschi, A.; Luna, J.M.; David, A.; Uchil, P.D.; Margottin-Goguet, F.; Mothes, W.; Hazan, U.; Transy, C.; Pancino, G.; et al. Human TRIM Gene Expression in Response to Interferons. PLoS ONE 2009, 4, e4894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagani, I.; Di Pietro, A.; Oteiza, A.; Ghitti, M.; Mechti, N.; Naffakh, N.; Vicenzi, E. Mutations Conferring Increased Sensitivity to Tripartite Motif 22 Restriction Accumulated Progressively in the Nucleoprotein of Seasonal Influenza A (H1N1) Viruses between 1918 and 2009. mSphere 2018, 3, e00110-18. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Wang, J.; Wang, S.; Wu, F.; Chen, Z.; Li, C.; Cheng, G.; Qin, F.X.-F. Inhibition of Influenza A Virus Replication by TRIM14 via Its Multifaceted Protein–Protein Interaction With NP. Front. Microbiol. 2019, 10, 344. [Google Scholar] [CrossRef] [Green Version]
- Patil, G.; Zhao, M.; Song, K.; Hao, W.; Bouchereau, D.; Wang, L.; Li, S. TRIM41-Mediated Ubiquitination of Nucleoprotein Limits Influenza A Virus Infection. J. Virol. 2018, 92, 15. [Google Scholar] [CrossRef] [Green Version]
- Fu, B.; Wang, L.; Ding, H.; Schwamborn, J.C.; Li, S.; Dorf, M.E. TRIM32 Senses and Restricts Influenza A Virus by Ubiquitination of PB1 Polymerase. PLoS Pathog. 2015, 11, e1004960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudryashova, E.; Wu, J.; Havton, L.A.; Spencer, M.J. Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum. Mol. Genet. 2009, 18, 1353–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicklas, S.; Otto, A.; Wu, X.; Miller, P.; Stelzer, S.; Wen, Y.; Kuang, S.; Wrogemann, K.; Patel, K.; Ding, H.; et al. TRIM32 Regulates Skeletal Muscle Stem Cell Differentiation and Is Necessary for Normal Adult Muscle Regeneration. PLoS ONE 2012, 7, e30445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, N.; Jiang, L.; Ye, M.; Wang, Y.; Wang, G.; Wan, X.; Zhao, Y.; Wen, X.; Liang, L.; Ma, S.; et al. TRIM35 mediates protection against influenza infection by activating TRAF3 and degrading viral PB2. Protein Cell 2020, 11, 894–914. [Google Scholar] [CrossRef]
- Choudhury, N.R.; Heikel, G.; Michlewski, G. TRIM25 and its emerging RNA-binding roles in antiviral defense. Wiley Interdiscip. Rev. RNA 2020, 11, e1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyerson, N.R.; Zhou, L.; Guo, Y.R.; Zhao, C.; Tao, Y.J.; Krug, R.M.; Sawyer, S.L. Nuclear TRIM25 Specifically Targets Influenza Virus Ribonucleoproteins to Block the Onset of RNA Chain Elongation. Cell Host Microbe 2017, 22, 627–638.e7. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Li, N.L.; Shen, Y.; Bao, X.; Fabrizio, T.; Elbahesh, H.; Webby, R.J.; Li, K. The C-Terminal Tail of TRIM56 Dictates Antiviral Restriction of Influenza A and B Viruses by Impeding Viral RNA Synthesis. J. Virol. 2016, 90, 4369–4382. [Google Scholar] [CrossRef] [Green Version]
- Seo, G.J.; Kim, C.; Shin, W.-J.; Sklan, E.H.; Eoh, H.; Jung, J.U. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chemudupati, M.; Kenney, A.D.; Bonifati, S.; Zani, A.; McMichael, T.M.; Wu, L.; Yount, J.S. From APOBEC to ZAP: Diverse mechanisms used by cellular restriction factors to inhibit virus infections. Biochim. Biophys. Acta Bioenerg. 2019, 1866, 382–394. [Google Scholar] [CrossRef]
- Li, M.M.H.; Aguilar, E.G.; Michailidis, E.; Pabon, J.; Park, P.; Wu, X.; De Jong, Y.P.; Schneider, W.M.; Molina, H.; Rice, C.M.; et al. Characterization of Novel Splice Variants of Zinc Finger Antiviral Protein (ZAP). J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meagher, J.L.; Takata, M.; Gonçalves-Carneiro, D.; Keane, S.C.; Rebendenne, A.; Ong, H.; Orr, V.K.; Macdonald, M.R.; Stuckey, J.A.; Bieniasz, P.D.; et al. Structure of the zinc-finger antiviral protein in complex with RNA reveals a mechanism for selective targeting of CG-rich viral sequences. Proc. Natl. Acad. Sci. USA 2019, 116, 24303–24309. [Google Scholar] [CrossRef]
- Takata, M.A.; Gonçalves-Carneiro, D.; Zang, T.M.; Soll, S.J.; York, A.; Blanco-Melo, D.; Bieniasz, P.D. CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nat. Cell Biol. 2017, 550, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-H.; Zhou, L.; Chen, G.; Krug, R.M. Battle between influenza A virus and a newly identified antiviral activity of the PARP-containing ZAPL protein. Proc. Natl. Acad. Sci. USA 2015, 112, 14048–14053. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Wang, X.; Gao, G. The Short Form of the Zinc Finger Antiviral Protein Inhibits Influenza A Virus Protein Expression and Is Antagonized by the Virus-Encoded NS1. J. Virol. 2016, 91. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Komano, J.; Saitoh, Y.; Yamaoka, S.; Kozaki, T.; Misawa, T.; Takahama, M.; Satoh, T.; Takeuchi, O.; Yamamoto, N.; et al. Zinc-finger antiviral protein mediates retinoic acid inducible gene I-like receptor-independent antiviral response to murine leukemia virus. Proc. Natl. Acad. Sci. USA 2013, 110, 12379–12384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, M.M.H.; Zhao, J.; Li, S.; Macdonald, M.R.; Rice, C.M.; Gao, X.; Gao, G. Sindbis Virus Can Exploit a Host Antiviral Protein To Evade Immune Surveillance. J. Virol. 2016, 90, 10247–10258. [Google Scholar] [CrossRef] [Green Version]
