
New World Cactaceae Plants Harbor Diverse Geminiviruses

 ,
,  ,
,  , , , ,
, , , ,  , , , , and
, , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collecting and Processing
2.2. Geminivirus Genome Identification and Recovery
2.3. Infectivity Assays
2.4. Southern Blot Analysis
2.5. Viral Particle Purification and Transmission Electron Microscopy
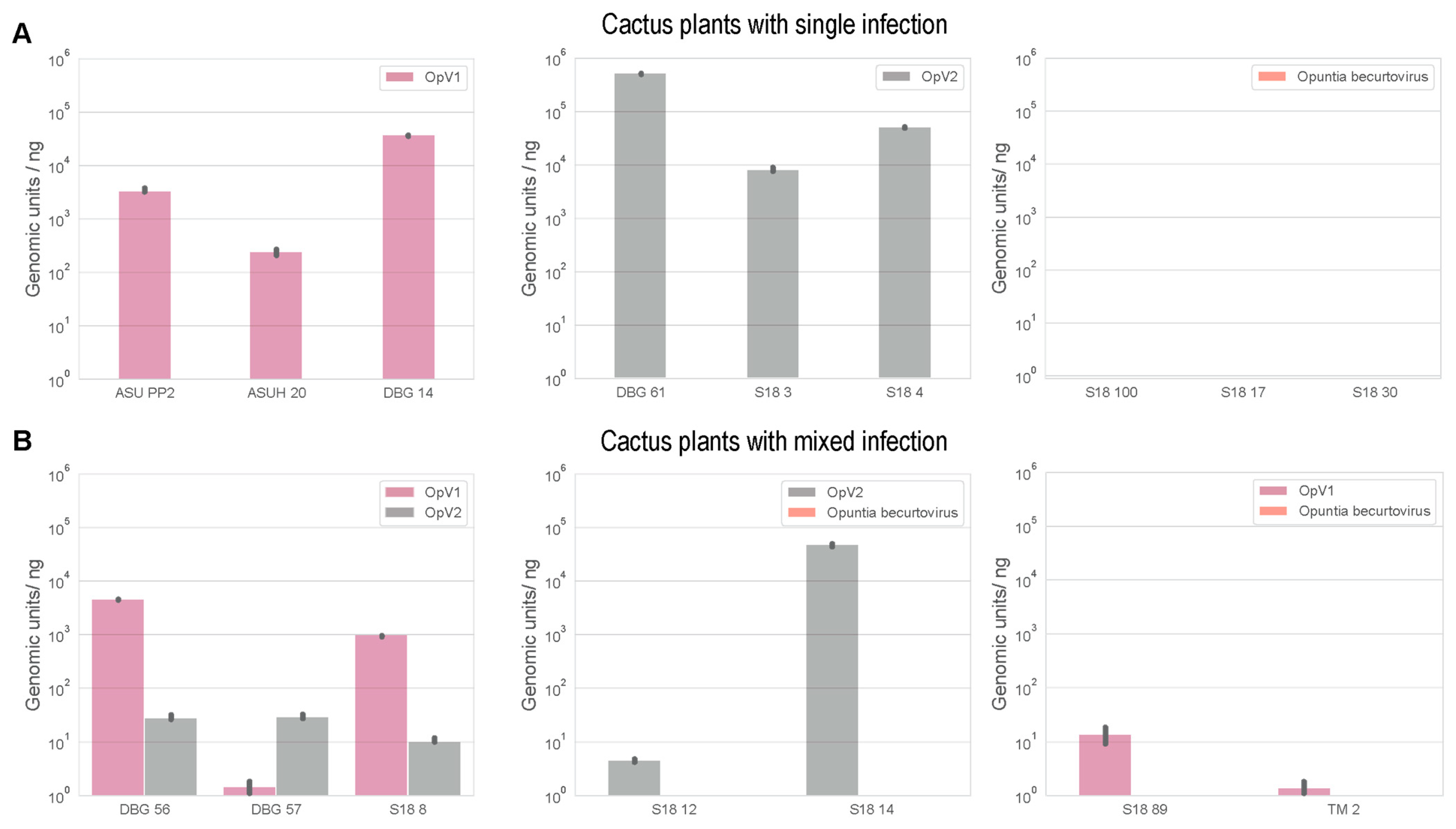
2.6. Viral Load Quantification
2.7. Pairwise Identity and Phylogenetic Analyses
2.8. Recombination Analyses
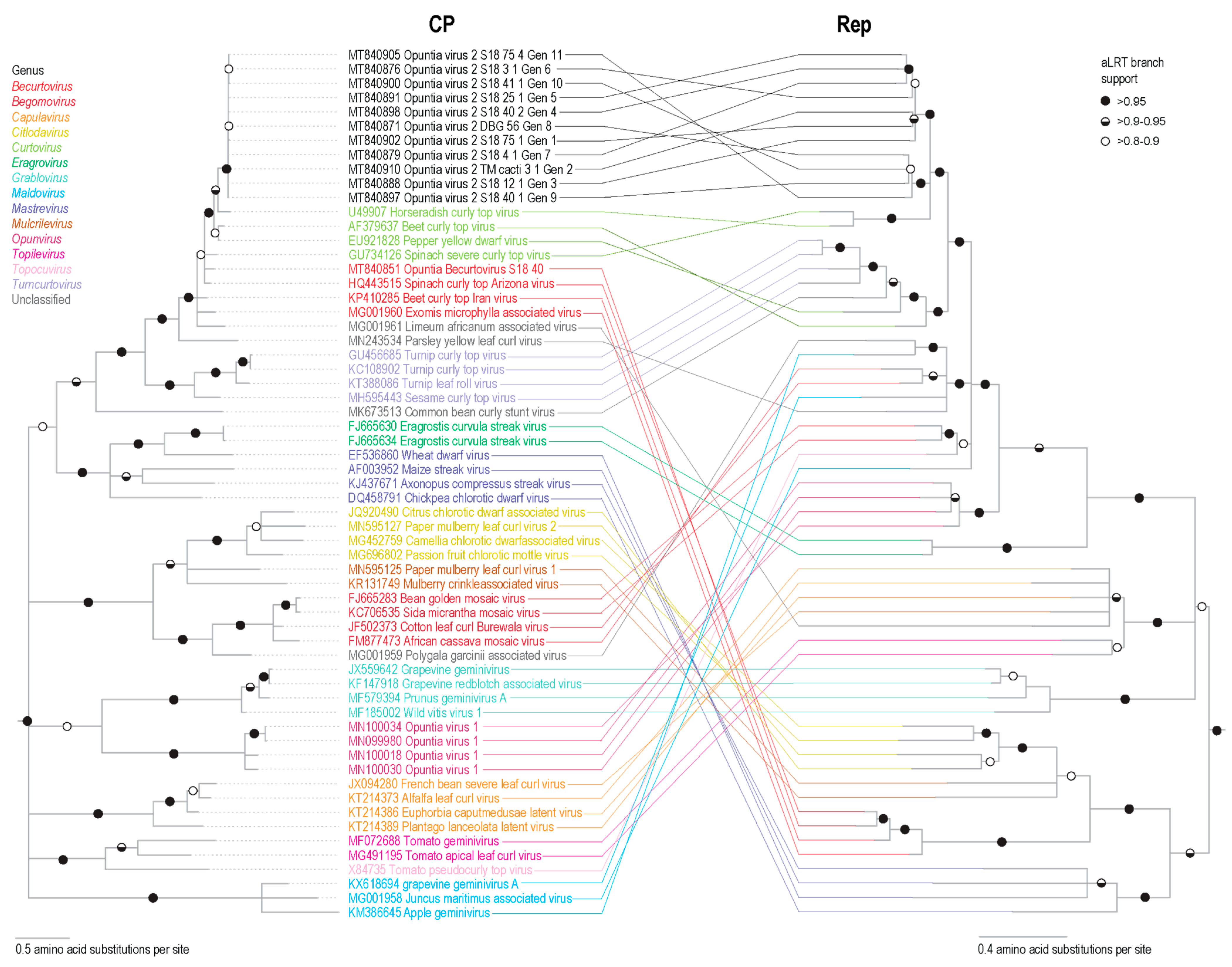
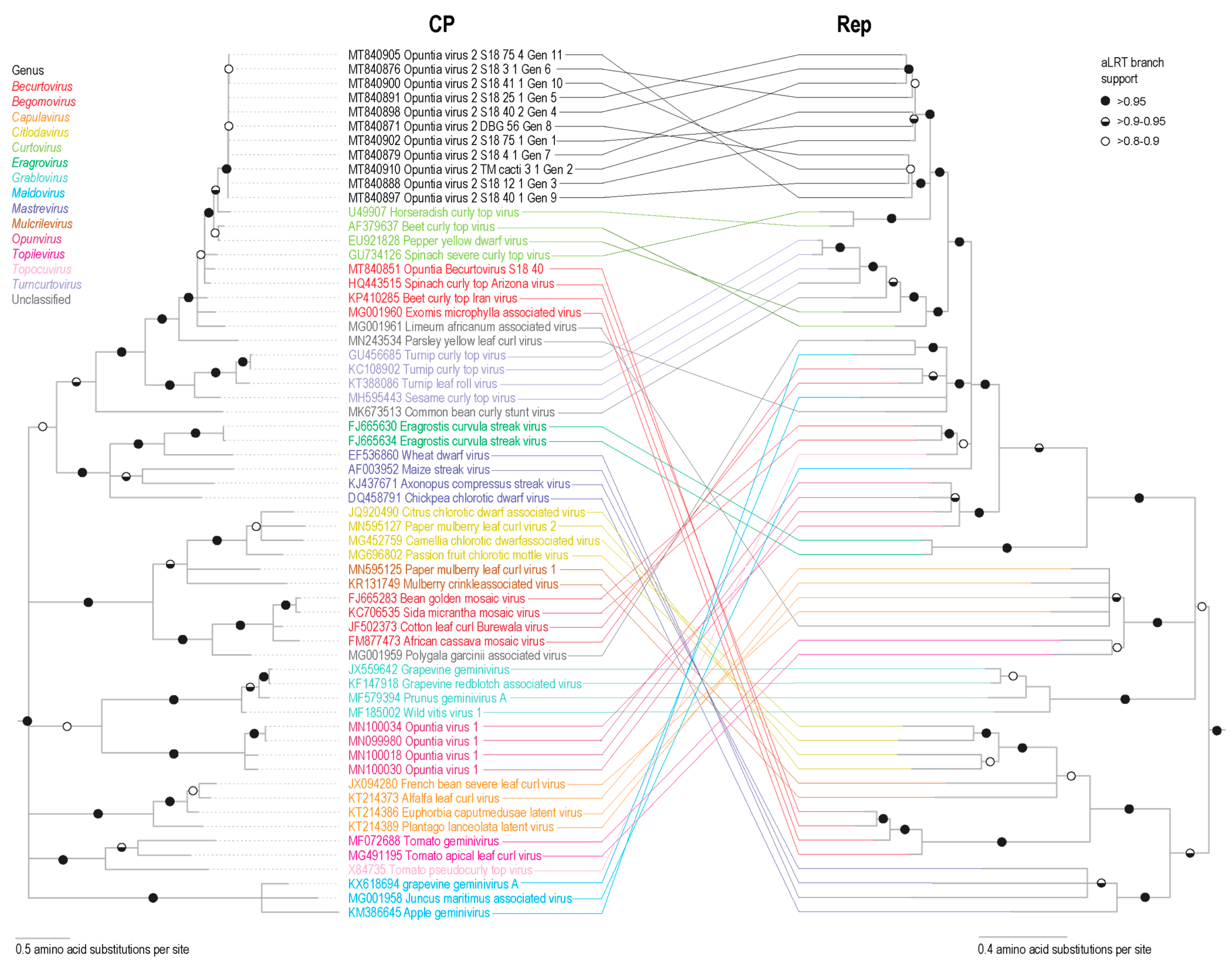
2.9. Capsid Protein Cluster Analysis
3. Results and Discussion
3.1. Geminiviruses Infecting Cacti
3.2. Opuntia Virus 2
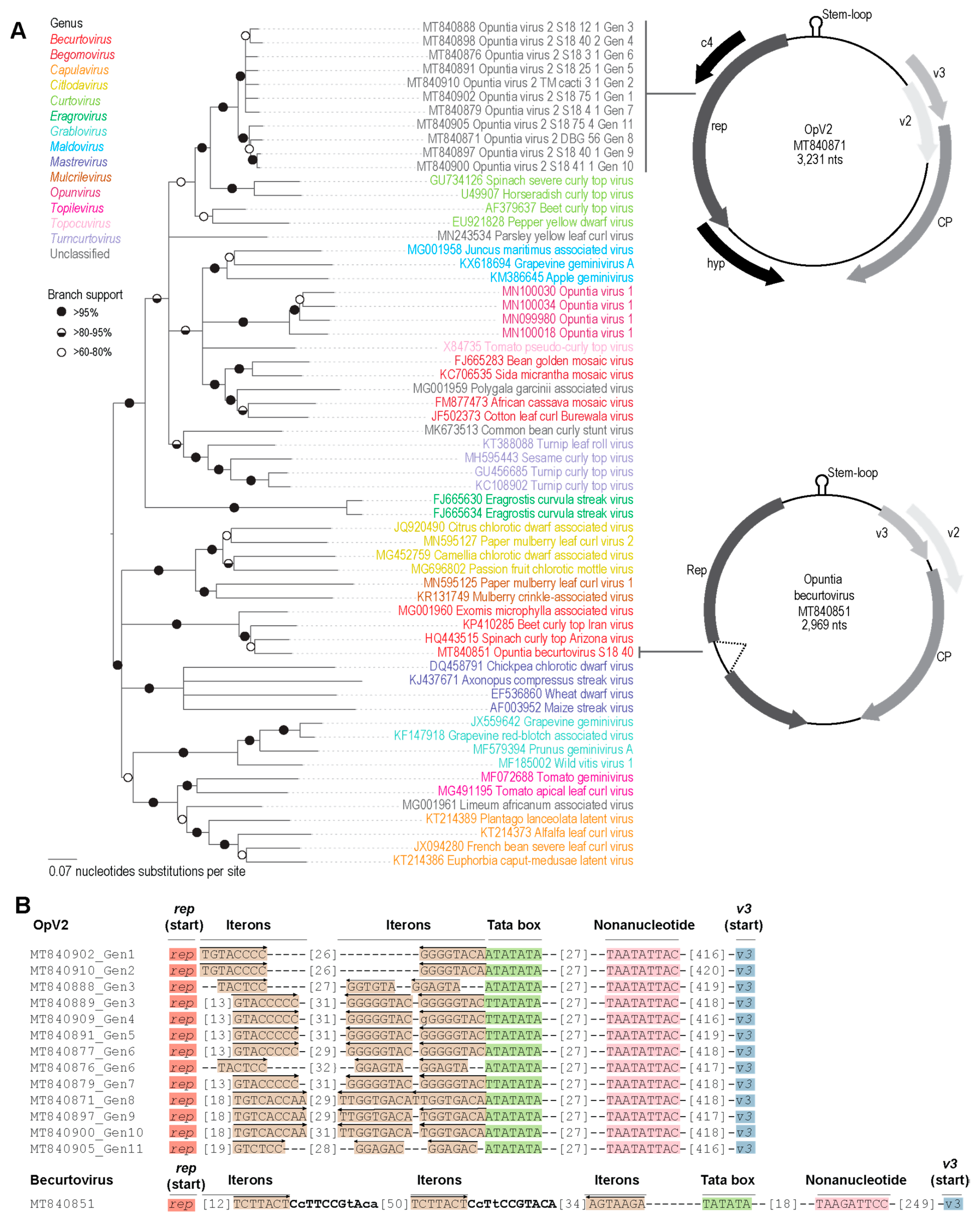
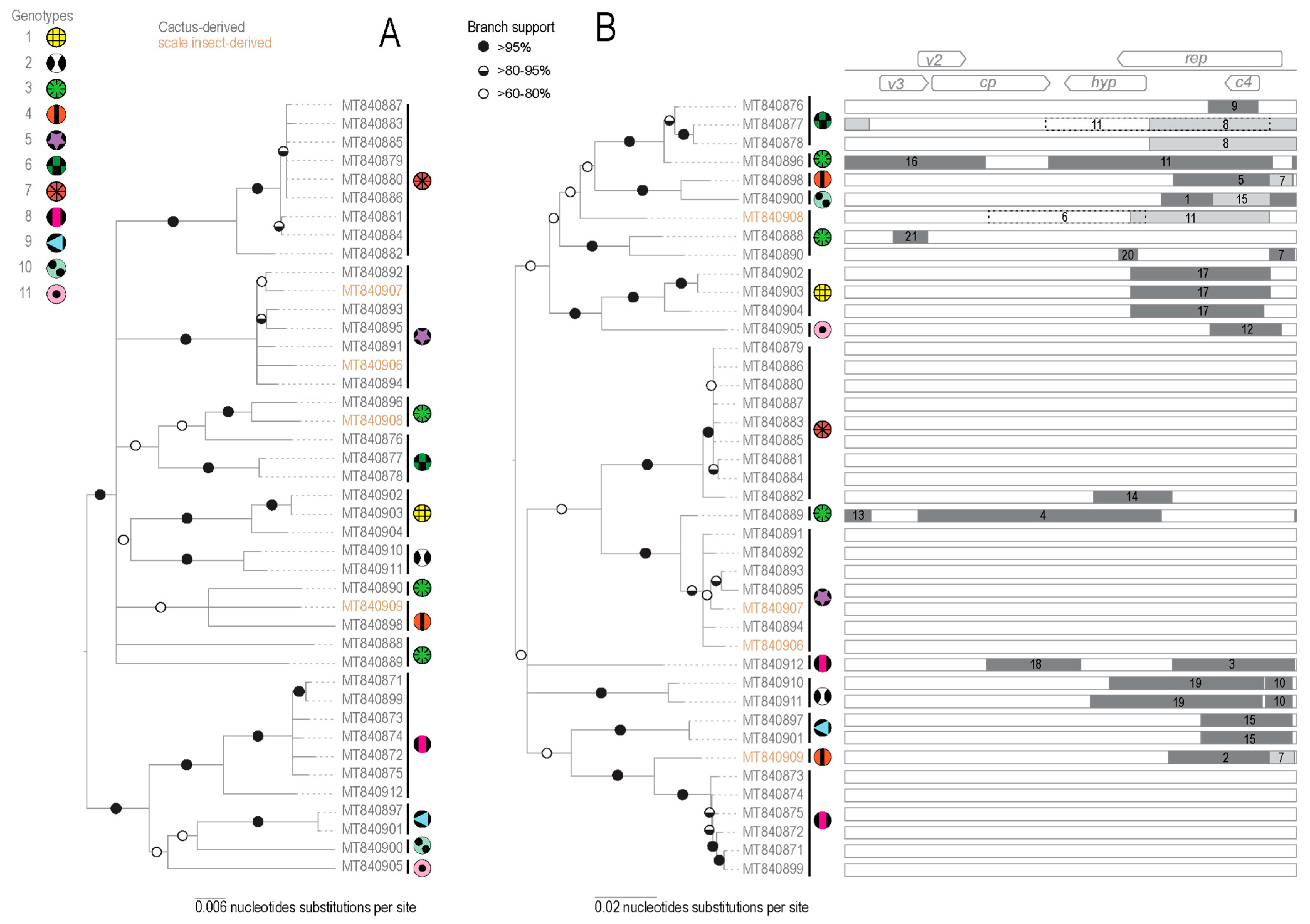
3.2.1. Genome Organization and Diversity
3.2.2. Identification of Recombination in OpV2
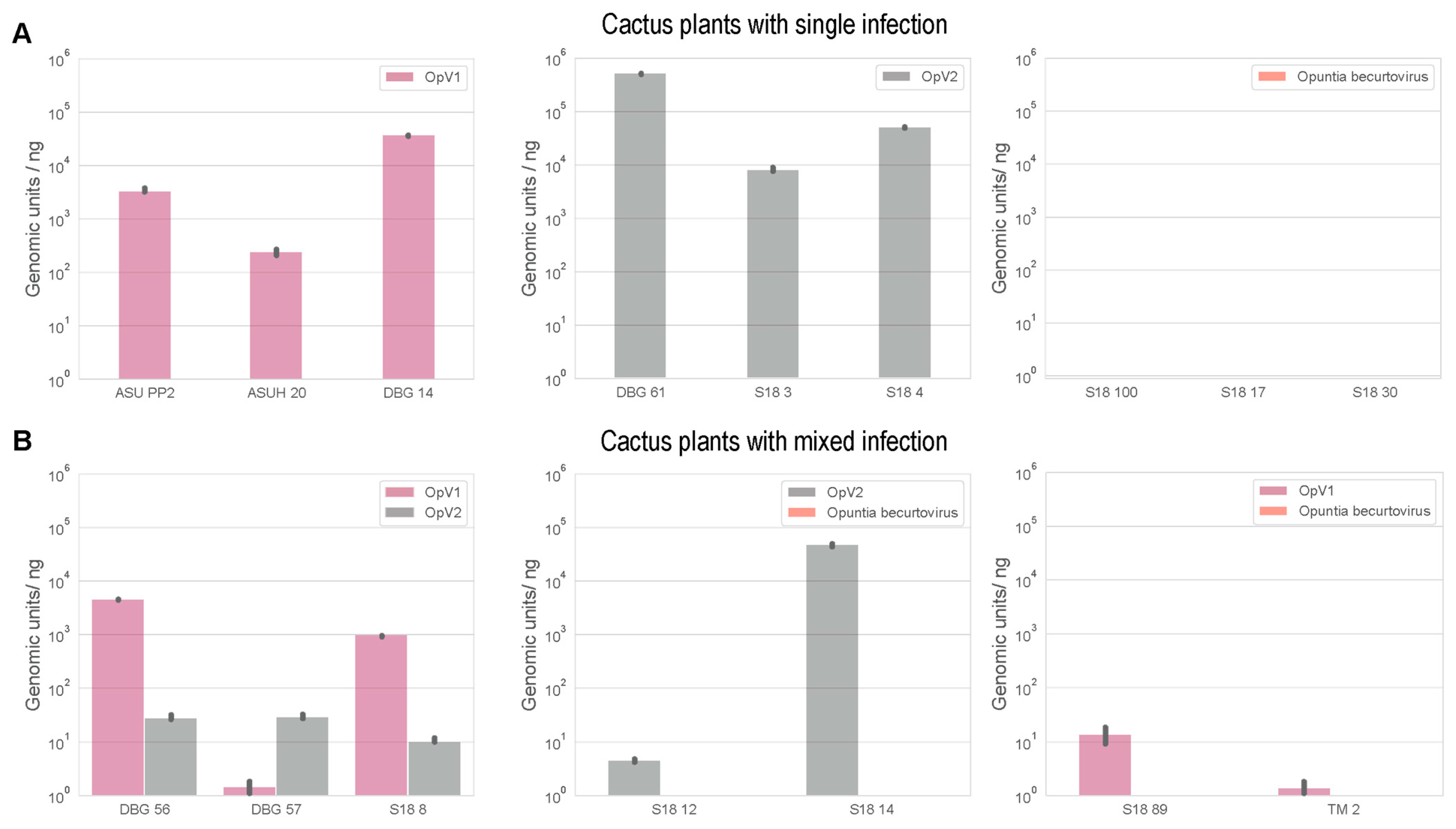
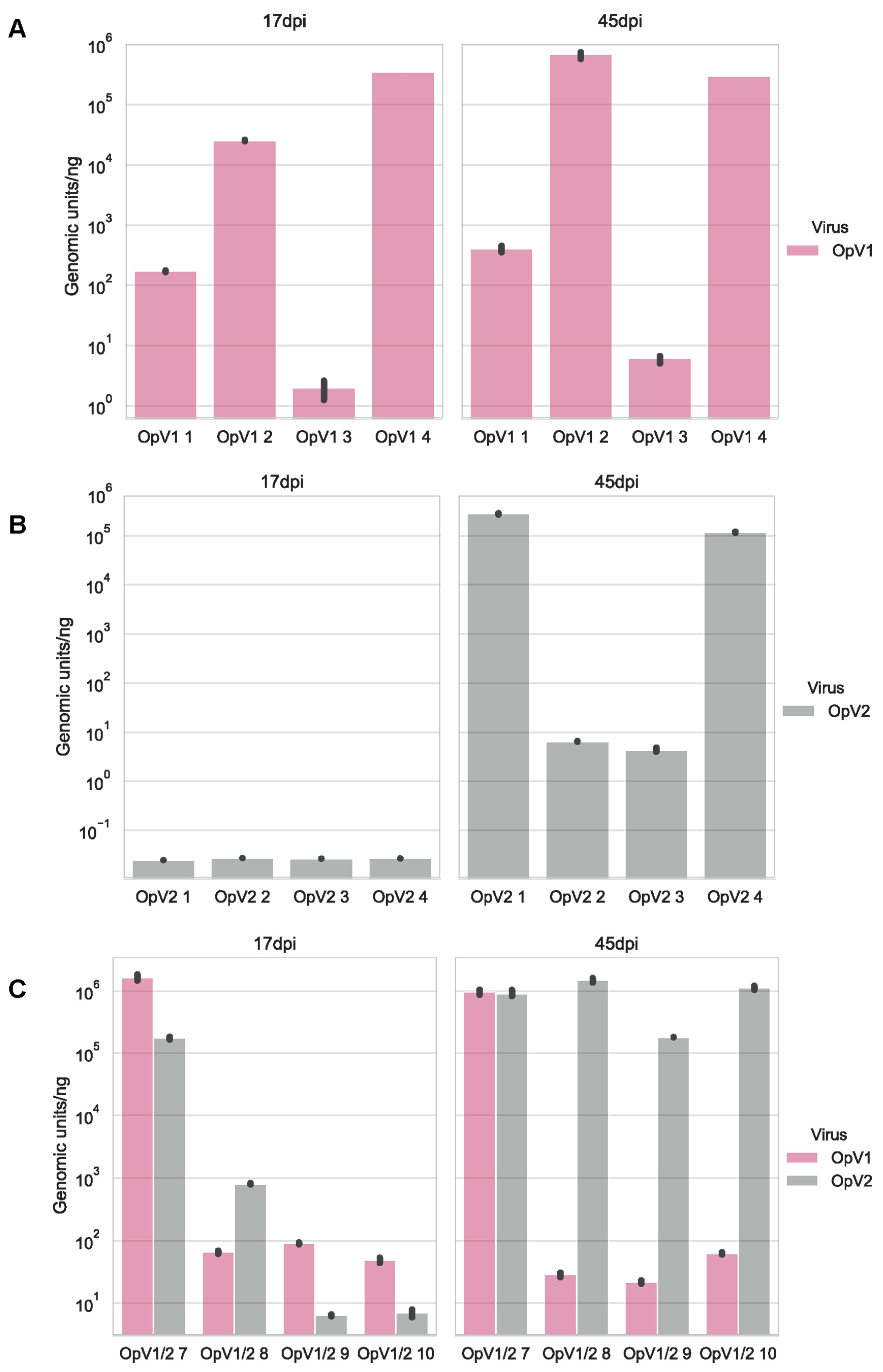
3.2.3. Infection Assays
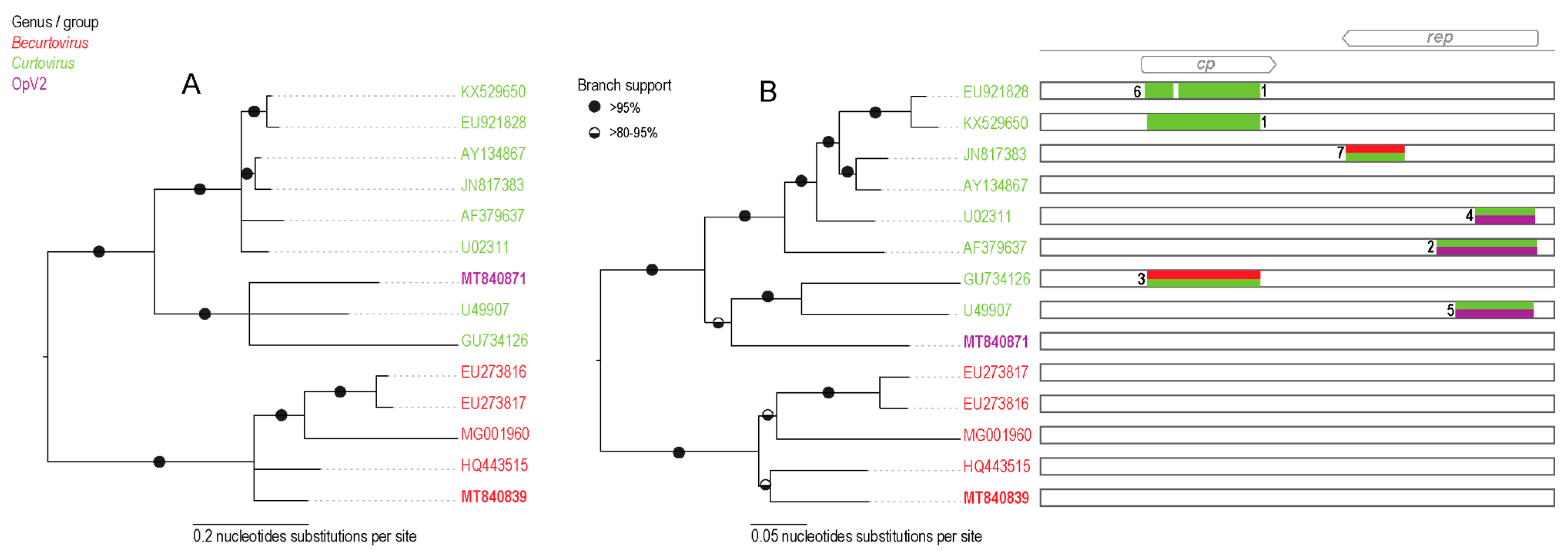
3.3. Opuntia Becurtovirus
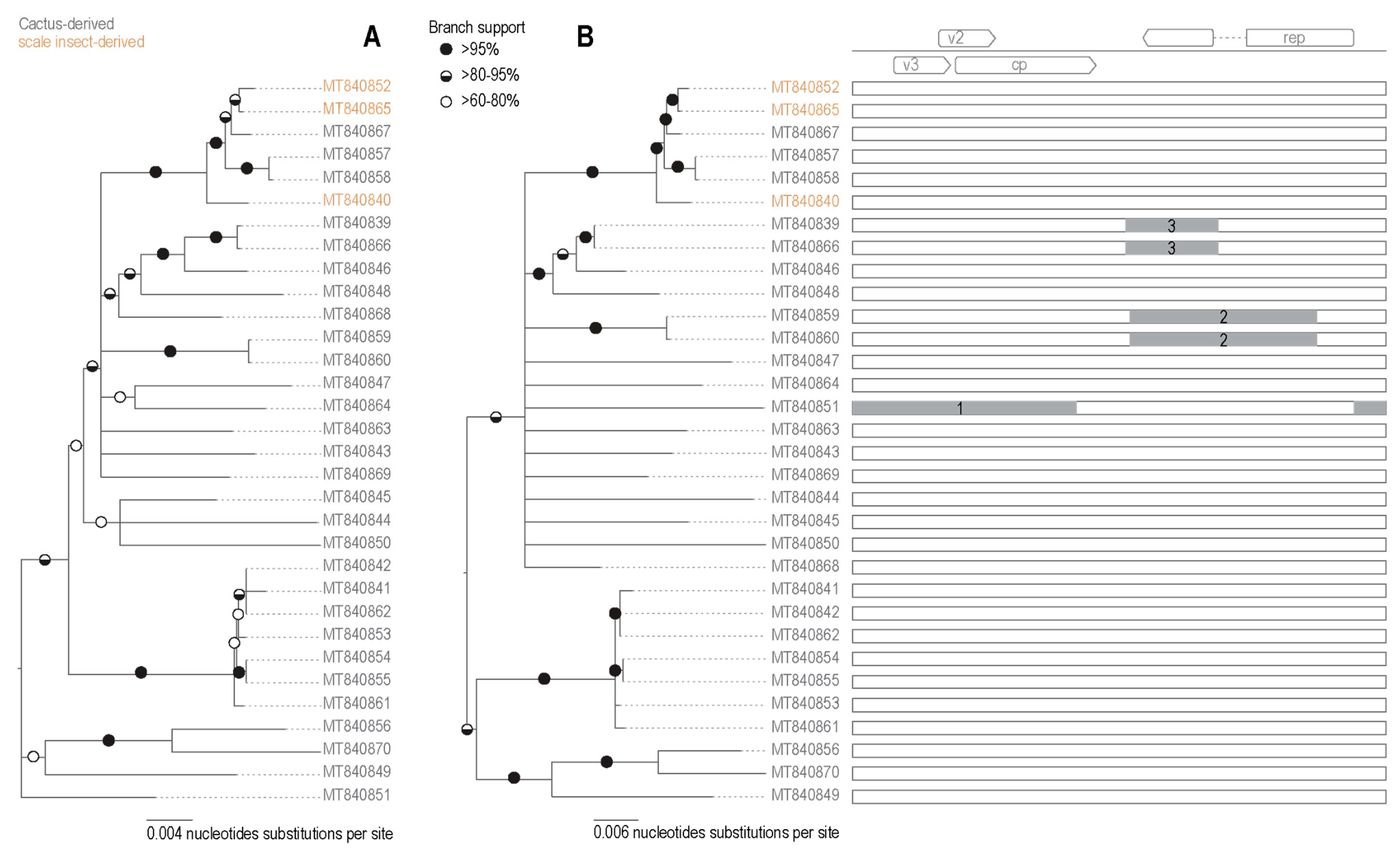
3.3.1. Diversity of Opuntia Becurtoviruses
3.3.2. Identification of Recombination in Becurtoviruses
3.3.3. Infectivity Assays
3.4. Inter-Genus Recombination
3.5. Mixed Infections of Geminiviruses in Cacti
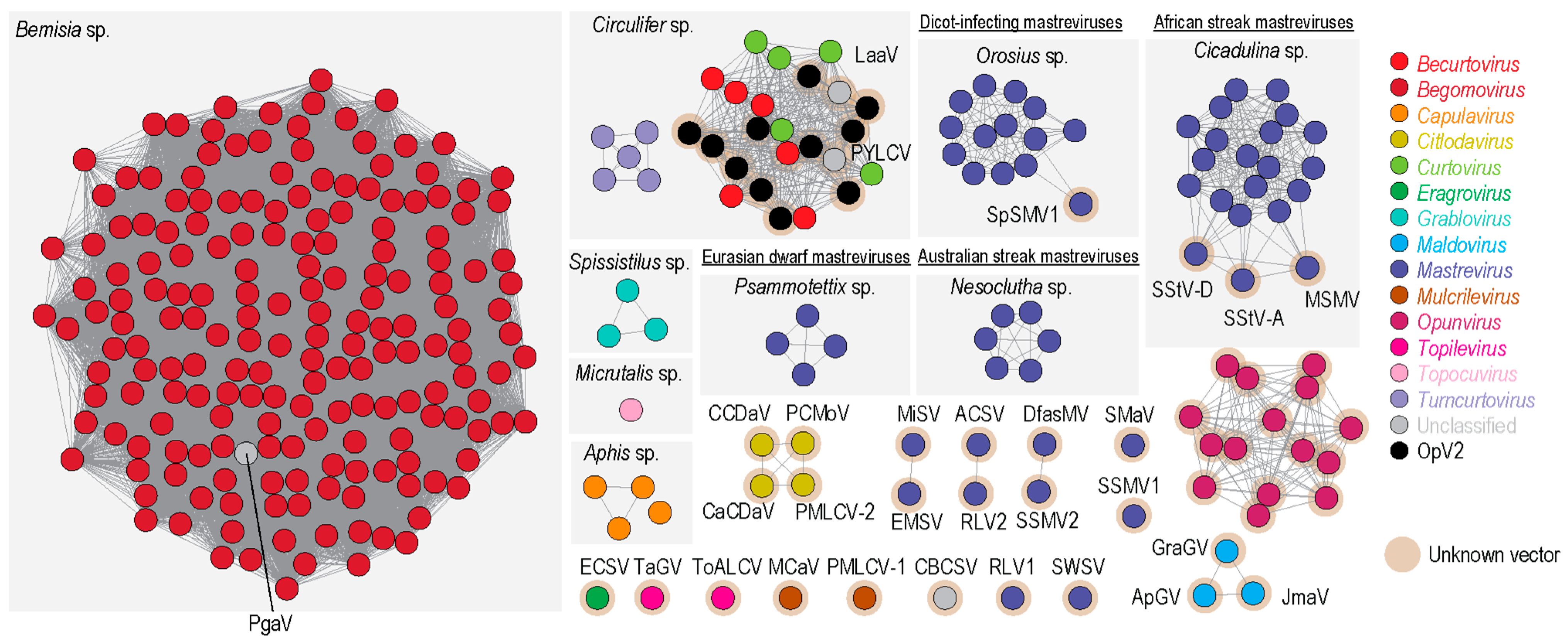
3.6. Cluster Analysis of Geminivirus Capsid Protein
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hernandez-Hernandez, T.; Brown, J.W.; Schlumpberger, B.O.; Eguiarte, L.E.; Magallon, S. Beyond aridification: Multiple explanations for the elevated diversification of cacti in the New World Succulent Biome. New Phytol. 2014, 202, 1382–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Hernandez, T.; Hernandez, H.M.; De-Nova, J.A.; Puente, R.; Eguiarte, L.E.; Magallon, S. Phylogenetic relationships and evolution of growth form in Cactaceae (Caryophyllales, Eudicotyledoneae). Am. J. Bot. 2011, 98, 44–61. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.F. The Cactus Family; Timber Press: Portland, OR, USA, 2001; p. 776. [Google Scholar]
