
Transcriptional Landscape of Vero E6 Cells during Early Swine Acute Diarrhea Syndrome Coronavirus Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
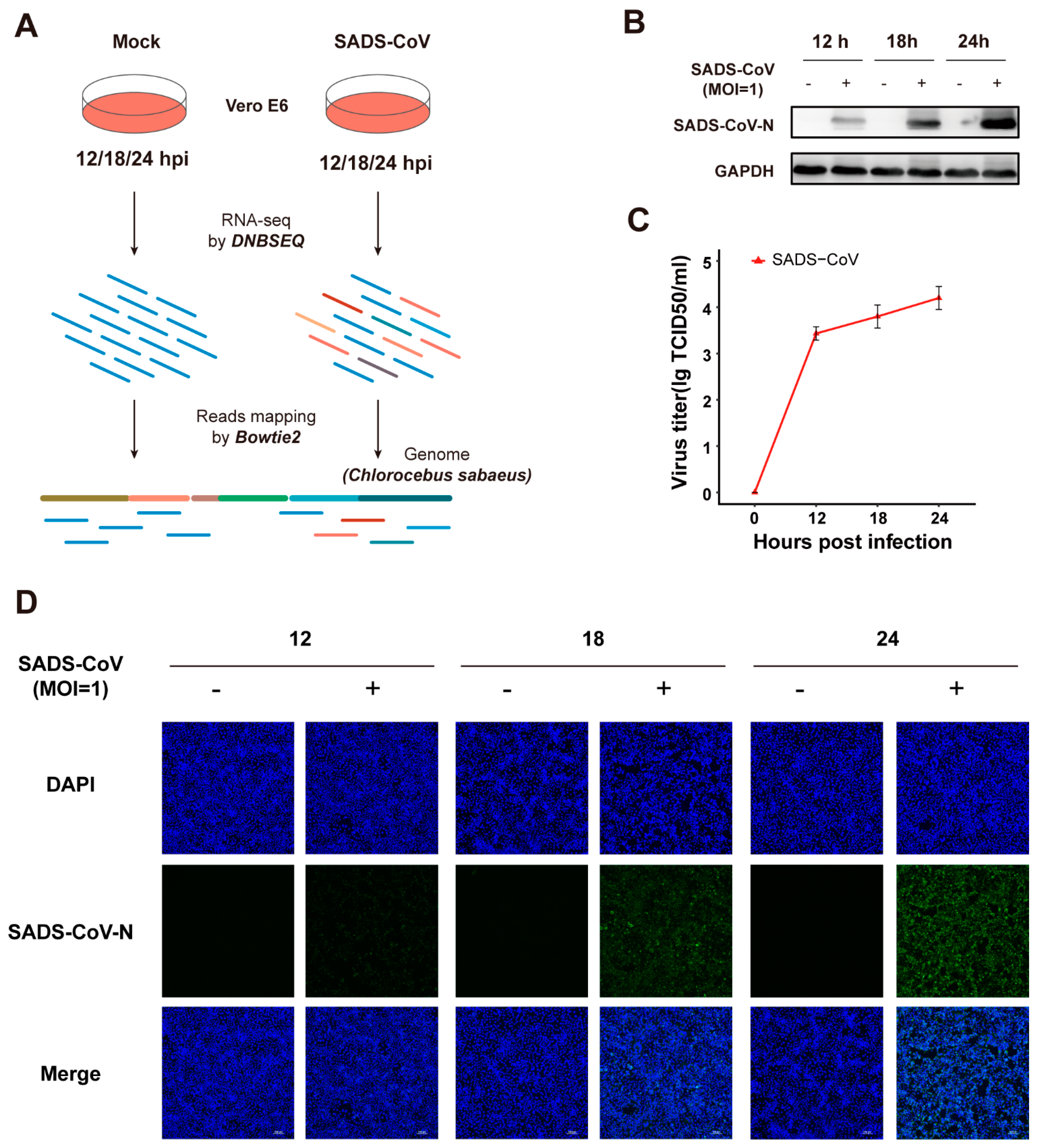
2.1. Cells and Viruses
2.2. Western Blotting and TCID50 (50% Tissue Culture Infectious Dose) Assay
2.3. Immunofluorescence Microscopy
2.4. Samples Preparation and RNA Extraction
2.5. Library Preparation and RNA-Sequencing
2.6. Sequencing Data Quality Control and Genome Mapping
2.7. Gene Expression Analysis
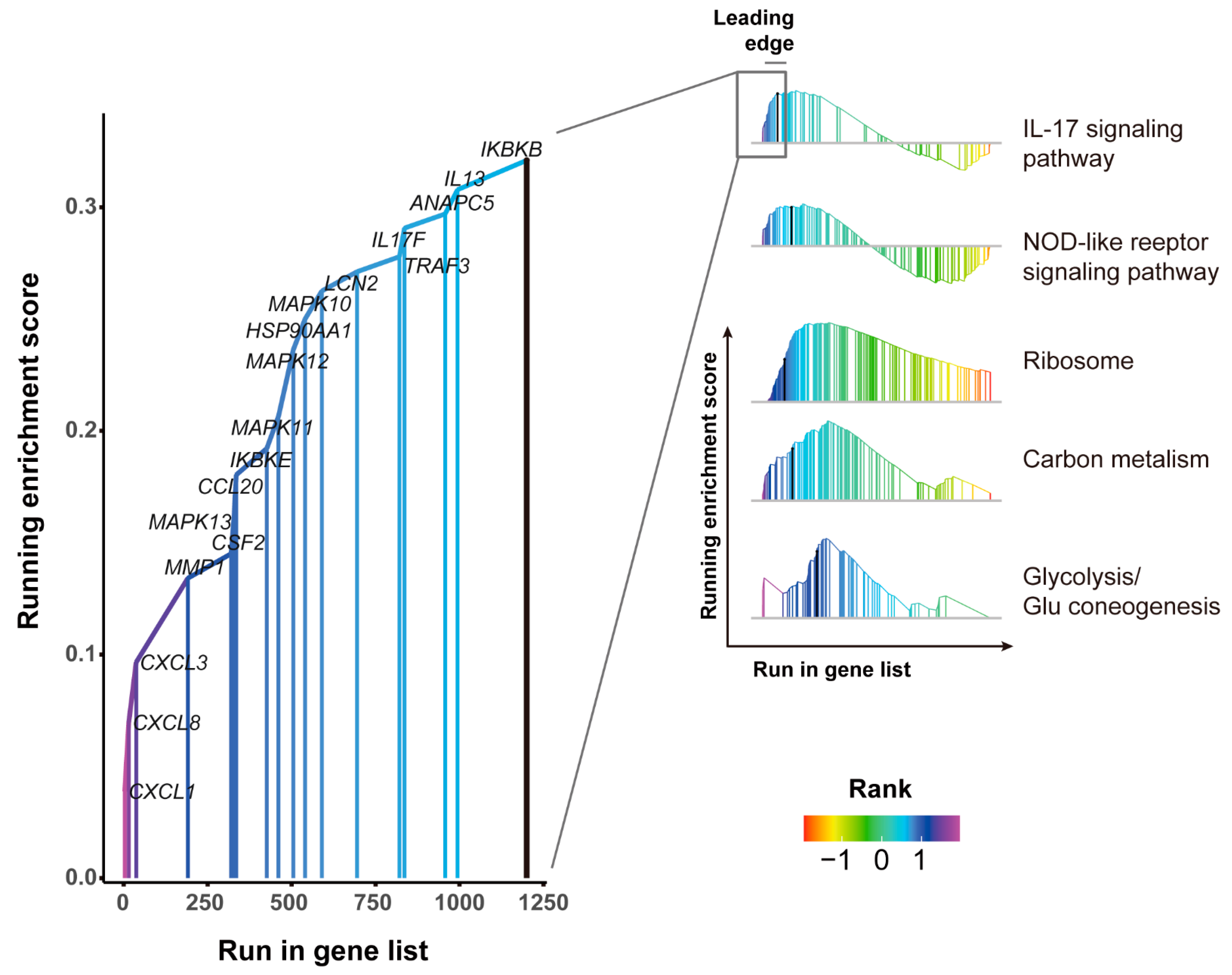
2.8. Gene Set Enrichment Analysis (GSEA)
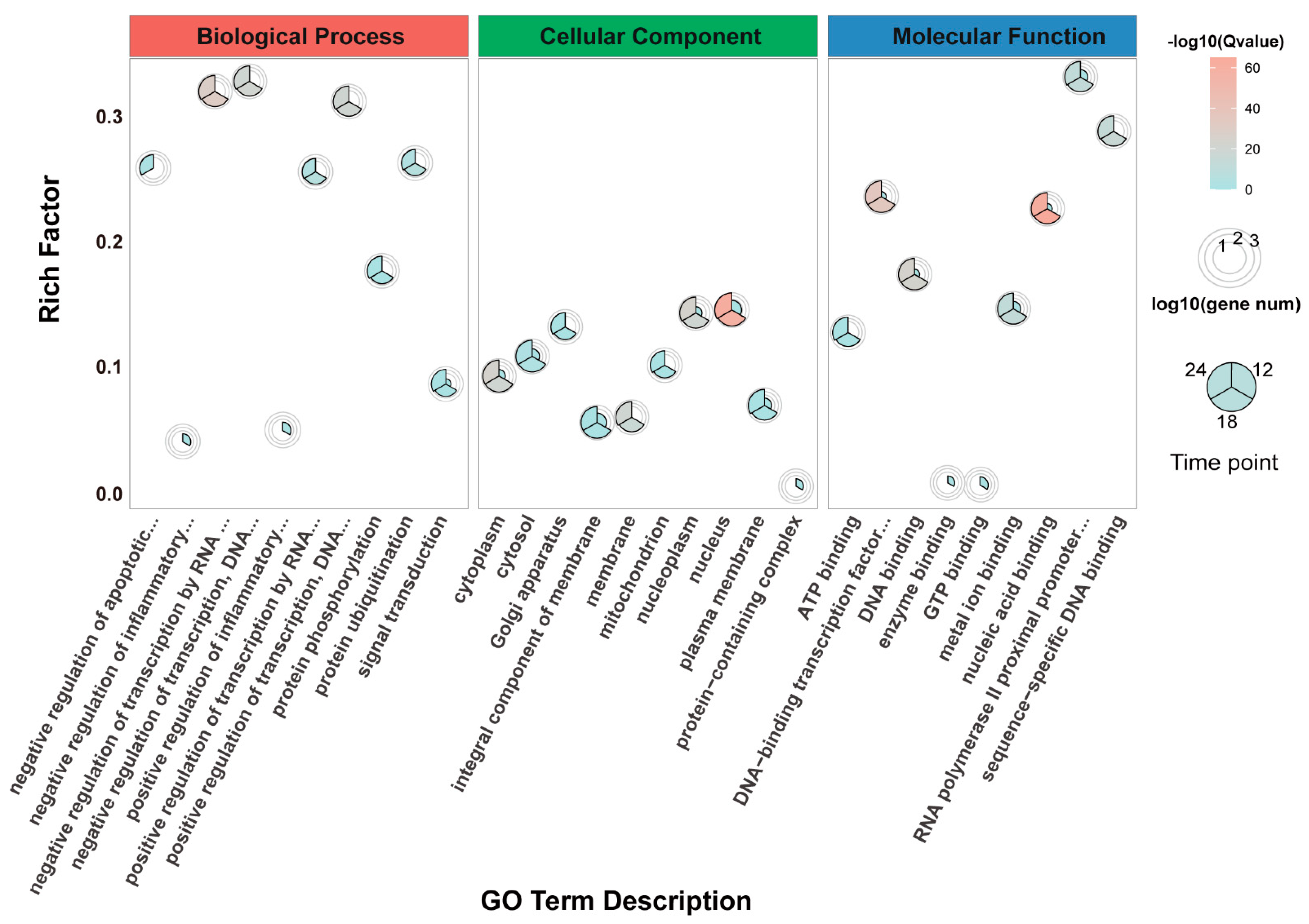
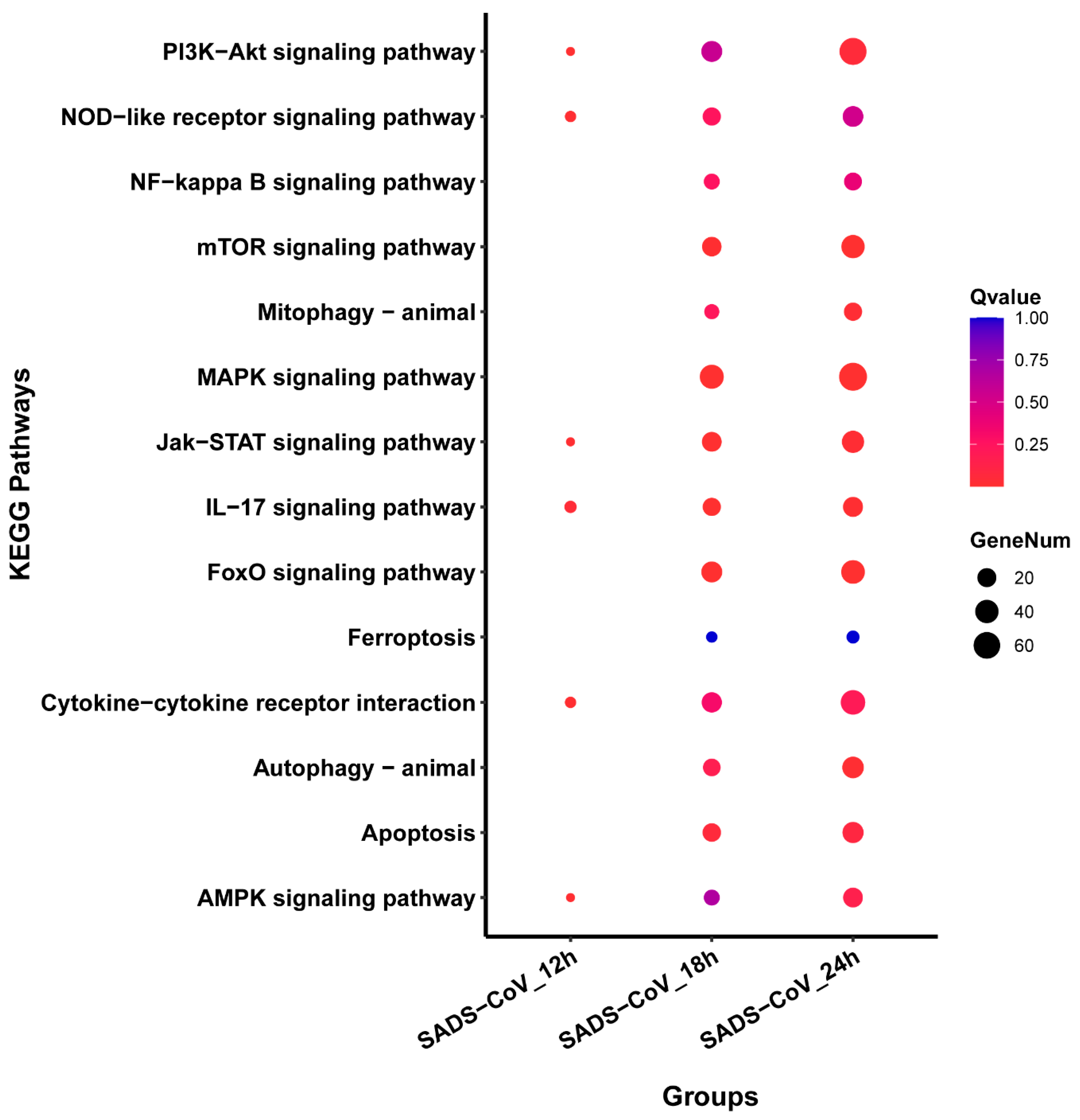
2.9. Gene Ontology (GO) Term and Pathway Enrichment Analysis of DEGs
2.10. Gene Correlation Analysis
2.11. Quantitative Real-Time PCR
3. Results
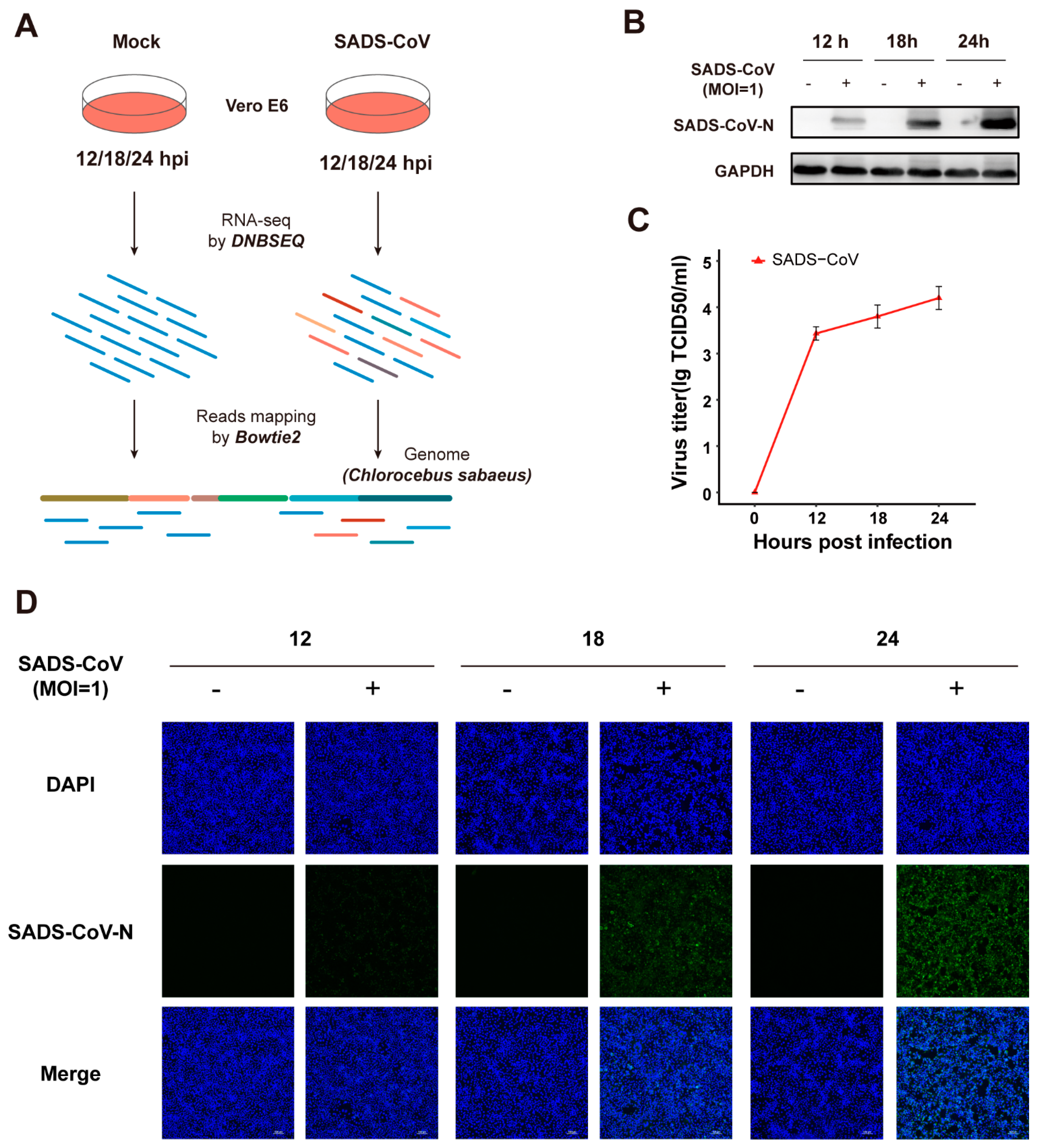
3.1. Vero E6 Cells Are Permissive to Infection Caused by SADS-CoV Strain GDS04
3.2. Transcriptomic Analysis of the SADS-CoVstrain GDS04-Infected Vero E6 Cells
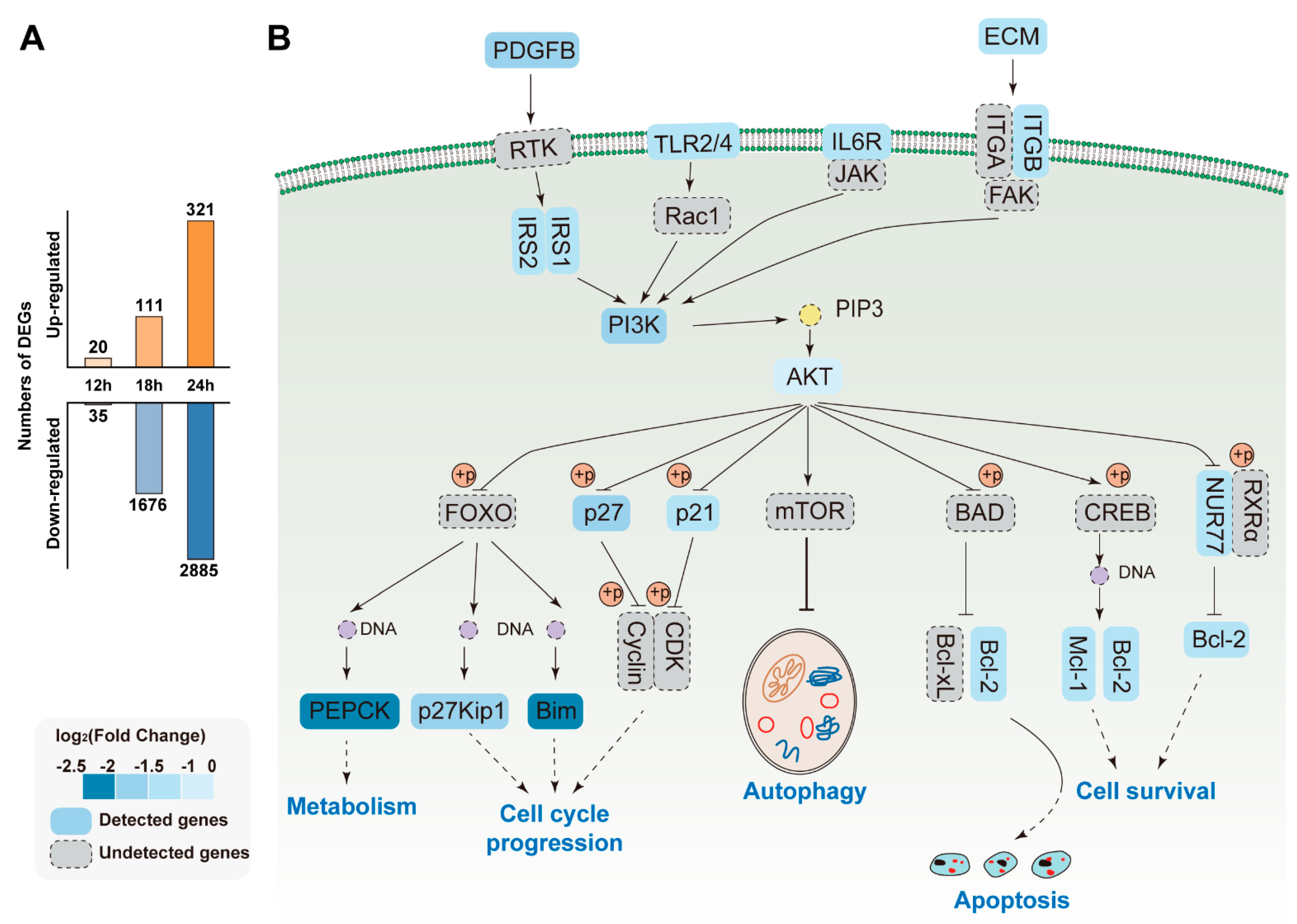