- Kerns, J.A.; Emerman, M.; Malik, H.S. Positive Selection and Increased Antiviral Activity Associated with the PARP-Containing Isoform of Human Zinc-Finger Antiviral Protein. PLoS Genet. 2008, 4, e21. [Google Scholar] [CrossRef]
- Gonçalves-Carneiro, D.; Takata, M.A.; Ong, H.; Shilton, A.; Bieniasz, P.D. Origin and evolution of the Zinc Finger Antiviral Protein. Microbiology 2020. [Google Scholar] [CrossRef]
- Goossens, K.E.; Karpala, A.J.; Ward, A.; Bean, A.G. Characterisation of chicken ZAP. Dev. Comp. Immunol. 2014, 46, 373–381. [Google Scholar] [CrossRef]
- Lin, R.-J.; Huang, C.-H.; Liu, P.-C.; Lin, I.-C.; Huang, Y.-L.; Chen, A.-Y.; Chiu, H.-P.; Shih, S.-R.; Lin, L.-H.; Lien, S.-P.; et al. Zinc finger protein ZFP36L1 inhibits influenza A virus through translational repression by targeting HA, M and NS RNA transcripts. Nucleic Acids Res. 2020, 48, 7371–7384. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.-O.; Kim, H.-E.; Choi, W.-S.; Chun, C.-H.; Chun, J.-S. RNA-binding protein ZFP36L1 regulates osteoarthritis by modulating members of the heat shock protein 70 family. Nat. Commun. 2019, 10, 77. [Google Scholar] [CrossRef]
- Diamond, M.S.; Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2012, 13, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Peters, K.L.; Sen, G.C. Induction of the Human Protein P56 by Interferon, Double-Stranded RNA, or Virus Infection. Virology 2000, 267, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichlmair, A.; Lassnig, C.; Eberle, C.-A.; Górna, M.W.; Baumann, C.L.; Burkard, T.R.; Bürckstümmer, T.; Stefanovic, A.; Krieger, S.; Bennett, K.L.; et al. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 2011, 12, 624–630. [Google Scholar] [CrossRef]
- Habjan, M.; Hubel, P.; Lacerda, L.; Benda, C.; Holze, C.; Eberl, C.H.; Mann, A.; Kindler, E.; Gil-Cruz, C.; Ziebuhr, J.; et al. Sequestration by IFIT1 Impairs Translation of 2′O-unmethylated Capped RNA. PLoS Pathog. 2013, 9, e1003663. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Katoh, H.; Kayama, H.; Saiga, H.; Okuyama, M.; Okamoto, T.; Umemoto, E.; Matsuura, Y.; Yamamoto, M.; Takeda, K. Ifit1 Inhibits Japanese Encephalitis Virus Replication through Binding to 5′ Capped 2′-O Unmethylated RNA. J. Virol. 2013, 87, 9997–10003. [Google Scholar] [CrossRef] [Green Version]
- Terenzi, F.; Saikia, P.; Sen, G.C. Interferon-inducible protein, P56, inhibits HPV DNA replication by binding to the viral protein E1. EMBO J. 2008, 27, 3311–3321. [Google Scholar] [CrossRef] [Green Version]
- Mears, H.V.; Sweeney, T.R. Better together: The role of IFIT protein–protein interactions in the antiviral response. J. Gen. Virol. 2018, 99, 1463–1477. [Google Scholar] [CrossRef]
- Pinto, A.K.; Williams, G.D.; Szretter, K.J.; White, J.P.; Proença-Módena, J.L.; Liu, G.; Olejnik, J.; Brien, J.D.; Ebihara, H.; Mühlberger, E.; et al. Human and Murine IFIT1 Proteins Do Not Restrict Infection of Negative-Sense RNA Viruses of the Orthomyxoviridae, Bunyaviridae, and Filoviridae Families. J. Virol. 2015, 89, 9465–9476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, V.; Ledwith, M.P.; Thamamongood, T.; Higgins, C.A.; Tripathi, S.; Chang, M.W.; Benner, C.; García-Sastre, A.; Schwemmle, M.; Boon, A.C.M.; et al. Influenza virus repurposes the antiviral protein IFIT2 to promote translation of viral mRNAs. Nat. Microbiol. 2020, 5, 1490–1503. [Google Scholar] [CrossRef]
- Rong, E.; Hu, J.; Yang, C.; Chen, H.; Wang, Z.; Liu, X.; Liu, W.; Lu, C.; He, P.; Wang, X.; et al. Broad-spectrum antiviral functions of duck interferon-induced protein with tetratricopeptide repeats (AvIFIT). Dev. Comp. Immunol. 2018, 84, 71–81. [Google Scholar] [CrossRef]
