1. Introduction
The first case of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection was reported in December 2019 in Wuhan, China, [
1,
2]. The virus spread rapidly thereafter, resulting in a viral pandemic in 2020. According to the coronavirus disease 2019 (COVID-19) dashboard designed by the Center for Systems Science and Engineering at Johns Hopkins University, in December 2020, there were more than 72 million confirmed COVID-19 cases worldwide and more than 1.6 million registered deaths. These numbers are constantly rising. COVID-19 is mainly transmitted via droplets and aerosols [
3], and, with the absence of vaccines, the only way to control the pandemic is to develop approaches for early and efficient diagnosis followed by patient isolation and treatment [
4].
The causative agent of COVID-19 is SARS-CoV-2, a single-stranded, plus-sense RNA virus in the
Coronaviridae family. The SARS-CoV-2 RNA genome encodes five major open reading frames that include non-structural replicase proteins as well as structural proteins [
5]. Among them, the nucleocapsid (NP) gene is highly conserved and stable, with more than 90% amino acid homology with SARS-CoV and a low mutation rate [
2,
6,
7]. As the NP is highly immunogenic, it is abundantly expressed in almost all coronavirus infections [
8,
9]. It is one of the early diagnostic markers of a SARS-CoV infection that can be detected up to one day prior to the onset of clinical symptoms [
8]. Thus, the SARS-CoV-2 NP is a potential biomarker for the early diagnosis of COVID-19.
COVID-19 diagnosis mainly relies on the real-time reverse transcription-polymerase chain reaction (RT-PCR) assay, which is the current gold standard test for laboratory diagnosis of SARS-CoV-2 infections. However, RT-PCR is time-consuming and requires skilled personnel and costly equipment. Therefore, rapid and accurate tests for SARS-CoV-2 screening are essential to expedite diagnosis and prevent further transmission [
10,
11]. Antigen assays are immunoassays which detect specific viral antigens; thus, confirming a current viral infection. These tests, aimed at COVID-19 detection, are currently granted for emergency use authorization by the U.S. Food and Drug Administration as they are relatively inexpensive and can be used at the point of care. Clinical evaluation of the sensitivity and specificity of these tests is necessary for their field application. WHO-recommended interim guidelines specify a minimum of 80% sensitivity and 97% specificity for antigen-related diagnostic tests, compared with a molecular test, to be used for diagnosing COVID-19 patients. Expectedly, antigen tests are emerging as a promising candidate for early and rapid diagnosis, which may help prevent COVID-19 cases.
In this study, we developed and attempted a clinical evaluation of a rapid SARS-CoV-2 NP antigen detection test, GenBody™ COVID-19 Ag test (COVAG025), through its sensitivity and specificity towards COVID-19 diagnosis in two separate assessments. The performance of this immunochromatographic lateral flow assay for the detection of the SARS-CoV-2 NP antigen was compared with EUA-approved RT-PCR tests, retrospectively involving pre-confirmed residual nasopharyngeal swabs in VTM, as well as prospectively involving unknown symptomatic and asymptomatic individuals. The results were then further compared with EUA-approved RT-PCR tests. This clinical evaluation is essential for the implementation of the rapid antigen test for the screening of SARS-CoV-2-infected individuals, ensuring proper COVID-19 surveillance and patient management.
2. Materials and Methods
2.1. Ethics Statement
Two different studies were conducted according to International Standards of Good Clinical Practice. The retrospective clinical study was conducted at Yeungnam University Medical Center (YUMC), South Korea on 29 June 2020 and submitted to a properly constituted institutional review board (IRB), in agreement with local legal and ethical standards for formal approval of candidate diagnostic tests (IRB No.: YUMC 2020-06-058). The prospective clinical study was conducted at the Indian Council of Medical Research (ICMR)-approved Rao’s pathlab, India from 25 January 2021 to 3 February 2021 and submitted to a properly constituted institutional review board (IRB), in agreement with local legal and ethical standards for formal approval of candidate diagnostic tests (IRB No.: IRB00012217).