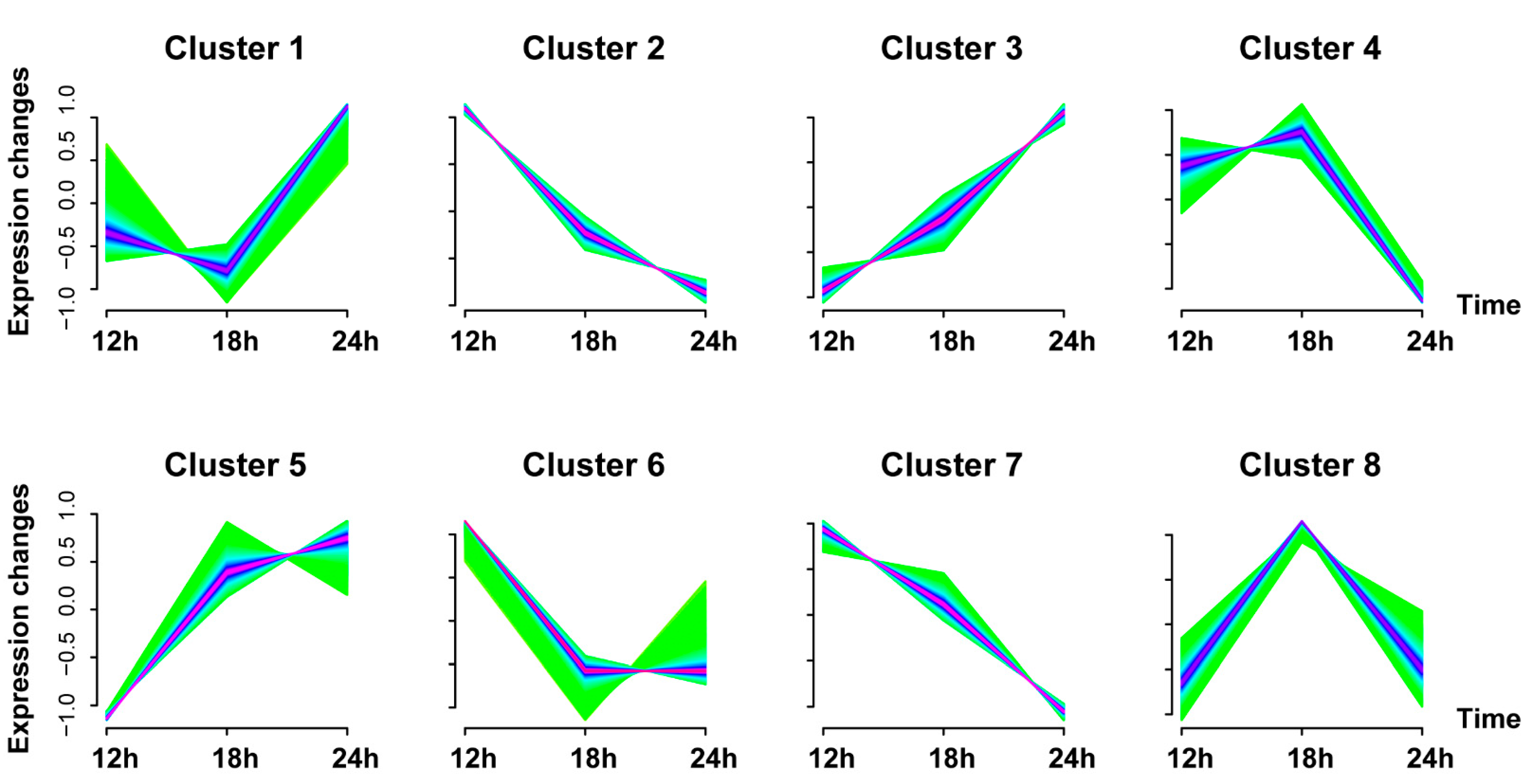
3.3. Gene Expression Trend Analysis
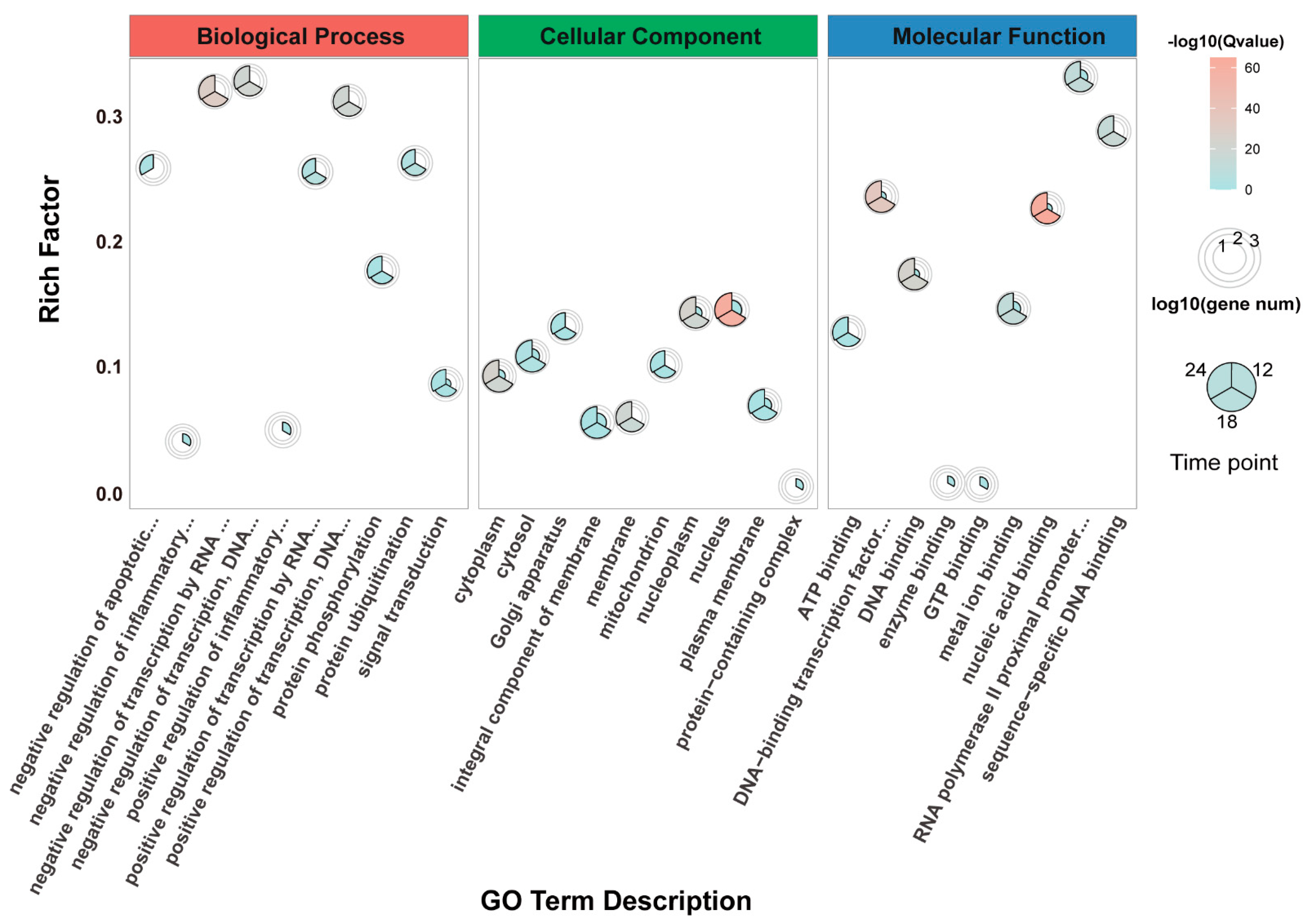
3.4. Gene Ontology (GO) Enrichment Analysis of DEGs
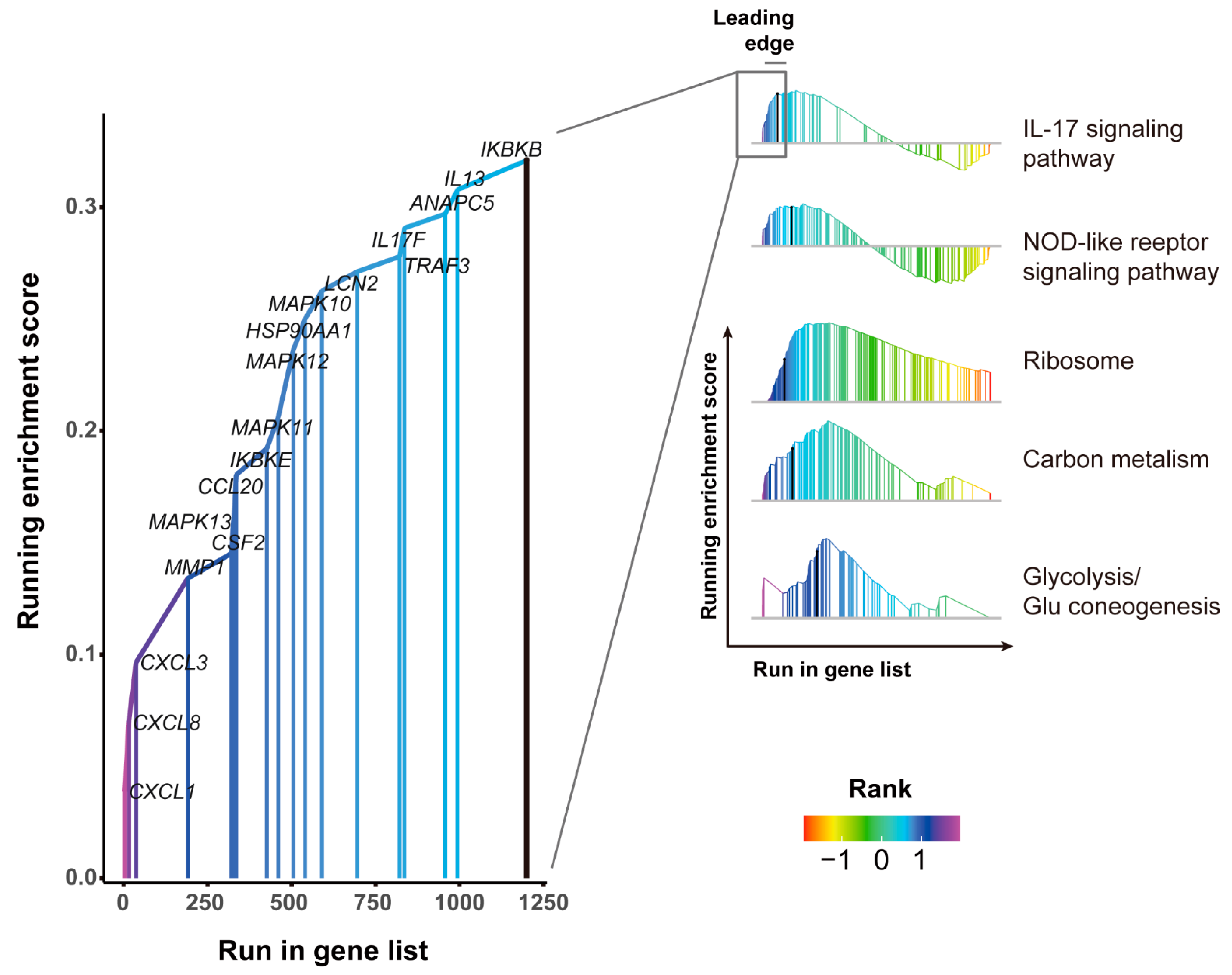
3.5. GSEA Pathways Enrichment Analysis of DEGs
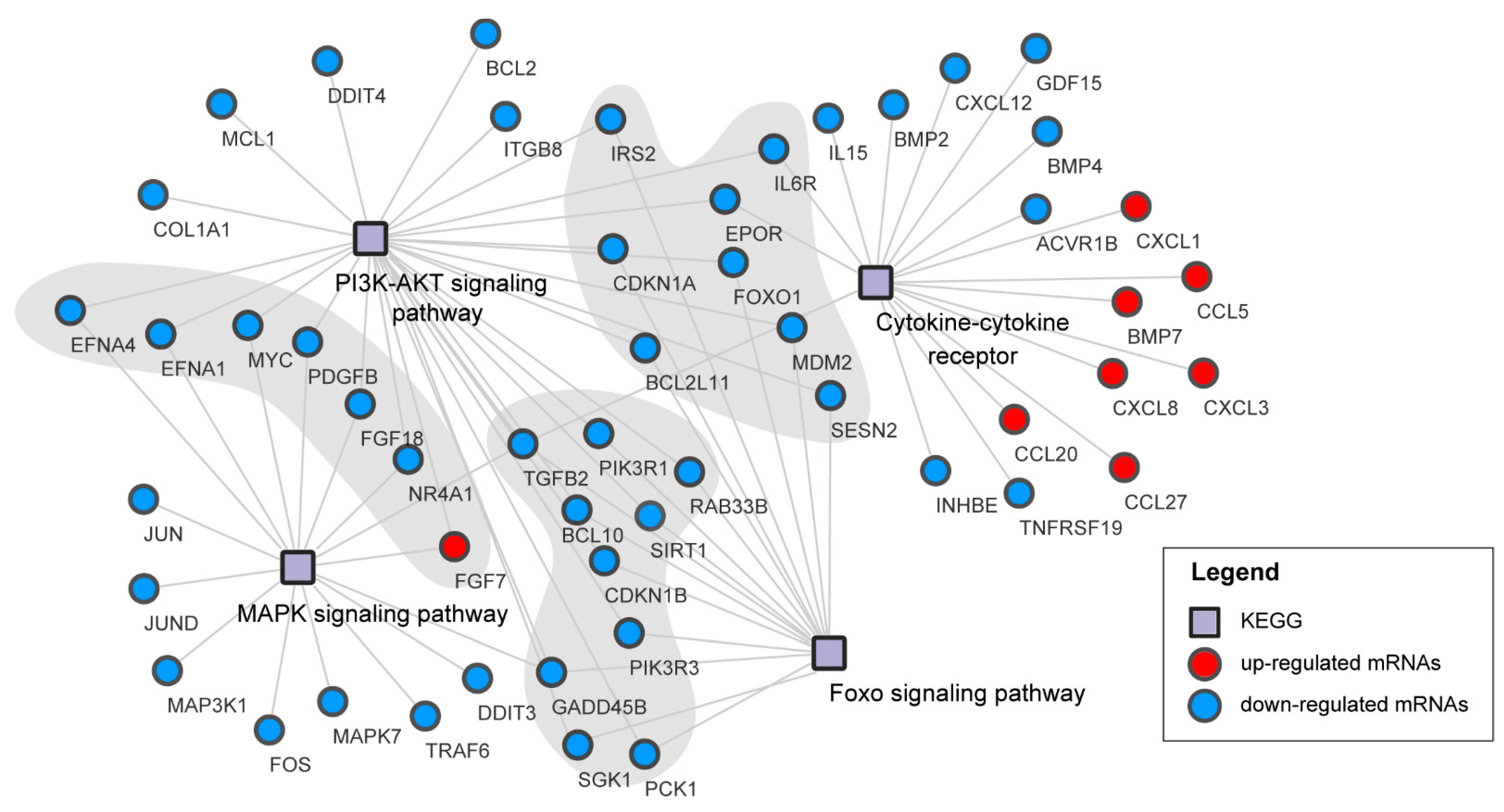
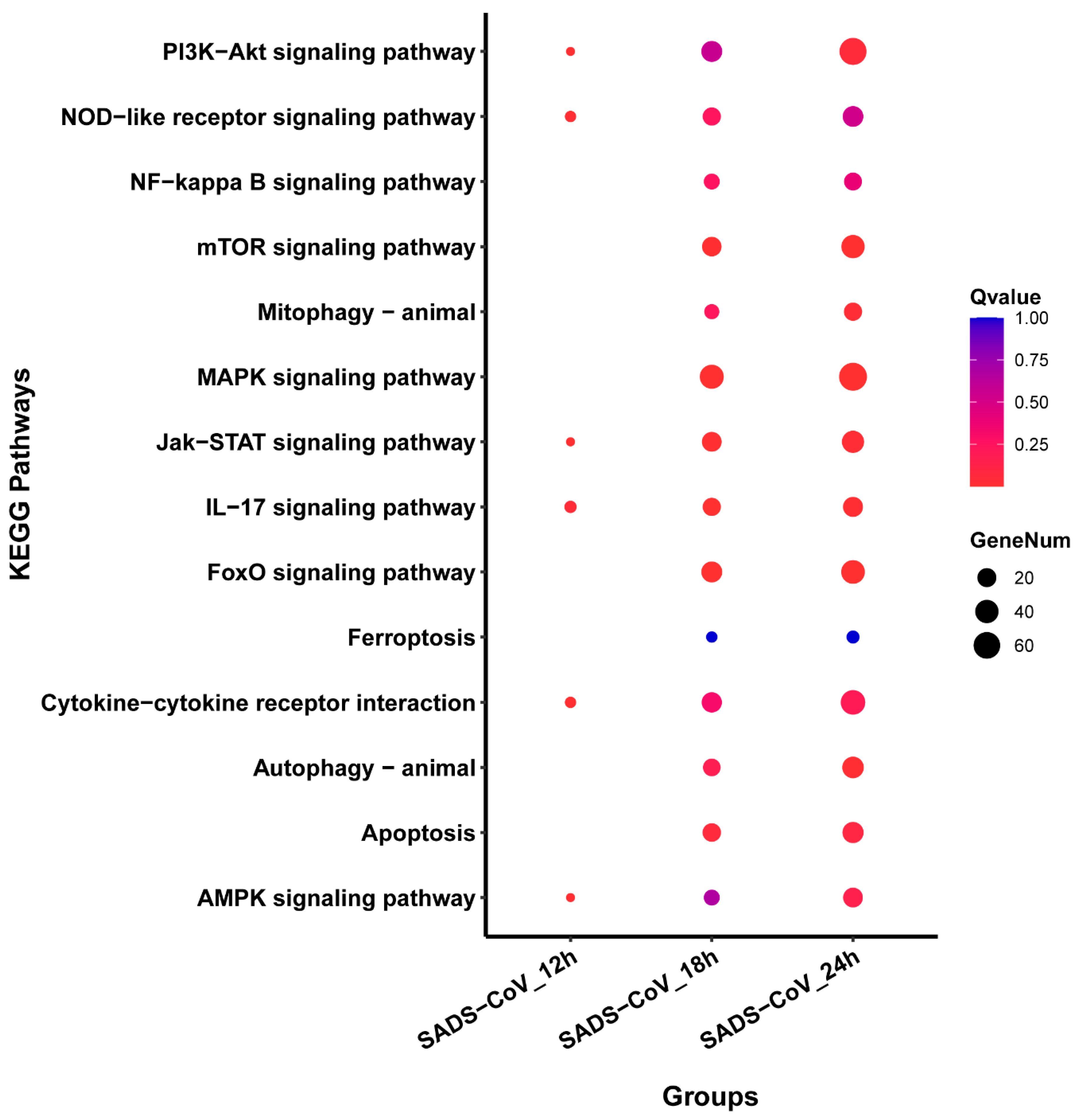
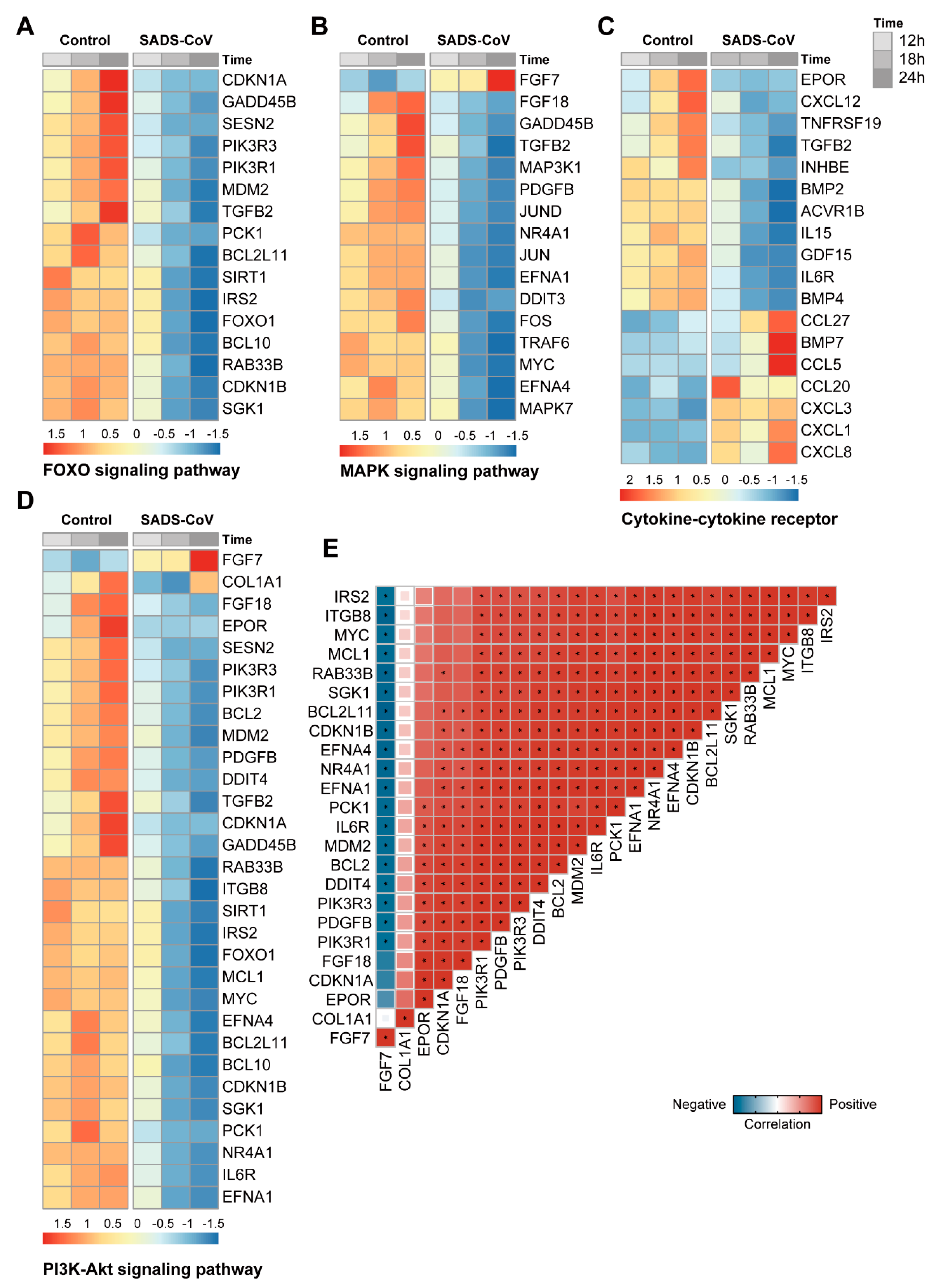
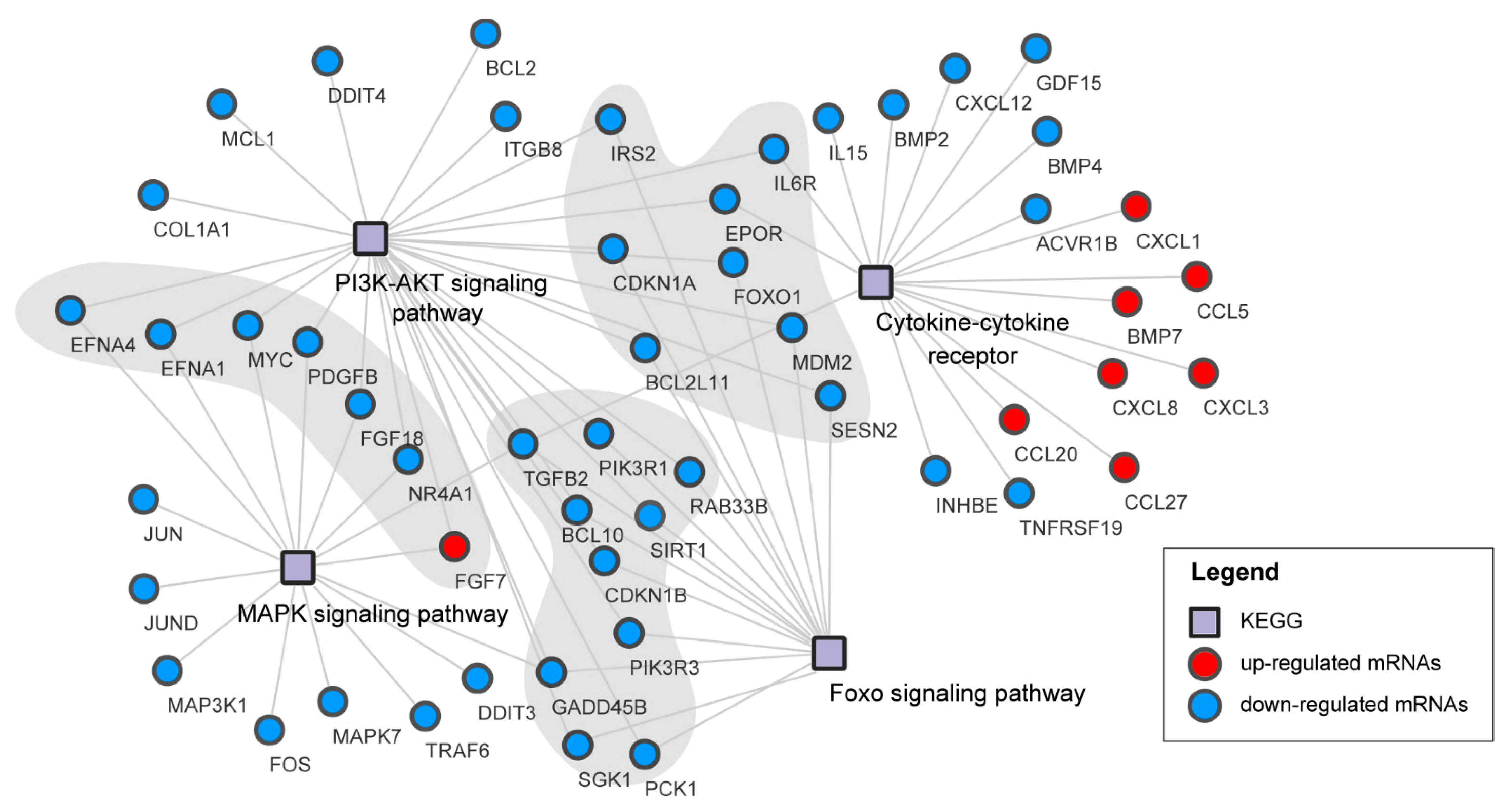
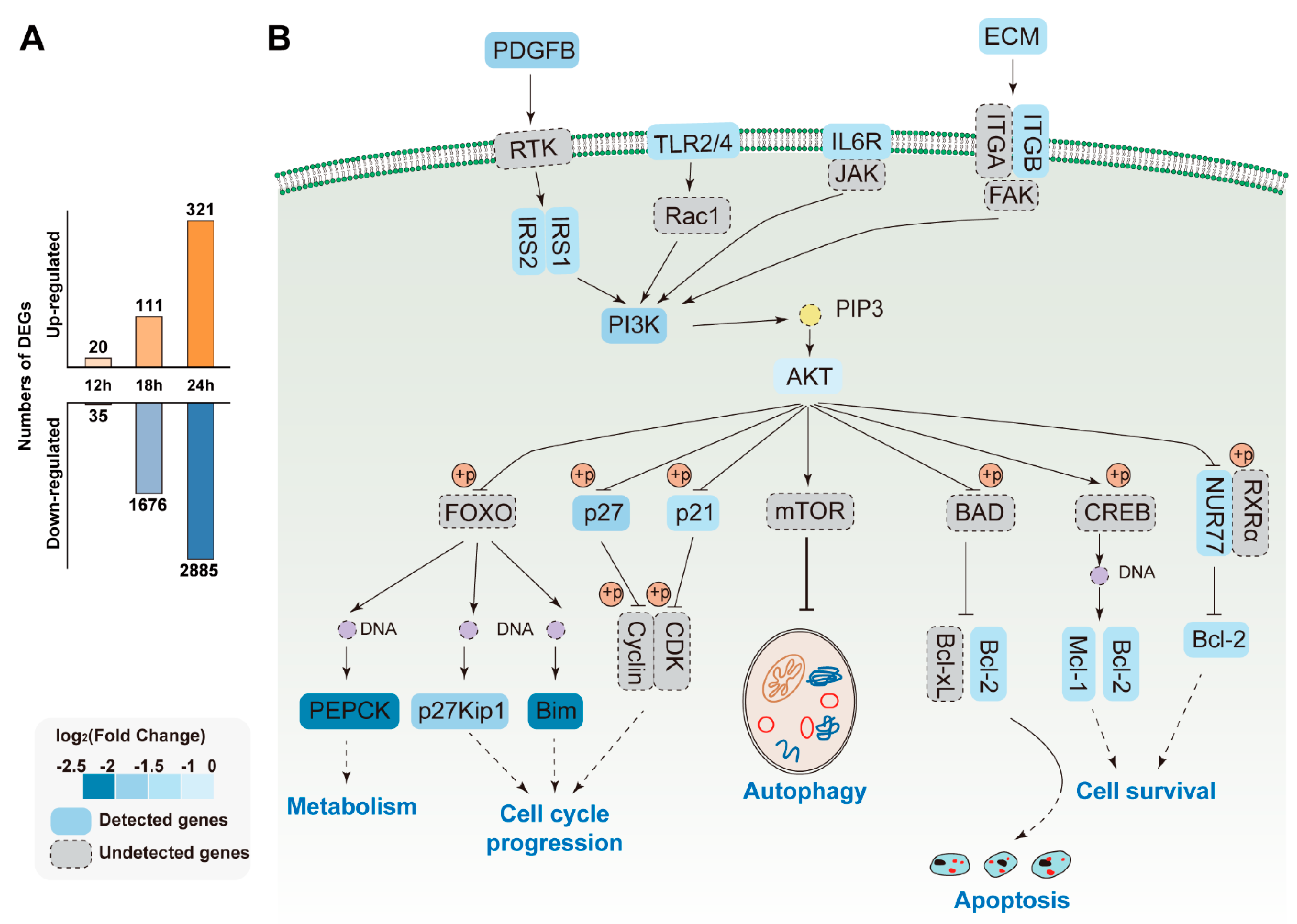
3.6. Pathways Analysis of DEGs
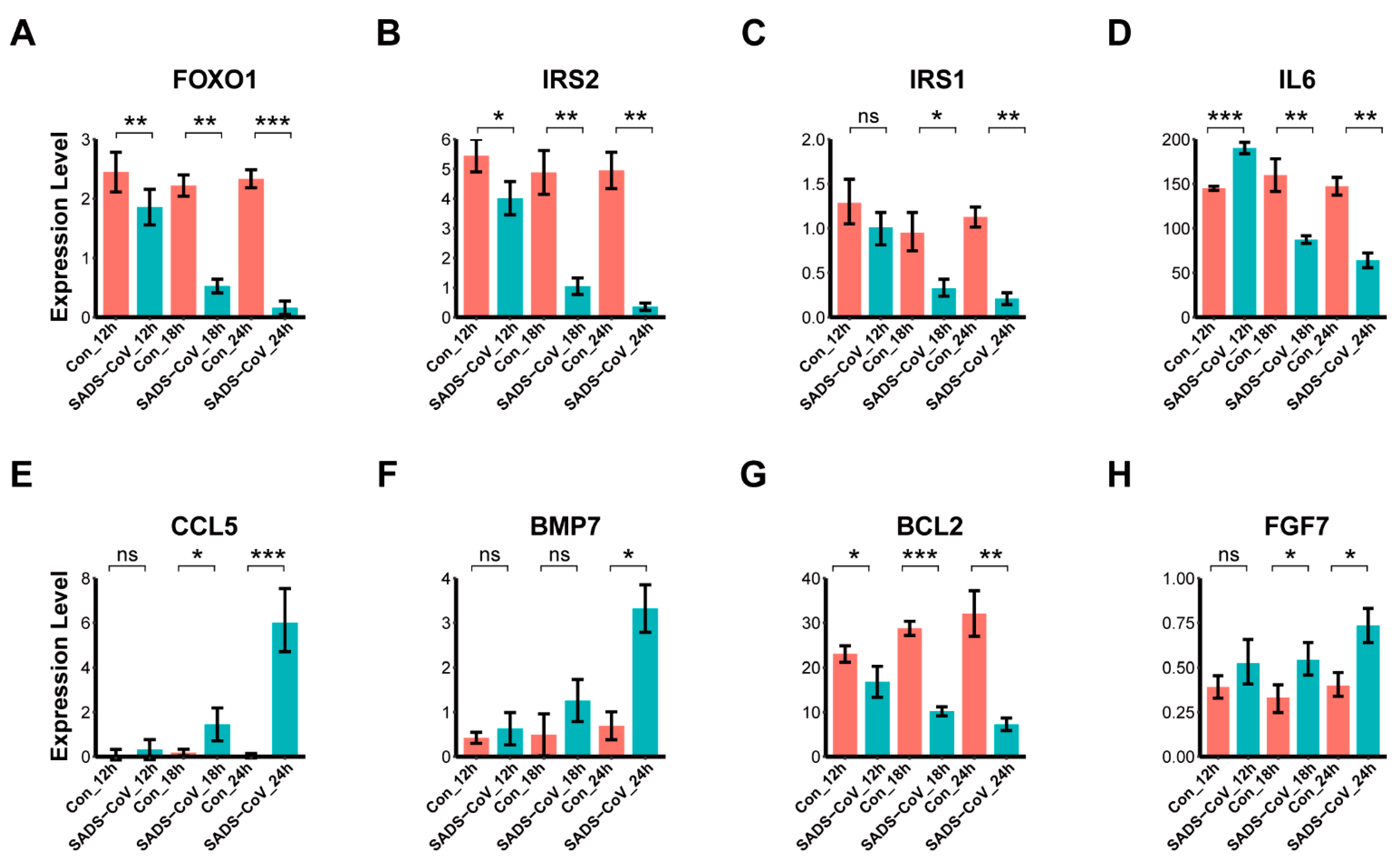
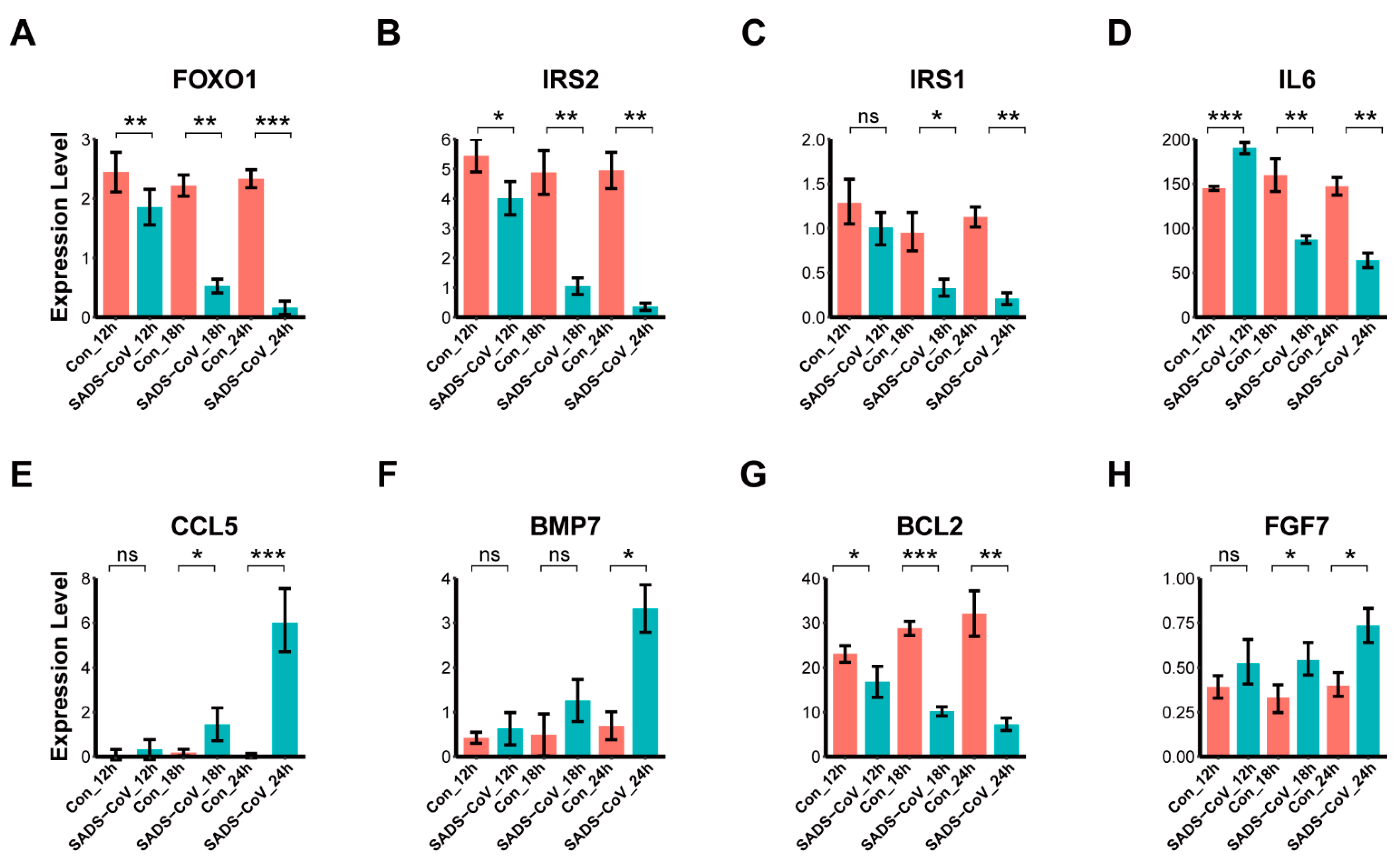
3.7. Confirmation of DEGs by Quantitative Real-Time PCR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gong, L.; Li, J.; Zhou, Q.; Xu, Z.; Chen, L.; Zhang, Y.; Xue, C.; Wen, Z.; Cao, Y. A New Bat-HKU2–like Coronavirus in Swine, China, 2017. Emerg. Infect. Dis. 2017, 23, 1607–1609. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Tian, X.; Qin, P.; Wang, B.; Zhao, P.; Yang, Y.L.; Wang, L.; Wang, D.; Song, Y.; Zhang, X.; et al. Discovery of a Novel Swine Enteric Alphacoronavirus (SeACoV) in Southern China. Vet. Microbiol. 