- Santhakumar, D.; Rohaim, M.A.M.S.; Hussein, H.A.; Hawes, P.; Ferreira, H.L.; Behboudi, S.; Iqbal, M.; Nair, V.; Arns, C.W.; Munir, M. Chicken Interferon-induced Protein with Tetratricopeptide Repeats 5 Antagonizes Replication of RNA Viruses. Sci. Rep. 2018, 8, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohaim, M.A.; Santhakumar, D.; El Naggar, R.F.; Iqbal, M.; Hussein, H.A.; Munir, M. Chickens Expressing IFIT5 Ameliorate Clinical Outcome and Pathology of Highly Pathogenic Avian Influenza and Velogenic Newcastle Disease Viruses. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Marié, I.; Svab, J.; Robert, N.; Galabru, J.; Hovanessian, A.G. Differential expression and distinct structure of 69- and 100-kDa forms of 2-5A synthetase in human cells treated with interferon. J. Biol. Chem. 1990, 265, 18601–18607. [Google Scholar] [CrossRef]
- Silverman, R.H.; Weiss, S.R. Viral Phosphodiesterases That Antagonize Double-Stranded RNA Signaling to RNase L by Degrading 2-5A. J. Interf. Cytokine Res. 2014, 34, 455–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malathi, K.; Saito, T.; Crochet, N.; Barton, D.J.; Gale, M.; Silverman, R.H. RNase L releases a small RNA from HCV RNA that refolds into a potent PAMP. RNA 2010, 16, 2108–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malathi, K.; Dong, B.; Gale, M.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Zhang, Y.; Ghosh, A.; Cuevas, R.A.; Forero, A.; Dhar, J.; Ibsen, M.S.; Schmid-Burgk, J.L.; Schmidt, T.; Ganapathiraju, M.K.; et al. Antiviral Activity of Human OASL Protein Is Mediated by Enhancing Signaling of the RIG-I RNA Sensor. Immunity 2014, 40, 936–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drappier, M.; Michiels, T. Inhibition of the OAS/RNase L pathway by viruses. Curr. Opin. Virol. 2015, 15, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Min, J.-Y.; Krug, R.M. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2′-5′ oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 7100–7105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Banerjee, S.; Wang, Y.; Goldstein, S.A.; Dong, B.; Gaughan, C.; Silverman, R.H.; Weiss, S.R. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc. Natl. Acad. Sci. USA 2016, 113, 2241–2246. [Google Scholar] [CrossRef] [Green Version]
- Zhou, A.; Paranjape, J.; Brown, T.L.; Nie, H.; Naik, S.; Dong, B.; Chang, A.; Trapp, B.; Fairchild, R.; Colmenares, C.; et al. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 1997, 16, 6355–6363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meurs, E.; Chong, K.; Galabru, J.; Thomas, N.B.; Kerr, I.M.; Williams, B.R.; Hovanessian, A.G. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 1990, 62, 379–390. [Google Scholar] [CrossRef]
- Dar, A.C.; Dever, T.E.; Sicheri, F. Higher-Order Substrate Recognition of eIF2α by the RNA-Dependent Protein Kinase PKR. Cell 2005, 122, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Dauber, B.; Wolff, T. Activation of the Antiviral Kinase PKR and Viral Countermeasures. Viruses 2009, 1, 523–544. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.G.; Smith, J.A.; Balachandran, S.; Perwitasari, O.; Proll, S.C.; Thomas, M.J.; Korth, M.J.; Barber, G.N.; Schiff, L.A.; Katze, M.G. The Cellular Protein P58IPK Regulates Influenza Virus mRNA Translation and Replication through a PKR-Mediated Mechanism. J. Virol. 2006, 81, 2221–2230. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Wambach, M.; Katze, M.G.; Krug, R.M. Binding of the Influenza Virus NS1 Protein to Double-Stranded RNA Inhibits the Activation of the Protein Kinase That Phosphorylates the eIF-2 Translation Initiation Factor. Virology 1995, 214, 222–228. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, M.; Garcia-Sastre, A.; Carnero, E.; Pehamberger, H.; Wolff, K.; Palese, P.; Muster, T. Influenza Virus NS1 Protein Counteracts PKR-Mediated Inhibition of Replication. J. Virol. 2000, 74, 6203–6206. [Google Scholar] [CrossRef] [Green Version]
- Hatada, E.; Saito, S.; Fukuda, R. Mutant Influenza Viruses with a Defective NS1 Protein Cannot Block the Activation of PKR in Infected Cells. J. Virol. 1999, 73, 2425–2433. [Google Scholar] [CrossRef] [Green Version]
- Schierhorn, K.L.; Jolmes, F.; Bespalowa, J.; Saenger, S.; Peteranderl, C.; Dzieciolowski, J.; Mielke, M.; Budt, M.; Pleschka, S.; Herrmann, A.; et al. Influenza A Virus Virulence Depends on Two Amino Acids in the N-Terminal Domain of Its NS1 Protein To Facilitate Inhibition of the RNA-Dependent Protein Kinase PKR. J. Virol. 2017, 91, e00198-17. [Google Scholar] [CrossRef] [Green Version]
- Balachandran, S.; Roberts, P.C.; Brown, E.L.; Truong, H.; Pattnaik, A.K.; Archer, D.R.; Barber, G.N. Essential Role for the dsRNA-Dependent Protein Kinase PKR in Innate Immunity to Viral Infection. Immunity 2000, 13, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Fleming-Canepa, X.; Aldridge, J.R.; Canniff, L.; Kobewka, M.; Jax, E.; Webster, R.G.; Magor, K.E. Duck innate immune responses to high and low pathogenicity H5 avian influenza viruses. Vet. Microbiol. 2019, 228, 101–111. [Google Scholar] [CrossRef]