- Guerrero, P.C.; Majure, L.C.; Cornejo-Romero, A.; Hernandez-Hernandez, T. Phylogenetic Relationships and Evolutionary Trends in the Cactus Family. J. Hered. 2019, 110, 4–21. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Baes, P.; Godínez-Alvarez, H. Global diversity and conservation priorities in the Cactaceae. Biodivers. Conserv. 2006, 15, 817–827. [Google Scholar] [CrossRef]
- Nobel, P.S. Cacti: Biology and Uses; University of California Press: Los Angeles, CA, USA, 2002; p. 280. [Google Scholar]
- Greenfield, A.B. A Perfect Red: Empire, Espionage, and the Quest for the Color of Desire; Harper Collins: New York, NY, USA, 2009; p. 352. [Google Scholar]
- Milbrath, G.M. Isolation and Characterization of a Virus from Saguaro Cactus. Phytopathology 1972, 62, 739. [Google Scholar] [CrossRef]
- Weng, Z.; Xiong, Z. Genome organization and gene expression of saguaro cactus carmovirus. J. Gen. Virol. 1997, 78, 525–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenig, R.; Pleij, C.W.; Loss, S.; Burgermeister, W.; Aust, H.; Schiemann, J. Molecular characterisation of potexviruses isolated from three different genera in the family Cactaceae. Arch. Virol. 2004, 149, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Min, B.E.; Song, Y.S.; Ryu, K.H. Complete sequence and genome structure of cactus mild mottle virus. Arch. Virol. 2009, 154, 1371–1374. [Google Scholar] [CrossRef]
- Min, B.E.; Chung, B.N.; Kim, M.J.; Ha, J.H.; Lee, B.Y.; Ryu, K.H. Cactus mild mottle virus is a new cactus-infecting tobamovirus. Arch. Virol. 2006, 151, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Liou, M.R.; Chen, Y.R.; Liou, R.F. Complete nucleotide sequence and genome organization of a Cactus virus X strain from Hylocereus undatus (Cactaceae). Arch. Virol. 2004, 149, 1037–1043. [Google Scholar] [CrossRef]