2017, 211, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.L.; Shi, W.F.; Zhang, W.; Zhu, Y.; Zhang, Y.W.; Xie, Q.M.; Mani, S.; et al. Fatal Swine Acute Diarrhoea Syndrome Caused by an HKU2-Related Coronavirus of Bat Origin. Nature 2018, 556, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Vlasova, A.N.; Kenney, S.P.; Saif, L.J. Emerging and Re-Emerging Coronaviruses in Pigs. Curr. Opin. Virol. 2019, 34, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Sun, Y.; Lan, T.; Wu, R.; Chen, J.; Wu, Z.; Xie, Q.; Zhang, X.; Ma, J. Retrospective Detection and Phylogenetic Analysis of Swine Acute Diarrhoea Syndrome Coronavirus in Pigs in Southern China. Transbound. Emerg. Dis. 2019, 66, 687–695. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Li, Q.N.; Su, J.N.; Chen, G.H.; Wu, Z.X.; Luo, Y.; Wu, R.T.; Sun, Y.; Lan, T.; Ma, J.Y. The Re-Emerging of SADS-CoV Infection in Pig Herds in Southern China. Transbound. Emerg. Dis. 2019, 66, 2180–2183. [Google Scholar] [CrossRef] [Green Version]
- Greenbaum, D.; Colangelo, C.; Williams, K.; Gerstein, M. Comparing Protein Abundance and MRNA Expression Levels on a Genomic Scale. Genome Biol. 2003, 4, 117. [Google Scholar] [CrossRef] [Green Version]
- Marcotte, E.M.; Pellegrini, M.; Thompson, M.J.; Yeates, T.O.; Eisenberg, D. A Combined Algorithm for Genome-Wide Prediction of Protein Function. Nature 1999, 402, 83–86. [Google Scholar] [CrossRef]
- Morin, R.D.; Bainbridge, M.; Fejes, A.; Hirst, M.; Krzywinski, M.; Pugh, T.J.; McDonald, H.; Varhol, R.; Jones, S.J.M.; Marra, M.A. Profiling the HeLa S3 Transcriptome Using Randomly Primed CDNA and Massively Parallel Short-Read Sequencing. Biotechniques 2008, 45, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Byrne, A.; Cole, C.; Volden, R.; Vollmers, C. Realizing the Potential of Full-Length Transcriptome Sequencing. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manger, I.D.; Relman, D.A. How the Host “sees” Pathogens: Global Gene Expression Responses to Infection. Curr. Opin. Immunol. 2000, 12, 215–218. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Yuan, W.; Li, Z.; Zhang, Y.; Ye, Y.; Li, K.; Ding, Z.; Chen, Y.; Cheng, T.; Wu, Q.; et al. RNA-Seq-Based Whole Transcriptome Analysis of IPEC-J2 Cells During Swine Acute Diarrhea Syndrome Coronavirus Infection. Front. Vet. Sci. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-L.; Qin, P.; Wang, B.; Liu, Y.; Xu, G.-H.; Peng, L.; Zhou, J.; Zhu, S.J.; Huang, Y.-W. Broad Cross-Species Infection of Cultured Cells by Bat HKU2-Related Swine Acute Diarrhea Syndrome Coronavirus and Identification of Its Replication in Murine Dendritic Cells In Vivo Highlight Its Potential for Diverse Interspecies Transmission. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Chen, Y.; Geng, R.; Li, B.; Chen, J.; Zhao, K.; Zheng, X.S.; Zhang, W.; Zhou, P.; Yang, X.L.; et al. Broad Cell Tropism of SADS-CoV In Vitro Implies Its Potential Cross-Species Infection Risk. Virol. Sin. 2020, 1–5. [Google Scholar] [CrossRef]
- Edwards, C.E.; Yount, B.L.; Graham, R.L.; Leist, S.R.; Hou, Y.J.; Dinnon, K.H.; Sims, A.C.; Swanstrom, J.; Gully, K.; Scobey, T.D.; et al. Swine Acute Diarrhea Syndrome Coronavirus Replication in Primary Human Cells Reveals Potential Susceptibility to Infection. Proc. Natl. Acad. Sci. USA 2020, 117, 26915–26925. [Google Scholar] [CrossRef]
- Wei, X.; She, G.; Wu, T.; Xue, C.; Cao, Y. PEDV Enters Cells through Clathrin-, Caveolae-, and Lipid Raft-Mediated Endocytosis and Traffics via the Endo-/Lysosome Pathway. Vet. Res. 2020, 51. [Google Scholar] [CrossRef] [Green Version]
- Lindenbach, B.D. Measuring HCV Infectivity Produced in Cell Culture and in Vivo. Methods Mol. Biol. 2009, 510, 329–336. [Google Scholar] [CrossRef]
- Huang, J.; Liang, X.; Xuan, Y.; Geng, C.; Li, Y.; Lu, H.; Qu, S.; Mei, X.; Chen, H.; Yu, T.; et al. A reference human genome dataset of the BGISEQ-500 sequencer. Gigascience 2017, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Mak, S.; Gopalakrishnan, S.; Caroe, C.; Geng, C.; Liu, S.; Sinding, M.S.; Kuderna, L.; Zhang, W.; Fu, S.; Vieira, F.G.; et al. Comparative performance of the BGISEQ-500 vs Illumina HiSeq2500 sequencing platforms for palaeogenomic sequencing. Gigascience 2017, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cock, P.J.A.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ File Format for Sequences with Quality Scores, and the Solexa/Illumina FASTQ Variants. Nucleic Acids Res. 2009, 38, 1767–1771. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The GO Term Finder. Available online: https://yeastgenome.org/goTermFinder (accessed on 20 January 2021).
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for Linking Genomes to Life and the Environment. Nucleic Acids Res. 2008, 36. [Google Scholar] [CrossRef] [PubMed]
- R Foundation for Statistical Computing. R: A Language and Environment for Statistical Computing; 4.0.4(2021-02-15); R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Wei, T.; Simko, V.M.; Levy, M.; Xie, Y.; Jin, Y.; Zemla, J. Package “Corrplot”: Visualization of a Correlation Matrix; 2017. Available online: https://github.com/taiyun/corrplot (accessed on 20 January 2021).