- Kao, P.N.; Chen, L.; Brock, G.; Ng, J.; Kenny, J.; Smith, A.J.; Corthesy, B. Cloning and expression of cyclosporin A- and FK506-sensitive nuclear factor of activated T-cells: NF45 and NF90. J. Biol. Chem. 1994, 269, 20691–20699. [Google Scholar] [CrossRef]
- Saunders, L.R.; Perkins, D.J.; Balachandran, S.; Michaels, R.; Ford, R.; Mayeda, A.; Barber, G.N. Characterization of Two Evolutionarily Conserved, Alternatively Spliced Nuclear Phosphoproteins, NFAR-1 and -2, That Function in mRNA Processing and Interact with the Double-stranded RNA-dependent Protein Kinase, PKR. J. Biol. Chem. 2001, 276, 32300–32312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patiño, C.; Haenni, A.-L.; Urcuqui-Inchima, S. NF90 isoforms, a new family of cellular proteins involved in viral replication? Biochimie 2015, 108, 20–24. [Google Scholar] [CrossRef]
- Li, T.; Li, X.; Zhu, W.; Wang, H.; Mei, L.; Wu, S.; Lin, X.; Han, X. NF90 is a novel influenza A virus NS1-interacting protein that antagonizes the inhibitory role of NS1 on PKR phosphorylation. FEBS Lett. 2016, 590, 2797–2810. [Google Scholar] [CrossRef]
- Wen, X.; Huang, X.; Mok, B.W.-Y.; Chen, Y.; Zheng, M.; Lau, S.-Y.; Wang, P.; Song, W.; Jin, D.-Y.; Yuen, K.-Y.; et al. NF90 Exerts Antiviral Activity through Regulation of PKR Phosphorylation and Stress Granules in Infected Cells. J. Immunol. 2014, 192, 3753–3764. [Google Scholar] [CrossRef] [PubMed]
- Bortz, E.; Westera, L.; Maamary, J.; Steel, J.; Albrecht, R.A.; Manicassamy, B.; Chase, G.; Martínez-Sobrido, L.; Schwemmle, M.; García-Sastre, A. Host- and Strain-Specific Regulation of Influenza Virus Polymerase Activity by Interacting Cellular Proteins. mBio 2011, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Song, W.; Mok, B.W.-Y.; Zhao, P.; Qin, K.; Lai, A.; Smith, G.J.D.; Zhang, J.; Lin, T.; Guan, Y.; et al. Nuclear Factor 90 Negatively Regulates Influenza Virus Replication by Interacting with Viral Nucleoprotein. J. Virol. 2009, 83, 7850–7861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Godfrey, W.R.; Lin, J.; Zhao, G.; Kao, P.N. NF90 regulates inducible IL-2 gene expression in T cells. J. Exp. Med. 2007, 204, 971–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, A.D.; Rao, B.G.; Jeang, K.-T. Viral and cellular RNA helicases as antiviral targets. Nat. Rev. Drug Discov. 2005, 4, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Ranji, A.; Boris-Lawrie, K. RNA helicases. RNA Biol. 2010, 7, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregersen, L.H.; Schueler, M.; Munschauer, M.; Mastrobuoni, G.; Chen, W.; Kempa, S.; Dieterich, C.; Landthaler, M. MOV10 Is a 5′ to 3′ RNA Helicase Contributing to UPF1 mRNA Target Degradation by Translocation along 3′ UTRs. Mol. Cell 2014, 54, 573–585. [Google Scholar] [CrossRef] [Green Version]
- Kenny, P.J.; Zhou, H.; Kim, M.; Skariah, G.; Khetani, R.S.; Drnevich, J.; Arcila, M.L.; Kosik, K.S.; Ceman, S. MOV10 and FMRP Regulate AGO2 Association with MicroRNA Recognition Elements. Cell Rep. 2014, 9, 1729–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodier, J.L.; Cheung, L.E.; Kazazian, H.H., Jr. MOV10 RNA Helicase Is a Potent Inhibitor of Retrotransposition in Cells. PLoS Genet. 2012, 8, e1002941. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.-H.; Jeang, K.-T.; Tokunaga, K. Host restriction factors in retroviral infection: Promises in virus-host interaction. Retrovirology 2012, 9, 112. [Google Scholar] [CrossRef] [Green Version]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.D.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nat. Cell Biol. 2011, 472, 481–485. [Google Scholar] [CrossRef]
- Cuevas, R.A.; Ghosh, A.; Wallerath, C.; Hornung, V.; Coyne, C.B.; Sarkar, S.N. MOV10 Provides Antiviral Activity against RNA Viruses by Enhancing RIG-I-MAVS-Independent IFN Induction. J. Immunol. 2016, 196, 3877–3886. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Huang, F.; Tan, L.; Bai, C.; Chen, B.; Liu, J.; Liang, J.; Liu, C.; Zhang, S.; Lu, G.; et al. Host Protein Moloney Leukemia Virus 10 (MOV10) Acts as a Restriction Factor of Influenza A Virus by Inhibiting the Nuclear Import of the Viral Nucleoprotein. J. Virol. 2016, 90, 3966–3980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Hu, S.; Xu, F.; Mei, S.; Liu, X.; Yin, L.; Zhao, F.; Zhao, X.; Sun, H.; Xiong, Z.; et al. MOV10 sequesters the RNP of influenza A virus in the cytoplasm and is antagonized by viral NS1 protein. Biochem. J. 2019, 476, 467–481. [Google Scholar] [CrossRef]