- Sanches, M.M.; Lamas, N.S.; Reis, M.B.; Arieta-Sosa, J.G.; Romano, E.; Melo, F.L.; Ribeiro, S.G. Genome Assembly of Schlumbergera Virus X Infecting Prickly Pear (Opuntia cochenillifera) in Brazil. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Fontenele, R.S.; Salywon, A.M.; Majure, L.C.; Cobb, I.N.; Bhaskara, A.; Avalos-Calleros, J.A.; Arguello-Astorga, G.R.; Schmidlin, K.; Khalifeh, A.; Smith, K.; et al. A Novel Divergent Geminivirus Identified in Asymptomatic New World Cactaceae Plants. Viruses 2020, 12, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontenele, R.S.; Roumagnac, P.; Richet, C.; Kraberger, S.; Stainton, D.; Aleamotu’a, M.; Filloux, D.; Bernardo, P.; Harkins, G.W.; McCarthy, J.; et al. Diverse genomoviruses representing twenty-nine species identified associated with plants. Arch. Virol. 2020, 165, 2891–2901. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Cao, M.; Wu, L.; Liu, H.; Chen, M.; Li, R. First identification and molecular characterization of a novel cavemovirus infecting Epiphyllum spp. Arch. Virol. 2020, 165, 2083–2086. [Google Scholar] [CrossRef] [PubMed]
- Lan, P.; Tian, T.; Pu, L.; Rao, W.; Li, F.; Li, R. Characterization and detection of a new badnavirus infecting Epiphyllum spp. Arch. Virol. 2019, 164, 1837–1841. [Google Scholar] [CrossRef] [PubMed]
- Zerbini, F.M.; Briddon, R.W.; Idris, A.; Martin, D.P.; Moriones, E.; Navas-Castillo, J.; Rivera-Bustamante, R.; Roumagnac, P.; Varsani, A.; Ictv Report, C. ICTV Virus Taxonomy Profile: Geminiviridae. J. Gen. Virol. 2017, 98, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.E.; Gronenborn, B.; Harding, R.M.; Mandal, B.; Grigoras, I.; Randles, J.W.; Sano, Y.; Timchenko, T.; Vetten, H.J.; Yeh, H.H.; et al. ICTV Virus Taxonomy Profile: Nanoviridae. J. Gen. Virol. 2021, 102. [Google Scholar] [CrossRef]
- Teycheney, P.Y.; Geering, A.D.W.; Dasgupta, I.; Hull, R.; Kreuze, J.F.; Lockhart, B.; Muller, E.; Olszewski, N.; Pappu, H.; Pooggin, M.M.; et al. ICTV Virus Taxonomy Profile: Caulimoviridae. J. Gen. Virol. 2020, 101, 1025–1026. [Google Scholar] [CrossRef]