2.2. Preparation of Target Antibody
Codon-optimized SARS-CoV-2 nucleocapsid protein (NP) DNA, synthesized by Bioneer, South Korea [
12], was cloned into
Escherichia coli for the expression and production of recombinant NP [
12] which was used for monoclonal antibody generation. Six-week-old BALB/c mice were injected subcutaneously with 50 μg of purified SARS-CoV-2 NP antigen in equal portions of complete Freund’s adjuvant (Sigma Aldrich, St. Louis, MO, USA) for initial immunization. Furthermore, three booster immunizations were administered at two-week intervals with a similar quantity of purified SARS-CoV-2 NP antigen in incomplete Freund’s adjuvant (Sigma, St. Louis, MO, USA). The mice received a final booster injection with 50 μg NP antigen intraperitoneally three days prior to cell fusion.
The immunized mice were sacrificed, and isolated spleen cells were fused with the myeloma cell line SP2/0-Ag14 at a ratio of 5:1 using PEG 1500, as described by Kohler and Milstein [
13]. The fused cells were then mixed with DMEM media supplemented with 20% (
v/
v) fetal bovine serum, 1% (
v/
v) HEPES, and 1% (
v/
v) HAT (Gibco; Grand Island, NY, USA) and cultured on 96-well plates (37 °C, and 5% CO2). Hybridoma culture supernatants were screened for high titer antibodies through indirect and novel antibody-capture ELISA, as described below.
2.3. Selection of Target MAb Pairs
2.3.1. Indirect Enzyme-Linked Immunosorbent Assay (ELISA) to Confirm Selectivity
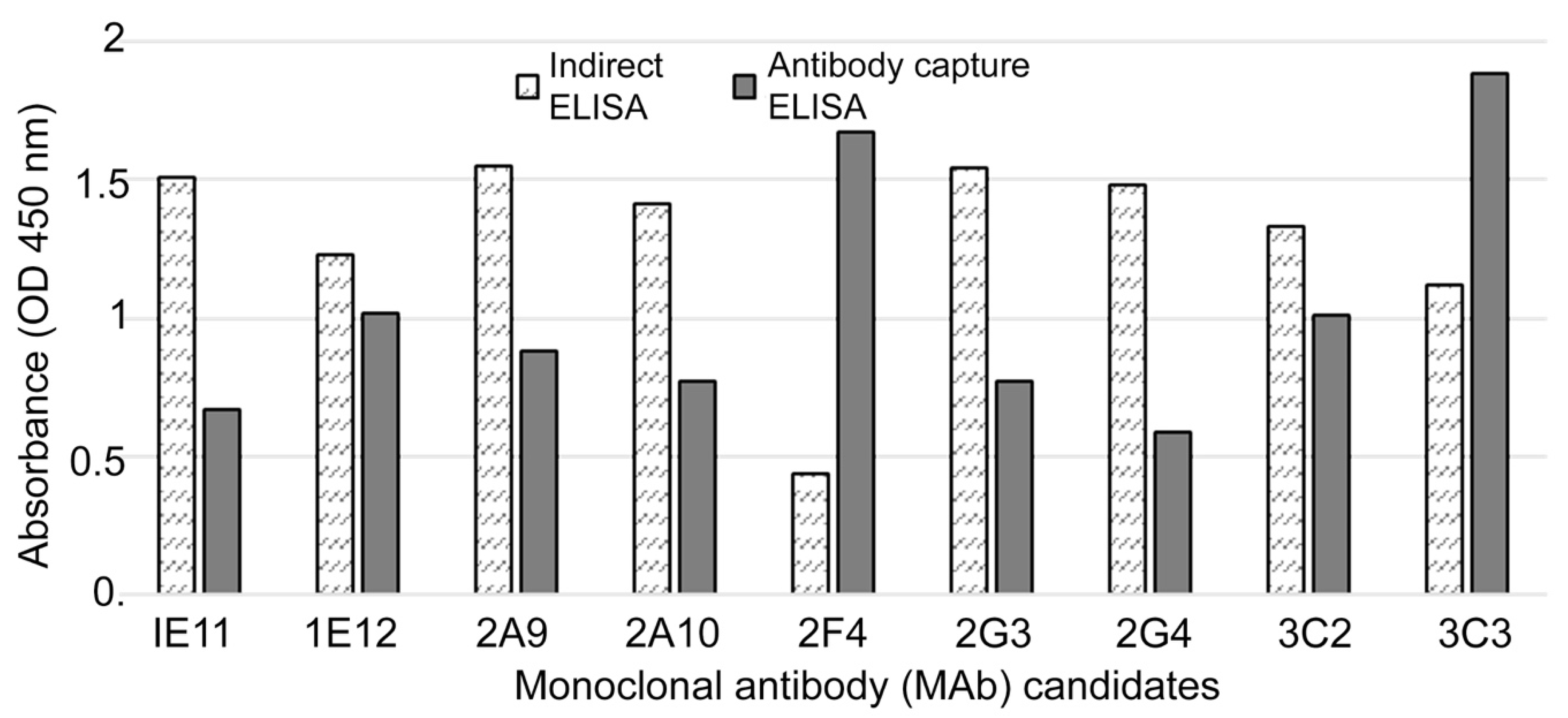
Indirect ELISA assays (
Figure 1) were used to confirm the selectivity of the MAbs against the purified recombinant NP of SARS-CoV-2. The purified NP (0.1 µg/well) was used to coat the wells of NUNC Maxisorp 96-well ELISA plates. The plates were washed three times with phosphate-buffered saline (PBS) + 0.05 % Tween 20 (PBST) and blocked with 0.5 % bovine serum albumin (BSA) for 2 h at 37 °C. Subsequently, 100 μL of hybridoma cell supernatants was added to the wells, which were then incubated for 30 min at 37 °C. The plates were washed with PBST three times and goat anti-mouse IgG-horseradish peroxidase (HRP)-conjugated secondary antibody (Sigma Aldrich, St. Louis, MO, USA) was added and incubated for 30 min at 37 °C, followed by a washing step. To detect the response, the substrate TMB (3,3′,5,5′-tetramethylbenzidine) solution (BioFX Laboratories Inc., Owings Mills, MD, USA) was added and the plates were incubated under similar conditions. The reaction was stopped with 0.5 N H