- Skariah, G.; Seimetz, J.; Norsworthy, M.; Lannom, M.C.; Kenny, P.J.; Elrakhawy, M.; Forsthoefel, C.; Drnevich, J.; Kalsotra, A.; Ceman, S. Mov10 suppresses retroelements and regulates neuronal development and function in the developing brain. BMC Biol. 2017, 15, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linder, P.; Jankowsky, E. From unwinding to clamping—The DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011, 12, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.; Hotz-Wagenblatt, A.; Voit, R.; Grummt, I. SIRT7 and the DEAD-box helicase DDX21 cooperate to resolve genomic R loops and safeguard genome stability. Genes Dev. 2017, 31, 1370–1381. [Google Scholar] [CrossRef]
- Taschuk, F.; Cherry, S. DEAD-Box Helicases: Sensors, Regulators, and Effectors for Antiviral Defense. Viruses 2020, 12, 181. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Liu, C.-H.; Zhou, L.; Krug, R.M. Cellular DDX21 RNA Helicase Inhibits Influenza A Virus Replication but Is Counteracted by the Viral NS1 Protein. Cell Host Microbe 2014, 15, 484–493. [Google Scholar] [CrossRef] [Green Version]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Boukari, H.; Banerjee, I.; Sbalzarini, I.F.; Horvath, P.; Helenius, A. Histone Deacetylase 8 Is Required for Centrosome Cohesion and Influenza A Virus Entry. PLoS Pathog. 2011, 7, e1002316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagesh, P.T.; Husain, M. Influenza A Virus Dysregulates Host Histone Deacetylase 1 That Inhibits Viral Infection in Lung Epithelial Cells. J. Virol. 2016, 90, 4614–4625. [Google Scholar] [CrossRef] [Green Version]
- Nagesh, P.T.; Hussain, M.; Galvin, H.D.; Husain, M. Histone Deacetylase 2 Is a Component of Influenza A Virus-Induced Host Antiviral Response. Front. Microbiol. 2017, 8, 1315. [Google Scholar] [CrossRef]
- Banerjee, I.; Miyake, Y.; Nobs, S.P.; Schneider, C.; Horvath, P.; Kopf, M.; Matthias, P.; Helenius, A.; Yamauchi, Y. Influenza A virus uses the aggresome processing machinery for host cell entry. Science 2014, 346, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.; Cheung, C.-Y. Histone Deacetylase 6 Inhibits Influenza A Virus Release by Downregulating the Trafficking of Viral Components to the Plasma Membrane via Its Substrate, Acetylated Microtubules. J. Virol. 2014, 88, 11229–11239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanin, M.; DeBeauchamp, J.; Vangala, G.; Webby, R.J.; Husain, M. Histone Deacetylase 6 Knockout Mice Exhibit Higher Susceptibility to Influenza A Virus Infection. Viruses 2020, 12, 728. [Google Scholar] [CrossRef]
- Galvin, H.D.; Husain, M. Influenza A virus-induced host caspase and viral PA-X antagonize the antiviral host factor, histone deacetylase 4. J. Biol. Chem. 2019, 294, 20207–20221. [Google Scholar] [CrossRef]
- Koyuncu, E.; Budayeva, H.G.; Miteva, Y.V.; Ricci, D.P.; Silhavy, T.J.; Shenk, T.; Cristea, I.M. Sirtuins Are Evolutionarily Conserved Viral Restriction Factors. mBio 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nutsford, A.N.; Galvin, H.D.; Ahmed, F.; Husain, M. The Class IV human deacetylase, HDAC11, exhibits anti-influenza A virus properties via its involvement in host innate antiviral response. Cell Microbiol. 2019, 21, e12989. [Google Scholar] [CrossRef]
- Reilly, P.T.; Yu, Y.; Hamiche, A.; Wang, L. Cracking the ANP32 whips: Important functions, unequal requirement, and hints at disease implications. BioEssays 2014, 36, 1062–1071. [Google Scholar] [CrossRef] [Green Version]
- Long, J.S.; Giotis, E.S.; Moncorgé, O.; Frise, R.; Mistry, B.; James, J.; Morisson, M.; Iqbal, M.; Vignal, A.; Skinner, M.A.; et al. Species difference in ANP32A underlies influenza A virus polymerase host restriction. Nat. Cell Biol. 2016, 529, 101–104. [Google Scholar] [CrossRef] [Green Version]
- Carrique, L.; Fan, H.; Walker, A.P.; Keown, J.R.; Sharps, J.; Staller, E.; Barclay, W.S.; Fodor, E.; Grimes, J.M. Host ANP32A mediates the assembly of the influenza virus replicase. Nat. Cell Biol. 2020, 587, 638–643. [Google Scholar] [CrossRef]
- Almond, J.W. A single gene determines the host range of influenza virus. Nat. Cell Biol. 1977, 270, 617–618. [Google Scholar] [CrossRef]
- Subbarao, E.K.; London, W.; Murphy, B.R. A single amino acid in the PB2 gene of influenza A virus is a determinant of host range. J. Virol. 1993, 67, 1761–1764. [Google Scholar] [CrossRef] [Green Version]
- Moncorgé, O.; Long, J.S.; Cauldwell, A.V.; Zhou, H.; Lycett, S.J.; Barclay, W.S. Investigation of Influenza Virus Polymerase Activity in Pig Cells. J. Virol. 2013, 87, 384–394. [Google Scholar] [CrossRef] [Green Version]