- Zhang, W.; Olson, N.H.; Baker, T.S.; Faulkner, L.; Agbandje-McKenna, M.; Boulton, M.I.; Davies, J.W.; McKenna, R. Structure of the Maize streak virus geminate particle. Virology 2001, 279, 471–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesketh, E.L.; Saunders, K.; Fisher, C.; Potze, J.; Stanley, J.; Lomonossoff, G.P.; Ranson, N.A. The 3.3 A structure of a plant geminivirus using cryo-EM. Nat. Commun. 2018, 9, 2369. [Google Scholar] [CrossRef] [Green Version]
- Varsani, A.; Roumagnac, P.; Fuchs, M.; Navas-Castillo, J.; Moriones, E.; Idris, A.; Briddon, R.W.; Rivera-Bustamante, R.; Murilo Zerbini, F.; Martin, D.P. Capulavirus and Grablovirus: Two new genera in the family Geminiviridae. Arch. Virol. 2017, 162, 1819–1831. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Navarro, B.; Zhang, Z.; Lu, M.; Zhou, X.; Chi, S.; Di Serio, F.; Li, S. Identification and molecular characterization of a novel monopartite geminivirus associated with mulberry mosaic dwarf disease. J. Gen. Virol. 2015, 96, 2421–2434. [Google Scholar] [CrossRef] [PubMed]
- Loconsole, G.; Saldarelli, P.; Doddapaneni, H.; Savino, V.; Martelli, G.P.; Saponari, M. Identification of a single-stranded DNA virus associated with citrus chlorotic dwarf disease, a new member in the family Geminiviridae. Virology 2012, 432, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, P.; Navarro, B.; Zhang, Z.; Wang, H.; Lu, M.; Xiao, H.; Wu, Q.; Zhou, X.; Di Serio, F.; Li, S. Identification and characterization of a novel geminivirus with a monopartite genome infecting apple trees. J. Gen. Virol. 2015, 96, 2411–2420. [Google Scholar] [CrossRef]
- Zhang, S.; Shen, P.; Li, M.; Tian, X.; Zhou, C.; Cao, M. Discovery of a novel geminivirus associated with camellia chlorotic dwarf disease. Arch. Virol. 