- Fu, X.; Fang, B.; Liu, Y.; Cai, M.; Jun, J.; Ma, J.; Bu, D.; Wang, L.; Zhou, P.; Wang, H.; et al. Newly Emerged Porcine Enteric Alphacoronavirus in Southern China: Identification, Origin and Evolutionary History Analysis. Infect. Genet. Evol. 2018, 62, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, Y.; Gong, L.; Huang, L.; Lin, Y.; Qin, J.; Du, Y.; Zhou, Q.; Xue, C.; Cao, Y. Isolation and Characterization of a Highly Pathogenic Strain of Porcine Enteric Alphacoronavirus Causing Watery Diarrhoea and High Mortality in Newborn Piglets. Transbound. Emerg. Dis. 2019, 66, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Kumari, S.; Ray, P.K.; Singh, R.; Desingu, P.A.; Sharma, G.T.; Saikumar, G. Development of a Taqman-Based Real-Time PCR Assay for Detection of Porcine Sapelovirus Infection in Pigs. Anim. Biotechnol. 2020, 31, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Desmyter, J.; Melnick, J.L.; Rawls, W.E. Defectiveness of Interferon Production and of Rubella Virus Interference in a Line of African Green Monkey Kidney Cells (Vero). J. Virol. 1968, 2, 955–961. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Sun, Y.; Yan, X.; Tang, X.; Li, Q.; Tan, Y.; Lan, T.; Ma, J. Swine acute diarrhea syndrome coronavirus (SADS- CoV) antagonizes interferon-beta production via blocking IPS-1 and RIG-I. Virus Res. 2020, 278, 197843. [Google Scholar] [CrossRef]
- Zhou, Z.; Sun, Y.; Xu, J.; Tang, X.; Zhou, L.; Li, Q.; Lan, T.; Ma, J. Swine Acute Diarrhea Syndrome Coronavirus Nucleocapsid Protein Antagonizes Interferon-beta Production via Blocking the Interaction Between TRAF3 and TBK1. Front. Immunol. 2021, 12, 573078. [Google Scholar] [CrossRef]
- Shen, Z.; Yang, Y.; Yang, S.; Zhang, G.; Xiao, S.; Fu, Z.F.; Peng, G. Structural and Biological Basis of Alphacoronavirus nsp1 Associated with Host Proliferation and Immune Evasion. Viruses 2020, 12, 812. [Google Scholar] [CrossRef]
- Honda, M.; Takehana, K.; Sakai, A.; Tagata, Y.; Shirasaki, T.; Nishitani, S.; Muramatsu, T.; Yamashita, T.; Nakamoto, Y.; Mi zukoshi, E.; et al. Malnutrition impairs interferon signaling through mTOR and FoxO pathways in patients with chronic hepatitis C. Gastroenterology 2011, 141, 128–140. [Google Scholar] [CrossRef]
- Oh, C.; Ryoo, J.; Park, K.; Kim, B.; Daly, M.B.; Cho, D.; Ahn, K. A central role for PI3K-AKT signaling pathway in linking SAMHD1-deficiency to the type I interferon signature. Sci. Rep. 2018, 8, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.J.; Hua, X.; He, S.F.; Ren, H.; Qi, Z.T. Interferon alpha regulates MAPK and STAT1 pathways in human hepatoma cells. Virol. J. 2011, 8, 157. [Google Scholar] [CrossRef] [Green Version]
- Krug, A.; Uppaluri, R.; Facchetti, F.; Dorner, B.G.; Sheehan, K.C.; Schreiber, R.D.; Cella, M.; Colonna, M. IFN-producing cells respond to CXCR3 ligands in the presence of CXCL12 and secrete inflammatory chemokines upon activation. J. Immunol. 2002, 169, 6079–6083. [Google Scholar] [CrossRef] [Green Version]
- Nakano, M.; Fujii, T.; Hashimoto, M.; Yukawa, N.; Yoshifuji, H.; Ohmura, K.; Nakaizumi, A.; Mimori, T. Type I interferon induces CX3CL1 (fractalkine) and CCL5 (RANTES) production in human pulmonary vascular endothelial cells. Clin. Exp. Immunol. 2012, 170, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.D.; Torres, V.J.; Cadwell, K. Autophagy and Microbial Pathogenesis. Cell Death Differ. 2020, 27, 872–886. [Google Scholar] [CrossRef] [PubMed]
- Prentice, E.; McAuliffe, J.; Lu, X.; Subbarao, K.; Denison, M.R. Identification and Characterization of Severe Acute Respiratory Syndrome Coronavirus Replicase Proteins. J. Virol. 2004, 78, 9977–9986. [Google Scholar] [CrossRef] [Green Version]
- Gassen, N.C.; Niemeyer, D.; Muth, D.; Corman, V.M.; Martinelli, S.; Gassen, A.; Hafner, K.; Papies, J.; Mösbauer, K.; Zellner, A.; et al. SKP2 Attenuates Autophagy through Beclin1-Ubiquitination and Its Inhibition Reduces MERS-Coronavirus Infection. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus Replication Complex Formation Utilizes Components of Cellular Autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar] [CrossRef] [Green Version]
- Cottam, E.M.; Whelband, M.C.; Wileman, T. Coronavirus NSP6 Restricts Autophagosome Expansion. Autophagy 2014, 10, 1426–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Mou, C.; Yang, X.; Lin, J.; Yang, Q. Mitophagy in TGEV Infection Counteracts Oxidative Stress and Apoptosis. Oncotarget 2016, 7, 27122–27141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, N.; Shan, T.; Wang, H.; Jiao, Y.; Zuo, Y.; Li, L.; Tong, W.; Yu, L.; Jiang, Y.; Zhou, Y.; et al. BST2 Suppresses Porcine Epidemic Diarrhea Virus Replication by Targeting and Degrading Virus Nucleocapsid Protein with Selective Autophagy. Autophagy 2020, 16, 1737–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appelberg, S.; Gupta, S.; Akusjärvi, S.S.; Ambikan, A.T.; Mikaeloff, F.; Saccon, E.; Végvári, Á.; Benfeitas, R.; Sperk, M.; Ståhlberg, M.; et al. Dysregulation in Akt/MTOR/HIF-1 Signaling Identified by Proteo-Transcriptomics of SARS-CoV-2 Infected Cells. Emerg. Microbes Infect. 2020, 9, 1–36. [Google Scholar] [CrossRef]
- Lin, H.; Li, B.; Liu, M.; Zhou, H.; He, K.; Fan, H. Nonstructural Protein 6 of Porcine Epidemic Diarrhea Virus Induces Autophagy to Promote Viral Replication via the PI3K/Akt/MTOR Axis. Vet. Microbiol. 2020, 244, 108684. [Google Scholar] [CrossRef]
- Hughes, C.E.; Nibbs, R.J.B. A Guide to Chemokines and Their Receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Hao, X.; Lv, Q.; Li, F.; Xu, Y.; Gao, H. The Characteristics of HDPP4 Transgenic Mice Subjected to Aerosol MERS Coronavirus Infection via an Animal Nose-only Exposure Device. Anim. Model. Exp. Med. 2019, 2, 269–281. [Google Scholar] [CrossRef]
- Reghunathan, R.; Jayapal, M.; Hsu, L.Y.; Chng, H.H.; Tai, D.; Leung, B.P.; Melendez, A.J. Expression Profile of Immune Response Genes in Patients with Severe Acute Respiratory Syndrome. BMC Immunol. 2005, 6. [Google Scholar] [CrossRef] [Green Version]
- Ng, L.F.P.; Hibberd, M.L.; Ooi, E.E.; Tang, K.F.; Neo, S.Y.; Tan, J.; Krishna Murthy, K.R.; Vega, V.B.; Chia, J.M.; Liu, E.T.; et al. A Human in Vitro Model System for Investigating Genome-Wide Host Responses to SARS Coronavirus Infection. BMC Infect. Dis. 2004, 4. [Google Scholar] [CrossRef]
- Fagone, P.; Ciurleo, R.; Lombardo, S.D.; Iacobello, C.; Palermo, C.I.; Shoenfeld, Y.; Bendtzen, K.; Bramanti, P.; Nicoletti, F. Transcriptional Landscape of SARS-CoV-2 Infection Dismantles Pathogenic Pathways Activated by the Virus, Proposes Unique Sex-Specific Differences and Predicts Tailored Therapeutic Strategies. Autoimmun. Rev. 2020, 19, 102571. [Google Scholar] [CrossRef]
- Sun, P.; Fahd, Q.; Li, Y.; Sun, Y.; Li, J.; Qaria, M.A.; He, Z.S.; Fan, Y.; Zhang, Q.; Xu, Q.; et al. Transcriptomic Analysis of Small Intestinal Mucosa from Porcine Epidemic Diarrhea Virus Infected Piglets. Microb. Pathog. 2019, 132, 73–79. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, S.; Peng, O.; Sun, R.; Xu, Q.; Hu, F.; Zhao, Y.; Xue, C.; Cao, Y.; Zhang, H. Transcriptional Landscape of Vero E6 Cells during Early Swine Acute Diarrhea Syndrome Coronavirus Infection. Viruses 2021, 13, 674. https://doi.org/10.3390/v13040674
Zeng S, Peng O, Sun R, Xu Q, Hu F, Zhao Y, Xue C, Cao Y, Zhang H. Transcriptional Landscape of Vero E6 Cells during Early Swine Acute Diarrhea Syndrome Coronavirus Infection. Viruses. 2021; 13(4):674. https://doi.org/10.3390/v13040674
Chicago/Turabian StyleZeng, Siying, Ouyang Peng, Ruipu Sun, Qiuping Xu, Fangyu Hu, Yan Zhao, Chunyi Xue, Yongchang Cao, and Hao Zhang. 2021. "Transcriptional Landscape of Vero E6 Cells during Early Swine Acute Diarrhea Syndrome Coronavirus Infection" Viruses 13, no. 4: 674. https://doi.org/10.3390/v13040674
APA StyleZeng, S., Peng, O., Sun, R., Xu, Q., Hu, F., Zhao, Y., Xue, C., Cao, Y., & Zhang, H. (2021). Transcriptional Landscape of Vero E6 Cells during Early Swine Acute Diarrhea Syndrome Coronavirus Infection. Viruses, 13(4), 674. https://doi.org/10.3390/v13040674