- Shinya, K.; Makino, A.; Ozawa, M.; Kim, J.H.; Sakai-Tagawa, Y.; Ito, M.; Le, Q.M.; Kawaoka, Y. Ostrich Involvement in the Selection of H5N1 Influenza Virus Possessing Mammalian-Type Amino Acids in the PB2 Protein. J. Virol. 2009, 83, 13015–13018. [Google Scholar] [CrossRef] [Green Version]
- Staller, E.; Sheppard, C.M.; Neasham, P.J.; Mistry, B.; Peacock, T.P.; Goldhill, D.H.; Long, J.S.; Barclay, W.S. ANP32 Proteins Are Essential for Influenza Virus Replication in Human Cells. J. Virol. 2019, 93, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.H.; Chungu, K.; Bin Lee, S.; Woo, S.J.; Cho, H.Y.; Lee, H.J.; Rengaraj, D.; Lee, J.-H.; Song, C.-S.; Lim, J.M.; et al. Host-Specific Restriction of Avian Influenza Virus Caused by Differential Dynamics of ANP32 Family Members. J. Infect. Dis. 2020, 221, 71–80. [Google Scholar] [CrossRef]
- Reilly, P.T.; Afzal, S.; Wakeham, A.; Haight, J.; You-Ten, A.; Zaugg, K.; Dembowy, J.; Young, A.; Mak, T.W. Generation and Characterization of the Anp32e-Deficient Mouse. PLoS ONE 2010, 5, e13597. [Google Scholar] [CrossRef] [Green Version]
- Beck, S.; Zickler, M.; Dos Reis, V.P.; Günther, T.; Grundhoff, A.; Reilly, P.T.; Mak, T.W.; Stanelle-Bertram, S.; Gabriel, G. ANP32B Deficiency Protects Mice From Lethal Influenza A Virus Challenge by Dampening the Host Immune Response. Front. Immunol. 2020, 11, 450. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.F.; Ledwith, M.P.; Mehle, A. Differential Splicing of ANP32A in Birds Alters Its Ability to Stimulate RNA Synthesis by Restricted Influenza Polymerase. Cell Rep. 2018, 24, 2581–2588.e4. [Google Scholar] [CrossRef] [Green Version]
- Domingues, P.; Eletto, D.; Magnus, C.; Turkington, H.L.; Schmutz, S.; Zagordi, O.; Lenk, M.; Beer, M.; Stertz, S.; Hale, B.G. Profiling host ANP32A splicing landscapes to predict influenza A virus polymerase adaptation. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fang, A.; Bi, Z.; Ye, H.; Yan, L. SRSF10 inhibits the polymerase activity and replication of avian influenza virus by regulating the alternative splicing of chicken ANP32A. Virus Res. 2020, 286, 198063. [Google Scholar] [CrossRef]
- Der, S.D.; Zhou, A.; Williams, B.R.G.; Silverman, R.H. Identification of genes differentially regulated by interferon, or using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar] [CrossRef] [Green Version]
- Dzimianski, J.V.; Scholte, F.E.; Bergeron, É.; Pegan, S.D. ISG15: It’s Complicated. J. Mol. Biol. 2019, 431, 4203–4216. [Google Scholar] [CrossRef]
- Shi, H.-X.; Yang, K.; Liu, X.; Liu, X.-Y.; Wei, B.; Shan, Y.-F.; Zhu, L.-H.; Wang, C. Positive Regulation of Interferon Regulatory Factor 3 Activation by Herc5 via ISG15 Modification. Mol. Cell. Biol. 2010, 30, 2424–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenschow, D.J.; Lai, C.; Frias-Staheli, N.; Giannakopoulos, N.V.; Lutz, A.; Wolff, T.; Osiak, A.; Levine, B.; Schmidt, R.E.; García-Sastre, A.; et al. From the cover: IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl. Acad. Sci. USA 2007, 104, 1371–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, D.J.; Monte, K.; Sun, L.; Struckhoff, J.J.; Agapov, E.; Holtzman, M.J.; Stappenbeck, T.S.; Lenschow, D.J. Novel Mode of ISG15-Mediated Protection against Influenza A Virus and Sendai Virus in Mice. J. Virol. 2014, 89, 337–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Hsiang, T.-Y.; Kuo, R.-L.; Krug, R.M. ISG15 conjugation system targets the viral NS1 protein in influenza A virus–infected cells. Proc. Natl. Acad. Sci. USA 2010, 107, 2253–2258. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Zhong, G.; Zhu, L.; Liu, X.; Shan, Y.; Feng, H.; Bu, Z.; Chen, H.; Wang, C. Herc5 Attenuates Influenza A Virus by Catalyzing ISGylation of Viral NS1 Protein. J. Immunol. 2010, 184, 5777–5790. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.H.; Espert, L.; Mechti, N.; Wilson, D.M. The Human Interferon- and Estrogen-RegulatedISG20/HEM45Gene Product Degrades Single-Stranded RNA and DNA in Vitro. Biochemistry 2001, 40, 7174–7179. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, L.; Pan, J. Interferon-stimulated gene 20-kDa protein (ISG20) in infection and disease: Review and outlook. Intractable Rare Dis. Res. 2017, 6, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Nguyen, X.-N.; Wang, L.; Appourchaux, R.; Zhang, C.; Panthu, B.; Gruffat, H.; Journo, C.; Alais, S.; Qin, J.; et al. The interferon stimulated gene 20 protein (ISG20) is an innate defense antiviral factor that discriminates self versus non-self translation. PLoS Pathog. 2019, 15, e1008093. [Google Scholar] [CrossRef]