2018, 163, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Al Rwahnih, M.; Alabi, O.J.; Westrick, N.M.; Golino, D.; Rowhani, A. Description of a Novel Monopartite Geminivirus and Its Defective Subviral Genome in Grapevine. Phytopathology 2017, 107, 240–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaghi Medina, C.G.; Teppa, E.; Bornancini, V.A.; Flores, C.R.; Marino-Buslje, C.; Lopez Lambertini, P.M. Tomato Apical Leaf Curl Virus: A Novel, Monopartite Geminivirus Detected in Tomatoes in Argentina. Front. Microbiol. 2017, 8, 2665. [Google Scholar] [CrossRef] [PubMed]
- Claverie, S.; Bernardo, P.; Kraberger, S.; Hartnady, P.; Lefeuvre, P.; Lett, J.M.; Galzi, S.; Filloux, D.; Harkins, G.W.; Varsani, A.; et al. From Spatial Metagenomics to Molecular Characterization of Plant Viruses: A Geminivirus Case Study. Adv. Virus Res. 2018, 101, 55–83. [Google Scholar] [CrossRef] [PubMed]
- Fontenele, R.S.; Abreu, R.A.; Lamas, N.S.; Alves-Freitas, D.M.T.; Vidal, A.H.; Poppiel, R.R.; Melo, F.L.; Lacorte, C.; Martin, D.P.; Campos, M.A.; et al. Passion Fruit Chlorotic Mottle Virus: Molecular Characterization of a New Divergent Geminivirus in Brazil. Viruses 2018, 10, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontenele, R.S.; Lamas, N.S.; Lacorte, C.; Lacerda, A.L.M.; Varsani, A.; Ribeiro, S.G. A novel geminivirus identified in tomato and cleome plants sampled in Brazil. Virus Res. 2017, 240, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wu, X.; Jiang, X.; Wu, X.; Luan, X.; Cheng, X. Molecular characterization of common bean curly stunt virus: A novel recombinant geminivirus in China. Arch. Virol. 2020, 165, 257–260. [Google Scholar] [CrossRef]
- Hasanvand, V.; Heydanejad, J.; Massumi, H.; Kleinow, T.; Jeske, H.; Varsani, A. Isolation and characterization of a novel geminivirus from parsley. Virus Res. 2020, 286, 198056. [Google Scholar] [CrossRef] [PubMed]
- Roumagnac, P.; Varsani, A.; Martin, D.P.; Lett, J.-M. 2020.008P.A.v1.Geminiviridae_5ng_11nsp. 2020. Available online: https://talk.ictvonline.org/files/proposals/taxonomy_proposals_plant1/m/plant04/10509 (accessed on 15 March 2021).