- Espert, L.; Degols, G.; Gongora, C.; Blondel, D.; Williams, B.R.; Silverman, R.H.; Mechti, N. ISG20, a New Interferon-induced RNase Specific for Single-stranded RNA, Defines an Alternative Antiviral Pathway against RNA Genomic Viruses. J. Biol. Chem. 2003, 278, 16151–16158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, H.; Li, J.; Yang, L.; Sun, L.; Liu, W.; He, H. Influenza A Virus-induced expression of ISG20 inhibits viral replication by interacting with nucleoprotein. Virus Genes 2016, 52, 759–767. [Google Scholar] [CrossRef]
- Chai, W.; Li, J.; Shangguan, Q.; Liu, Q.; Li, X.; Qi, D.; Tong, X.; Liu, W.; Ye, X. Lnc-ISG20 Inhibits Influenza A Virus Replication by Enhancing ISG20 Expression. J. Virol. 2018, 92, e00539-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, C.M.; Trobaugh, D.W.; Sun, C.; Lucas, T.M.; Diamond, M.S.; Ryman, K.D.; Klimstra, W.B. The Interferon-Induced Exonuclease ISG20 Exerts Antiviral Activity through Upregulation of Type I Interferon Response Proteins. mSphere 2018, 3, e00209-18. [Google Scholar] [CrossRef] [Green Version]
- Simon, V.; Bloch, N.; Landau, N.R. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat. Immunol. 2015, 16, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nat. Cell Biol. 2008, 451, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin Inhibits HIV-1 Release by Directly Tethering Virions to Cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Winkler, M.; Bertram, S.; Gnirß, K.; Nehlmeier, I.; Gawanbacht, A.; Kirchhoff, F.; Ehrhardt, C.; Ludwig, S.; Kiene, M.; Moldenhauer, A.-S.; et al. Influenza A Virus Does Not Encode a Tetherin Antagonist with Vpu-Like Activity and Induces IFN-Dependent Tetherin Expression in Infected Cells. PLoS ONE 2012, 7, e43337. [Google Scholar] [CrossRef] [Green Version]
- Bruce, E.A.; Abbink, T.E.; Wise, H.M.; Rollason, R.; Galao, R.P.; Banting, G.; Neil, S.J.; Digard, P. Release of filamentous and spherical influenza A virus is not restricted by tetherin. J. Gen. Virol. 2012, 93, 963–969. [Google Scholar] [CrossRef]
- Londrigan, S.L.; Tate, M.D.; Job, E.R.; Moffat, J.M.; Wakim, L.M.; Gonelli, C.A.; Purcell, D.F.J.; Brooks, A.G.; Villadangos, J.A.; Reading, P.C.; et al. Endogenous Murine BST-2/Tetherin Is Not a Major Restriction Factor of Influenza A Virus Infection. PLoS ONE 2015, 10, e0142925. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Leser, G.P.; Lamb, R.A. Influenza virus is not restricted by tetherin whereas influenza VLP production is restricted by tetherin. Virology 2011, 417, 50–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnirß, K.; Zmora, P.; Blazejewska, P.; Winkler, M.; Lins, A.; Nehlmeier, I.; Gärtner, S.; Moldenhauer, A.-S.; Hofmann-Winkler, H.; Wolff, T.; et al. Tetherin Sensitivity of Influenza A Viruses Is Strain Specific: Role of Hemagglutinin and Neuraminidase. J. Virol. 2015, 89, 9178–9188. [Google Scholar] [CrossRef] [Green Version]
- Mangeat, B.; Cavagliotti, L.; Lehmann, M.; Gers-Huber, G.; Kaur, I.; Thomas, Y.; Kaiser, L.; Piguet, V. Influenza Virus Partially Counteracts Restriction Imposed by Tetherin/BST-2. J. Biol. Chem. 2012, 287, 22015–22029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leyva-Grado, V.H.; Hai, R.; Fernandes, F.; Belicha-Villanueva, A.; Carter, C.; Yondola, M.A. Modulation of an Ectodomain Motif in the Influenza A Virus Neuraminidase Alters Tetherin Sensitivity and Results in Virus Attenuation In Vivo. J. Mol. Biol. 2014, 426, 1308–1321. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Yin, L.; Mei, S.; Li, J.; Xu, F.; Sun, H.; Liu, X.; Cen, S.; Liang, C.; Li, A.; et al. BST-2 restricts IAV release and is countered by the viral M2 protein. Biochem. J. 2017, 474, 715–730. [Google Scholar] [CrossRef] [PubMed]
- Yi, E.; Oh, J.; Giao, N.Q.; Oh, S.; Park, S.-H. Enhanced production of enveloped viruses in BST-2-deficient cell lines. Biotechnol. Bioeng. 2017, 114, 2289–2297. [Google Scholar] [CrossRef] [PubMed]
- Yondola, M.A.; Fernandes, F.; Belicha-Villanueva, A.; Uccelini, M.; Gao, Q.; Carter, C.; Palese, P. Budding Capability of the Influenza Virus Neuraminidase Can Be Modulated by Tetherin. J. Virol. 2011, 85, 2480–2491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Melo, D.; Venkatesh, S.; Bieniasz, P.D. Origins and Evolution of tetherin, an Orphan Antiviral Gene. Cell Host Microbe 2016, 20, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Heusinger, E.; Kluge, S.F.; Kirchhoff, F.; Sauter, D. Early Vertebrate Evolution of the Host Restriction Factor Tetherin. J. Virol. 2015, 89, 12154–12165. [Google Scholar] [CrossRef] [Green Version]