- Rojas, M.R.; Macedo, M.A.; Maliano, M.R.; Soto-Aguilar, M.; Souza, J.O.; Briddon, R.W.; Kenyon, L.; Rivera Bustamante, R.F.; Zerbini, F.M.; Adkins, S.; et al. World Management of Geminiviruses. Annu. Rev. Phytopathol. 2018, 56, 637–677. [Google Scholar] [CrossRef]
- Moffat, A.S. PLANT PATHOLOGY: Geminiviruses Emerge as Serious Crop Threat. Science 1999, 286, 1835. [Google Scholar] [CrossRef]
- Bernardo, P.; Golden, M.; Akram, M.; Nadarajan, N.; Fernandez, E.; Granier, M.; Rebelo, A.G.; Peterschmitt, M.; Martin, D.P.; Roumagnac, P.; et al. Identification and characterisation of a highly divergent geminivirus: Evolutionary and taxonomic implications. Virus Res. 2013, 177, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Kraberger, S.; Geering, A.D.W.; Walters, M.; Martin, D.P.; Varsani, A. Novel mastreviruses identified in Australian wild rice. Virus Res. 2017, 238, 193–197. [Google Scholar] [CrossRef] [Green Version]
- Susi, H.; Laine, A.L.; Filloux, D.; Kraberger, S.; Farkas, K.; Bernardo, P.; Frilander, M.J.; Martin, D.P.; Varsani, A.; Roumagnac, P. Genome sequences of a capulavirus infecting Plantago lanceolata in the Aland archipelago of Finland. Arch. Virol. 2017, 162, 2041–2045. [Google Scholar] [CrossRef]
- Rocha, C.S.; Castillo-Urquiza, G.P.; Lima, A.T.; Silva, F.N.; Xavier, C.A.; Hora-Junior, B.T.; Beserra-Junior, J.E.; Malta, A.W.; Martin, D.P.; Varsani, A.; et al. Brazilian begomovirus populations are highly recombinant, rapidly evolving, and segregated based on geographical location. J. Virol. 2013, 87, 5784–5799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varsani, A.; Shepherd, D.N.; Monjane, A.L.; Owor, B.E.; Erdmann, J.B.; Rybicki, E.P.; Peterschmitt, M.; Briddon, R.W.; Markham, P.G.; Oluwafemi, S.; et al. Recombination, decreased host specificity and increased mobility may have driven the emergence of maize streak virus as an agricultural pathogen. J. Gen. Virol. 2008, 89, 2063–2074. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, P.; Charles-Dominique, T.; Barakat, M.; Ortet, P.; Fernandez, E.; Filloux, D.; Hartnady, P.; Rebelo, T.A.; Cousins, S.R.; Mesleard, F.; et al. Geometagenomics illuminates the impact of agriculture on the distribution and prevalence of plant viruses at the ecosystem scale. ISME J. 2018, 12, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Sallinen, S.; Norberg, A.; Susi, H.; Laine, A.L. Intraspecific host variation plays a key role in virus community assembly. Nat. Commun. 2020, 11, 5610. [Google Scholar] [CrossRef] [PubMed]
- Susi, H.; Laine, A.L. Agricultural land use disrupts biodiversity mediation of virus infections in wild plant populations. New Phytol. 2020. [Google Scholar] [CrossRef]
- McLeish, M.J.; Fraile, A.; Garcia-Arenal, F. Population Genomics of Plant Viruses: The Ecology and Evolution of Virus Emergence. Phytopathology 2021, 111, 32–39. [Google Scholar] [CrossRef]
- Lefeuvre, P.; Martin, D.P.; Elena, S.F.; Shepherd, D.N.; Roumagnac, P.; Varsani, A. Evolution and ecology of plant viruses. Nat. Rev. Microbiol. 2019, 17, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Gaur, R.K.; Petrov, N.M.; Patil, B.L.; Stoyanova, M.I. Plant Viruses: Evolution and Management; Springer: Berlin/Heidelberg, Germany, 2016; p. 312. [Google Scholar]
- Van Mölken, T.; Stuefer, J.F. Virulence in clonal plants: Conflicting selection pressures at work? Evol. Ecol. 2007, 22, 467–470. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Arenal, F.; Zerbini, F.M. Life on the Edge: Geminiviruses at the Interface between Crops and Wild Plant Hosts. Annu. Rev. Virol. 2019, 6, 411–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindbo, J.A. TRBO: A high-efficiency tobacco mosaic virus RNA-based overexpression vector. Plant. Physiol. 2007, 145, 1232–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A., 3rd; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Ferro, M.M.M.; Ramos-Sobrinho, R.; Xavier, C.A.D.; Zerbini, F.M.; Lima, G.S.A.; Nagata, T.; Assuncao, I.P. New approach for the construction of infectious clones of a circular DNA plant virus using Gibson Assembly. J. Virol. Methods 2019, 263, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stover, B.C.; Muller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 2010, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [Green Version]
- Varsani, A.; Krupovic, M. Sequence-based taxonomic framework for the classification of uncultured single-stranded DNA viruses of the family Genomoviridae. Virus Evol. 2017, 3, vew037. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analysing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2020. [Google Scholar] [CrossRef]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef] [PubMed]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retrovir. 2005, 21, 98–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Gerlt, J.A.; Bouvier, J.T.; Davidson, D.B.; Imker, H.J.; Sadkhin, B.; Slater, D.R.; Whalen, K.L. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta 2015, 1854, 1019–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Varsani, A.; Navas-Castillo, J.; Moriones, E.; Hernandez-Zepeda, C.; Idris, A.; Brown, J.K.; Murilo Zerbini, F.; Martin, D.P. Establishment of three new genera in the family Geminiviridae: Becurtovirus, Eragrovirus and Turncurtovirus. Arch. Virol. 2014, 159, 2193–2203. [Google Scholar] [CrossRef] [PubMed]
- Wren, J.D.; Roossinck, M.J.; Nelson, R.S.; Scheets, K.; Palmer, M.W.; Melcher, U. Plant virus biodiversity and ecology. PLoS Biol. 2006, 4, e80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Negrete, E.A.; Morales-Aguilar, J.J.; Dominguez-Duran, G.; Torres-Devora, G.; Camacho-Beltran, E.; Leyva-Lopez, N.E.; Voloudakis, A.E.; Bejarano, E.R.; Mendez-Lozano, J. High-Throughput Sequencing Reveals Differential Begomovirus Species Diversity in Non-Cultivated Plants in Northern-Pacific Mexico. Viruses 2019, 11, 594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varsani, A.; Shepherd, D.N.; Dent, K.; Monjane, A.L.; Rybicki, E.P.; Martin, D.P. A highly divergent South African geminivirus species illuminates the ancient evolutionary history of this family. Virol. J. 2009, 6, 36. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.A.; Coutts, B.A. Spread of introduced viruses to new plants in natural ecosystems and the threat this poses to plant biodiversity. Mol. Plant. Pathol. 2015, 16, 541–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, H.M.; Mauck, K.E.; Whitfield, A.E.; Garrett, K.A.; Malmstrom, C.M. Plant-virus interactions and the agro-ecological interface. Eur. J. Plant. Pathol. 2013, 138, 529–547. [Google Scholar] [CrossRef]
- Jones, R.A. Plant virus emergence and evolution: Origins, new encounter scenarios, factors driving emergence, effects of changing world conditions, and prospects for control. Virus Res. 2009, 141, 113–130. [Google Scholar] [CrossRef]
- Nash, T.E.; Dallas, M.B.; Reyes, M.I.; Buhrman, G.K.; Ascencio-Ibanez, J.T.; Hanley-Bowdoin, L. Functional analysis of a novel motif conserved across geminivirus Rep proteins. J. Virol. 2011, 85, 1182–1192. [Google Scholar] [CrossRef] [Green Version]
- Lefeuvre, P.; Moriones, E. Recombination as a motor of host switches and virus emergence: Geminiviruses as case studies. Curr. Opin. Virol. 2015, 10, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Lefeuvre, P.; Varsani, A.; Hoareau, M.; Semegni, J.Y.; Dijoux, B.; Vincent, C.; Reynaud, B.; Lett, J.M. Complex recombination patterns arising during geminivirus coinfections preserve and demarcate biologically important intra-genome interaction networks. PLoS Pathog. 2011, 7, e1002203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefeuvre, P.; Martin, D.P.; Hoareau, M.; Naze, F.; Delatte, H.; Thierry, M.; Varsani, A.; Becker, N.; Reynaud, B.; Lett, J.M. Begomovirus ‘melting pot’ in the south-west Indian Ocean islands: Molecular diversity and evolution through recombination. J. Gen. Virol. 2007, 88, 3458–3468. [Google Scholar] [CrossRef]
- Garcia-Andres, S.; Tomas, D.M.; Sanchez-Campos, S.; Navas-Castillo, J.; Moriones, E. Frequent occurrence of recombinants in mixed infections of tomato yellow leaf curl disease-associated begomoviruses. Virology 2007, 365, 210–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefeuvre, P.; Lett, J.M.; Varsani, A.; Martin, D.P. Widely conserved recombination patterns among single-stranded DNA viruses. J. Virol. 2009, 83, 2697–2707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharouni Kardani, S.; Heydarnejad, J.; Zakiaghl, M.; Mehrvar, M.; Kraberger, S.; Varsani, A. Diversity of beet curly top Iran virus isolated from different hosts in Iran. Virus Genes 2013, 46, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Mandal, B.; Rao, G.P.; Baranwal, V.K.; Jain, R.K. A Century of Plant. Virology in India; Springer: Berlin/Heidelberg, Germany, 2017; p. 805. [Google Scholar]
- Eini, O.; Ebadzad Sahraei, G.; Behjatnia, S.A. Molecular characterization and construction of an infectious clone of a pepper isolate of Beet curly top Iran virus. Mol. Biol. Res. Commun. 2016, 5, 101–113. [Google Scholar]
- Heydarnejad, J.; Keyvani, N.; Razavinejad, S.; Massumi, H.; Varsani, A. Fulfilling Koch’s postulates for beet curly top Iran virus and proposal for consideration of new genus in the family Geminiviridae. Arch. Virol. 2013, 158, 435–443. [Google Scholar] [CrossRef]
- Yazdi, H.R.; Heydarnejad, J.; Massumi, H. Genome characterization and genetic diversity of beet curly top Iran virus: A geminivirus with a novel nonanucleotide. Virus Genes 2008, 36, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, R.; Matic, S.; Taheri, H.; Behjatnia, S.A.A.; Vecchiati, M.; Izadpanah, K.; Accotto, G.P. The unconventional geminivirusBeet curly top Iran virus: Satisfying Koch’s postulates and determining vector and host range. Ann. Appl. Biol. 2013, 162, 174–181. [Google Scholar] [CrossRef]
- Hernández-Zepeda, C.; Varsani, A.; Brown, J.K. Intergeneric recombination between a new, spinach-infecting curtovirus and a new geminivirus belonging to the genus Becurtovirus: First New World exemplar. Arch. Virol. 2013, 158, 2245–2254. [Google Scholar] [CrossRef] [PubMed]
- Tahan, V.; Heydarnejad, J.; Jafarpour, B. Characterization of Beet curly top Iran virus infecting eggplant and pepper in north-eastern Iran. Indian Phytopathol. 2020, 73, 577–581. [Google Scholar] [CrossRef]
- Briddon, R.W.; Bedford, I.D.; Tsai, J.H.; Markham, P.G. Analysis of the nucleotide sequence of the treehopper-transmitted geminivirus, tomato pseudo-curly top virus, suggests a recombinant origin. Virology 1996, 219, 387–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybicki, E.P. A phylogenetic and evolutionary justification for three genera of Geminiviridae. Arch. Virol. 1994, 139, 49–77. [Google Scholar] [CrossRef] [PubMed]
- Jeske, H.; Lutgemeier, M.; Preiss, W. DNA forms indicate rolling circle and recombination-dependent replication of Abutilon mosaic virus. EMBO J. 2001, 20, 6158–6167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barboza, N.; Blanco-Meneses, M.; Esker, P.; Moriones, E.; Inoue-Nagata, A.K. Distribution and diversity of begomoviruses in tomato and sweet pepper plants in Costa Rica. Ann. Appl. Biol. 2018, 172, 20–32. [Google Scholar] [CrossRef]
- Alabi, O.J.; Ogbe, F.O.; Bandyopadhyay, R.; Lava Kumar, P.; Dixon, A.G.; Hughes, J.; Naidu, R.A. Alternate hosts of African cassava mosaic virus and East African cassava mosaic Cameroon virus in Nigeria. Arch. Virol. 2008, 153, 1743–1747. [Google Scholar] [CrossRef]
- Ferro, C.G.; Silva, J.P.; Xavier, C.A.D.; Godinho, M.T.; Lima, A.T.M.; Mar, T.B.; Lau, D.; Zerbini, F.M. The ever increasing diversity of begomoviruses infecting non-cultivated hosts: New species from Sida spp. and Leonurus sibiricus, plus two New World alphasatellites. Ann. Appl. Biol. 2017, 170, 204–218. [Google Scholar] [CrossRef]
- Fondong, V.N.; Pita, J.S.; Rey, M.E.; de Kochko, A.; Beachy, R.N.; Fauquet, C.M. Evidence of synergism between African cassava mosaic virus and a new double-recombinant geminivirus infecting cassava in Cameroon. J. Gen. Virol. 2000, 81, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Macedo, M.A.; Albuquerque, L.C.; Maliano, M.R.; Souza, J.O.; Rojas, M.R.; Inoue-Nagata, A.K.; Gilbertson, R.L. Characterization of tomato leaf curl purple vein virus, a new monopartite New World begomovirus infecting tomato in Northeast Brazil. Arch. Virol. 2018, 163, 737–743. [Google Scholar] [CrossRef]
- Mendez-Lozano, J.; Torres-Pacheco, I.; Fauquet, C.M.; Rivera-Bustamante, R.F. Interactions Between Geminiviruses in a Naturally Occurring Mixture: Pepper huasteco virus and Pepper golden mosaic virus. Phytopathology 2003, 93, 270–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pita, J.S.; Fondong, V.N.; Sangare, A.; Otim-Nape, G.W.; Ogwal, S.; Fauquet, C.M. Recombination, pseudorecombination and synergism of geminiviruses are determinant keys to the epidemic of severe cassava mosaic disease in Uganda. J. Gen. Virol. 2001, 82, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Oyeniran, K.A.; Hartnady, P.; Claverie, S.; Lefeuvre, P.; Monjane, A.L.; Donaldson, L.; Lett, J.M.; Varsani, A.; Martin, D.P. How virulent are emerging maize-infecting mastreviruses? Arch. Virol. 2021, 166, 955–959. [Google Scholar] [CrossRef]
- De la Higuera, I.; Kasun, G.W.; Torrance, E.L.; Pratt, A.A.; Maluenda, A.; Colombet, J.; Bisseux, M.; Ravet, V.; Dayaram, A.; Stainton, D.; et al. Unveiling Crucivirus Diversity by Mining Metagenomic Data. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Levy, H.; Fontenele, R.S.; Harding, C.; Suazo, C.; Kraberger, S.; Schmidlin, K.; Djurhuus, A.; Black, C.E.; Hart, T.; Smith, A.L.; et al. Identification and Distribution of Novel Cressdnaviruses and Circular molecules in Four Penguin Species in South Georgia and the Antarctic Peninsula. Viruses 2020, 12, 1029. [Google Scholar] [CrossRef] [PubMed]
- Tisza, M.J.; Pastrana, D.V.; Welch, N.L.; Stewart, B.; Peretti, A.; Starrett, G.J.; Pang, Y.S.; Krishnamurthy, S.R.; Pesavento, P.A.; McDermott, D.H.; et al. Discovery of several thousand highly diverse circular DNA viruses. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Host Species | ID | Accession Number | Genotype | Collection Year | Associated Scale Insect | Region of Collection |
|---|---|---|---|---|---|---|---|
| Opuntia becurtovirus | Opuntia spp. | ASU_PP13 | MT840840 | 2 | 2018 | SI_47 | Arizona, USA |
| Opuntia spp. | ASU_PP7 | MT840839 | 2 | 2018 | Arizona, USA | ||
| Opuntia martiniana | DBG_38 | MT840843 | 2 | 2018 | Arizona, USA | ||
| Cylindropuntia echinocarpa | DBG_80 | MT840841 | 2 | 2018 | Arizona, USA | ||
| Cylindropuntia spinosior | DBG_86 | MT840842 | 2 | 2018 | Arizona, USA | ||
| Opuntia phaecantha | LCM_23 | MT840844 | 2 | 2006 | Texas, USA | ||
| Opuntia stenopetala | 2014 | MT840845 | 2 | 2015 | Arizona, USA | ||
| Opuntia spp. | S18_100 | MT840861 | 2 | 2018 | Arizona, USA | ||
| Opuntia spp. | S18_101 | MT840862 | 2 | 2018 | Arizona, USA | ||
| Opuntia microdasys | S18_12 | MT840846 | 2 | 2018 | Arizona, USA | ||
| Opuntia microdasys | S18_14 | MT840847 | 2 | 2018 | Arizona, USA | ||
| Opuntia santa-rita | S18_17 | MT840870 | 2 | 2018 | Arizona, USA | ||
| Opuntia basilaris | S18_24 | MT840848 | 2 | 2018 | Arizona, USA | ||
| Opuntia engelmannii var. linguiformis | S18_30 | MT840849 | 2 | 2018 | Arizona, USA | ||
| Opuntia spp. | S18_34 | MT840850 | 2 | 2018 | Arizona, USA | ||
| Opuntia aciculata | S18_40 | MT840851 | 2 | 2018 | Arizona, USA | ||
| Opuntia aciculata | S18_54 | MT840863 | 2 | 2018 | Arizona, USA | ||
| Opuntia microdasys | S18_56 | MT840864 | 2 | 2018 | Arizona, USA | ||
| Opuntia spp. | S18_59_1 | MT840852 | 2 | 2018 | SI_68 | Arizona, USA | |
| S18_59_2 | MT840865 | 2 | 2018 | SI_68 | Arizona, USA | ||
| Opuntia engelmannii var. lindheimeri | S18_69 | MT840853 | 2 | 2018 | Arizona, USA | ||
| Opuntia santa-rita | S18_71_1 | MT840854 | 2 | 2018 | Arizona, USA | ||
| S18_71_2 | MT840855 | 2 | 2018 | Arizona, USA | |||
| Opuntia phaeacantha | S18_77 | MT840856 | 2 | 2018 | Arizona, USA | ||
| Opuntia microdasys | S18_84_1 | MT840857 | 2 | 2018 | Arizona, USA | ||
| S18_84_2 | MT840858 | 2 | 2018 | Arizona, USA | |||
| Opuntia engelmannii | S18_89_1 | MT840859 | 2 | 2018 | Arizona, USA | ||