- Krchlíková, V.; Fábryová, H.; Hron, T.; Young, J.M.; Koslová, A.; Hejnar, J.; Strebel, K.; Elleder, D. Antiviral Activity and Adaptive Evolution of Avian Tetherins. J. Virol. 2020, 94, 01. [Google Scholar] [CrossRef]
- Chin, K.-C.; Cresswell, P. Viperin (cig5), an IFN-inducible antiviral protein directly induced by human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2001, 98, 15125–15130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panayiotou, C.; Lindqvist, R.; Kurhade, C.; Vonderstein, K.; Pasto, J.; Edlund, K.; Upadhyay, A.S.; Överby, A.K. Viperin Restricts Zika Virus and Tick-Borne Encephalitis Virus Replication by Targeting NS3 for Proteasomal Degradation. J. Virol. 2018, 92, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.-B.; Lu, Z.-L.; Wei, X.-K.; Zhong, T.-Z.; Zhong, Y.-Z.; Ouyang, L.-X.; Luo, Y.; Xing, X.-W.; Liao, F.; Peng, K.-K.; et al. Viperin inhibits rabies virus replication via reduced cholesterol and sphingomyelin and is regulated upstream by TLR4. Sci. Rep. 2016, 6, 30529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Feng, L.; Chen, P.; Li, A.; Guo, S.; Jiao, X.; Zhang, C.; Zhao, Y.; Jin, X.; Zhong, K.; et al. Viperin inhibits classical swine fever virus replication by interacting with viral nonstructural 5A protein. J. Med. Virol. 2020, 92, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hinson, E.R.; Cresswell, P. The Interferon-Inducible Protein Viperin Inhibits Influenza Virus Release by Perturbing Lipid Rafts. Cell Host Microbe 2007, 2, 96–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, K.S.; Olfat, F.; Phoon, M.C.; Hsu, J.P.; Howe, J.L.C.; Seet, J.E.; Chin, K.C.; Chow, V.T.K. In vivo and in vitro studies on the antiviral activities of viperin against influenza H1N1 virus infection. J. Gen. Virol. 2012, 93, 1269–1277. [Google Scholar] [CrossRef]
- Cesari, M.; Pahor, M.; Incalzi, R.A. Review: Plasminogen Activator Inhibitor-1 (PAI-1): A Key Factor Linking Fibrinolysis and Age-Related Subclinical and Clinical Conditions. Cardiovasc. Ther. 2010, 28, e72–e91. [Google Scholar] [CrossRef] [Green Version]
- Dittmann, M.; Hoffmann, H.-H.; Scull, M.A.; Gilmore, R.H.; Bell, K.L.; Ciancanelli, M.; Wilson, S.J.; Crotta, S.; Yu, Y.; Flatley, B.; et al. A Serpin Shapes the Extracellular Environment to Prevent Influenza A Virus Maturation. Cell 2015, 160, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhu, X.; Liu, J.; Ding, X.; Han, M.; Hu, W.; Wang, X.; Zhou, Z.; Wang, S. Inhibition of Hepatitis B virus replication by Phospholipid scramblase 1 in vitro and in vivo. Antivir. Res. 2012, 94, 9–17. [Google Scholar] [CrossRef]
- Kusano, S.; Eizuru, Y. Human phospholipid scramblase 1 interacts with and regulates transactivation of HTLV-1 Tax. Virology 2012, 432, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Zhang, J.; Liang, L.; Wang, G.; Li, Q.; Zhu, P.; Zhou, Y.; Li, J.; Zhao, Y.; Sun, N.; et al. Phospholipid scramblase 1 interacts with influenza A virus NP, impairing its nuclear import and thereby suppressing virus replication. PLoS Pathog. 2018, 14, e1006851. [Google Scholar] [CrossRef]
- Sun, N.; Li, C.; Li, X.-F.; Deng, Y.-Q.; Jiang, T.; Zhang, N.-N.; Zu, S.; Zhang, R.-R.; Li, L.; Chen, X.; et al. Type-IInterferon-Inducible SERTAD3 Inhibits Influenza A Virus Replication by Blocking the Assembly of Viral RNA Polymerase Complex. Cell Rep. 2020, 33, 108342. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Yang, C.; Ren, C.; Zhang, S.; Gao, X.; Jin, M.; Chen, H.; Ma, W.; Zhou, H. Eukaryotic Translation Elongation Factor 1 Delta Inhibits the Nuclear Import of the Nucleoprotein and PA-PB1 Heterodimer of Influenza A Virus. J. Virol. 2020, 95. [Google Scholar] [CrossRef]
- Tafforeau, L.; Chantier, T.; Pradezynski, F.; Pellet, J.; Mangeot, P.E.; Vidalain, P.-O.; Andre, P.; Rabourdin-Combe, C.; Lotteau, V. Generation and Comprehensive Analysis of an Influenza Virus Polymerase Cellular Interaction Network. J. Virol. 2011, 85, 13010–13018. [Google Scholar] [CrossRef] [PubMed] [Green Version]

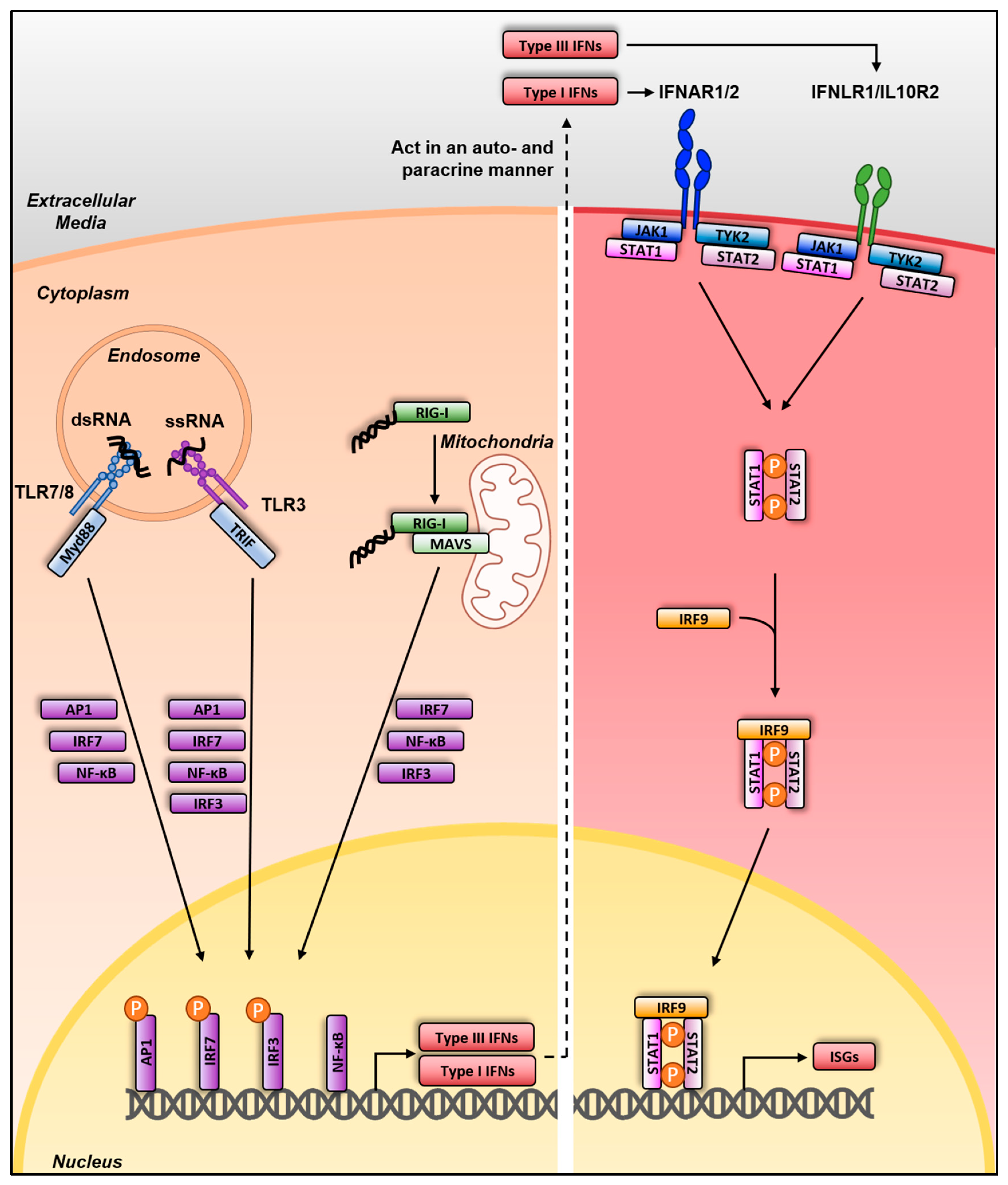

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McKellar, J.; Rebendenne, A.; Wencker, M.; Moncorgé, O.; Goujon, C. Mammalian and Avian Host Cell Influenza A Restriction Factors. Viruses 2021, 13, 522. https://doi.org/10.3390/v13030522
McKellar J, Rebendenne A, Wencker M, Moncorgé O, Goujon C. Mammalian and Avian Host Cell Influenza A Restriction Factors. Viruses. 2021; 13(3):522. https://doi.org/10.3390/v13030522
Chicago/Turabian StyleMcKellar, Joe, Antoine Rebendenne, Mélanie Wencker, Olivier Moncorgé, and Caroline Goujon. 2021. "Mammalian and Avian Host Cell Influenza A Restriction Factors" Viruses 13, no. 3: 522. https://doi.org/10.3390/v13030522