| S18_89_2 | MT840860 | 2 | 2018 | Arizona, USA | |||
| Scale insect | SI_47 | MT840866 | 2 | 2018 | Arizona, USA | ||
| Scale insect | SI_68 | MT840867 | 2 | 2018 | Arizona, USA | ||
| Opuntia sp. | TM3_2 | MT840868 | 2 | 2018 | Arizona, USA | ||
| Opuntia engelmannii | UTH_RH6 | MT840869 | 2 | 2018 | Utah, USA | ||
| Opuntia virus 2 | Opuntia basilaris | DBG_56 | MT840871 | 8 | 2018 | Arizona, USA | |
| Opuntia basilaris | DBG_57 | MT840872 | 8 | 2018 | Arizona, USA | ||
| Opuntia santa-rita | DBG_61 | MT840873 | 8 | 2018 | Arizona, USA | ||
| Opuntia santa-rita | DBG_62 | MT840874 | 8 | 2018 | Arizona, USA | ||
| Opuntia santa-rita | DBG_63 | MT840875 | 8 | 2018 | Arizona, USA | ||
| Opuntia microdasys | S18_12_1 | MT840888 | 3 | 2018 | Arizona, USA | ||
| S18_12_2 | MT840889 | 3 | 2018 | Arizona, USA | |||
| Opuntia microdasys | S18_14 | MT840890 | 3 | 2018 | Arizona, USA | ||
| Opuntia phaeacantha | S18_25_1 | MT840891 | 5 | 2018 | Arizona, USA | ||
| S18_25_2 | MT840892 | 5 | 2018 | Arizona, USA | |||
| Opuntia phaeacantha | S18_26_1 | MT840893 | 5 | 2018 | SI_63 | Arizona, USA | |
| S18_26_2 | MT840894 | 5 | 2018 | SI_63 | Arizona, USA | ||
| S18_26_3 | MT840895 | 5 | 2018 | SI_63 | Arizona, USA | ||
| Opuntia phaeacantha | S18_27 | MT840896 | 3 | 2018 | SI_64 | Arizona, USA | |
| Opuntia engelmannii | S18_3_1 | MT840876 | 6 | 2018 | Arizona, USA | ||
| S18_3_2 | MT840877 | 6 | 2018 | Arizona, USA | |||
| S18_3_3 | MT840878 | 6 | 2018 | Arizona, USA | |||
| Opuntia engelmannii | S18_4_1 | MT840879 | 7 | 2018 | Arizona, USA | ||
| S18_4_2 | MT840880 | 7 | 2018 | Arizona, USA | |||
| S18_4_3 | MT840881 | 7 | 2018 | Arizona, USA | |||
| Opuntia aciculata | S18_40_1 | MT840897 | 9 | 2018 | Arizona, USA | ||
| S18_40_2 | MT840898 | 4 | 2018 | Arizona, USA | |||
| S18_40_3 | MT840899 | 8 | 2018 | Arizona, USA | |||
| Opuntia microdasys | S18_41_1 | MT840900 | 10 | 2018 | Arizona, USA | ||
| S18_41_2 | MT840901 | 9 | 2018 | Arizona, USA | |||
| Opuntia basilaris | S18_5_1 | MT840882 | 7 | 2018 | Arizona, USA | ||
| S18_5_2 | MT840883 | 7 | 2018 | Arizona, USA | |||
| Opuntia santa-rita | S18_75_1 | MT840902 | 1 | 2018 | Arizona, USA | ||
| S18_75_2 | MT840903 | 1 | 2018 | Arizona, USA | |||
| S18_75_3 | MT840904 | 1 | 2018 | Arizona, USA | |||
| S18_75_4 | MT840905 | 11 | 2018 | Arizona, USA | |||
| Opuntia santa-rita | S18_8_1 | MT840884 | 7 | 2018 | Arizona, USA | ||
| S18_8_2 | MT840885 | 7 | 2018 | Arizona, USA | |||
| S18_8_3 | MT840886 | 7 | 2018 | Arizona, USA | |||
| S18_8_4 | MT840887 | 7 | 2018 | Arizona, USA | |||
| Scale insect | SI_63_1 | MT840906 | 5 | 2018 | Arizona, USA | ||
| SI_63_2 | MT840907 | 5 | 2018 | Arizona, USA | |||
| Scale insect | SI_64 | MT840908 | 3 | 2018 | Arizona, USA | ||
| Scale insect | SI_70 | MT840909 | 4 | 2018 | Arizona, USA | ||
| Opuntia sp. | TM_3_1 | MT840910 | 2 | 2018 | Arizona, USA | ||
| TM_3_2 | MT840911 | 2 | 2018 | Arizona, USA | |||
| Opuntia santa-rita | UTH_RH4 | MT840912 | 8 | 2018 | Utah, USA |
| Recombination EVENT | Region | Recombinant Sequence(s) | Minor Parental Sequence(s) | Major Parental Sequence(s) | Detection Methods | p-Value |
|---|---|---|---|---|---|---|
| 1 | 2242–3220 | Genotype 10 | Genotype 9 | Genotype 4 | GBMCST | 3.15 × 10−42 |
| 2~ | 2294–3203 | Genotype 4 | Genotype 3 | Genotype 8 | RGBMCST | 1.91 × 10−34 |
| 3 | 2318–3210 | Genotype 8 | Genotype 8 | Genotype 7 | RGBMCT | 1.12 × 10−17 |
| Genotype 7 | Genotype 8 | |||||
| 4 | 531–2241 | Genotype 3 | Genotype 3 | Genotype 5 | RGBMCST | 7.72 × 10−22 |
| 5 | 2325–3200 | ^Genotype 4 | Genotype 3 | Genotype 8 | RBMCT | 1.52 × 10−15 |
| Genotype 8 | Genotype 3 | |||||
| 6 | 1051–2144 | ^Genotype 3 | Unknown(Genotype 7) | Genotype 3 | RBMCT | 3.23 × 10−8 |
| 7 | 3014–3186 | ^Genotype 3 | Genotype 5 | Genotype 3 | GMCST | 7.31 × 10−14 |
| Genotype 4 | ||||||
| 8 | 2147–173 | ^Genotype 6 | Genotype 7 | Genotype 6 | RGBMCST | 7.09 × 10−18 |
| 9 | 2585–2950 | Genotype 6 | Genotype 6 | Genotype 3 | RGBMCST | 4.51 × 10−13 |
| 10 | 3009–3178 | ^Genotype 6 | Genotype 1 | Unknown (Genotype 7) | RGBMCS | 9.77 × 10−16 |
| 11 | 1466–3037 | Genotype 3 | Genotype 5 | Genotype 3 | MCT | 3.69 × 10−6 |
| Genotype 6[T] | ||||||
| Genotype 3[P] | ||||||
| 12 | 2611–3142 | Genotype 11 | Genotype 9 | Genotype 1 | RBMCST | 7.37 × 10−15 |
| 13 | 3204–192 | Genotype 3 | Genotype 1 | Genotype 5 | RBT | 1.93 × 10−3 |
| 14 | 1740–2322 | ^Genotype 7 | Unknown (Genotype 5) | Genotype 7 | RGMCST | 1.78 × 10−11 |
| 15 | 2493–3161 | ^Genotype 9 | Genotype 8 | Unknown (Genotype 7) | GBMCS | 2.69 × 10−18 |
| Genotype 10 | ||||||
| 16 | 3169 *–1027 | ^Genotype 3 | Genotype 3 | Genotype 6 | RGBMCST | 2.95 × 10−9 |
| 17 | 2008–2991 | ^Genotype 1 | Genotype 5 | Genotype 8 | RGBMCS | 5.19 × 10−15 |
| 18 | 1034–1650 * | ^Genotype 8 | Genotype 6 | Genotype 6 | RGMCS | 9.12 × 10−11 |
| 19 | 1862–2995 * | ^Genotype 6 | Genotype 5 | Genotype 8 | RGMCST | 2.68 × 10−12 |
| 20 | 1907–2049 | ^Genotype 3 | Genotype 4 | Genotype 4 | RGMCS | 1.11 × 10−6 |
| Genotype 10 | ||||||
| 21 | 349–606 | ^Genotype 3 | Genotype 2 | Genotype 3 | RBT | 5.23 × 10−4 |
| Recombination Event | Region | Recombinant Sequence(s) | Minor Parental Sequence(s) | Major Parental Sequence(s) | Detection Methods | p-Value |
|---|---|---|---|---|---|---|
| 1 | 2790–1165 | ^MT840851 | MT840856 | Unknown | RBMCST | 1.29 × 10−7 |
| 2 | 1546–2590 * | ^MT840860 | MT840870 | MT840848 | MCST | 2.86 × 10−11 |
| MT840859 | MT840856 | |||||
| 3~ | 1525–2036 | ^MT840839 | MT840841 | MT840868 | RBCS | 4.86 × 10−7 |
| MT840866 |
| Recombination Event | Region | Recombinant Sequence(s) | Minor Parental Sequence(s) | Major Parental Sequence(s) | Detection Methods | p-Value |
|---|---|---|---|---|---|---|
| 1 | 726–1474 | ^KX529650 curtovirus | Unknown (AF379637 curtovirus) | AY134867 curtovirus | RGBMCST | 8.79 × 10−33 |
| EU921828 curtovirus | JN817383 curtovirus | |||||
| 2~ | 2719–3418 * | ^AF379637 curtovirus | MT840871 OpV2 | AY134867 curtovirus | RGBMCS | 2.56 × 10−24 |
| JN817383 curtovirus | ||||||
| U02311 curtovirus | ||||||
| 3 | 729 *–1498 * | ^GU734126 curtovirus | HQ443515 becurtovirus | U49907 curtovirus | RMCST | 1.22 × 10−14 |
| 4 | 2978–3374 | ^U02311 curtovirus | MT840871 OpV2 | JN817383 curtovirus | RGBMCST | 5.13 × 10−46 |
| 5 | 2843 *–3375 | ^U49907 curtovirus | Unknown (GU734126 curtovirus) | MT840871 OpV2 | RMCT | 1.08 × 10−11 |
| Unknown (MT840871 OpV2) | GU734126 curtovirus | |||||
| 6 | 727 *–928 | EU921828 curtovirus | JN817383 curtovirus | KX529650 curtovirus | GBMCST | 4.57 × 10−12 |
| AY134867 curtovirus | ||||||
| 7 | 2073–2502 | ^JN817383 curtovirus | U02311 curtovirus | AY134867 Becurtovirus | RGBMST | 4.11 × 10−12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontenele, R.S.; Salywon, A.M.; Majure, L.C.; Cobb, I.N.; Bhaskara, A.; Avalos-Calleros, J.A.; Argüello-Astorga, G.R.; Schmidlin, K.; Khalifeh, A.; Smith, K.; et al. New World Cactaceae Plants Harbor Diverse Geminiviruses. Viruses 2021, 13, 694. https://doi.org/10.3390/v13040694
Fontenele RS, Salywon AM, Majure LC, Cobb IN, Bhaskara A, Avalos-Calleros JA, Argüello-Astorga GR, Schmidlin K, Khalifeh A, Smith K, et al. New World Cactaceae Plants Harbor Diverse Geminiviruses. Viruses. 2021; 13(4):694. https://doi.org/10.3390/v13040694
Chicago/Turabian StyleFontenele, Rafaela S., Andrew M. Salywon, Lucas C. Majure, Ilaria N. Cobb, Amulya Bhaskara, Jesús A. Avalos-Calleros, Gerardo R. Argüello-Astorga, Kara Schmidlin, Anthony Khalifeh, Kendal Smith, and et al. 2021. "New World Cactaceae Plants Harbor Diverse Geminiviruses" Viruses 13, no. 4: 694. https://doi.org/10.3390/v13040694
APA StyleFontenele, R. S., Salywon, A. M., Majure, L. C., Cobb, I. N., Bhaskara, A., Avalos-Calleros, J. A., Argüello-Astorga, G. R., Schmidlin, K., Khalifeh, A., Smith, K., Schreck, J., Lund, M. C., Köhler, M., Wojciechowski, M. F., Hodgson, W. C., Puente-Martinez, R., Van Doorslaer, K., Kumari, S., Oyeniran, K. A., ... Varsani, A. (2021). New World Cactaceae Plants Harbor Diverse Geminiviruses. Viruses, 13(4), 694. https://doi.org/10.3390/v13040694