2SO
4 and absorbance was measured at 450 nm in a microplate reader (microplate spectrophotometer; Bio-Rad Laboratories Inc., Hercules, CA, USA).
2.3.2. Antibody-Capture ELISA for Selection of Mab Pairs Against NP
In the novel antibody-capture ELISA assay (
Figure 1), NUNC Maxisorp 96-well ELISA plates were coated with goat anti-mouse IgG (1 μg/mL) and blocked with 0.5% BSA. Next, 100 μL hybridoma cell supernatants were added to each well and the plates were incubated for 30 min at 37 °C. After washing, HRP-conjugated recombinant NP antigen was added, and the plates were incubated as before. The enzymatic reaction was visualized as described above. HRP-conjugated recombinant NP antigen was prepared according to the manufacturer’s instructions (Peroxidase Labeling Kit; Sigma Aldrich, St. Louis, MO, USA).
2.4. Assembly of Rapid Diagnosis Test (RDT) Kit
Several RDT kits have been designed and are awaiting clinical approval for use in COVID-19 diagnosis. With slight modifications from earlier studies [
14,
15,
16], we assembled the GenBody™ COVID-19 Ag test as follows. The MAbs against SARS-CoV-2 NP (1 mg) were conjugated with previously prepared colloidal gold particles (100 mL) [
12]. The MAb-gold conjugates were precipitated by centrifugation and dissolved with PBS containing 0.1% BSA to adjust the OD
450 to 10. The conjugates were then treated on a glass fiber and dried to prepare the conjugator pads. The MAbs against SARS-CoV-2 NP were dispensed and immobilized at the appropriate positions on a nitrocellulose membrane (2.5 mg/mL). Goat anti-mouse IgG (1 mg/mL; Arista Biologicals Inc., Allentown, PA, USA) was dispensed and immobilized on the control line of the membrane. The buffer pad was prepared by treating cellulose paper (Grade 319; Ahlstrom Inc., Alpharetta, GA, USA) with 0.1 M carbonate (pH 9.0). The absorbance pad consisted of untreated cotton paper. All pads were partially overlapped to enable the migration of the sample and buffer solution along the strip.
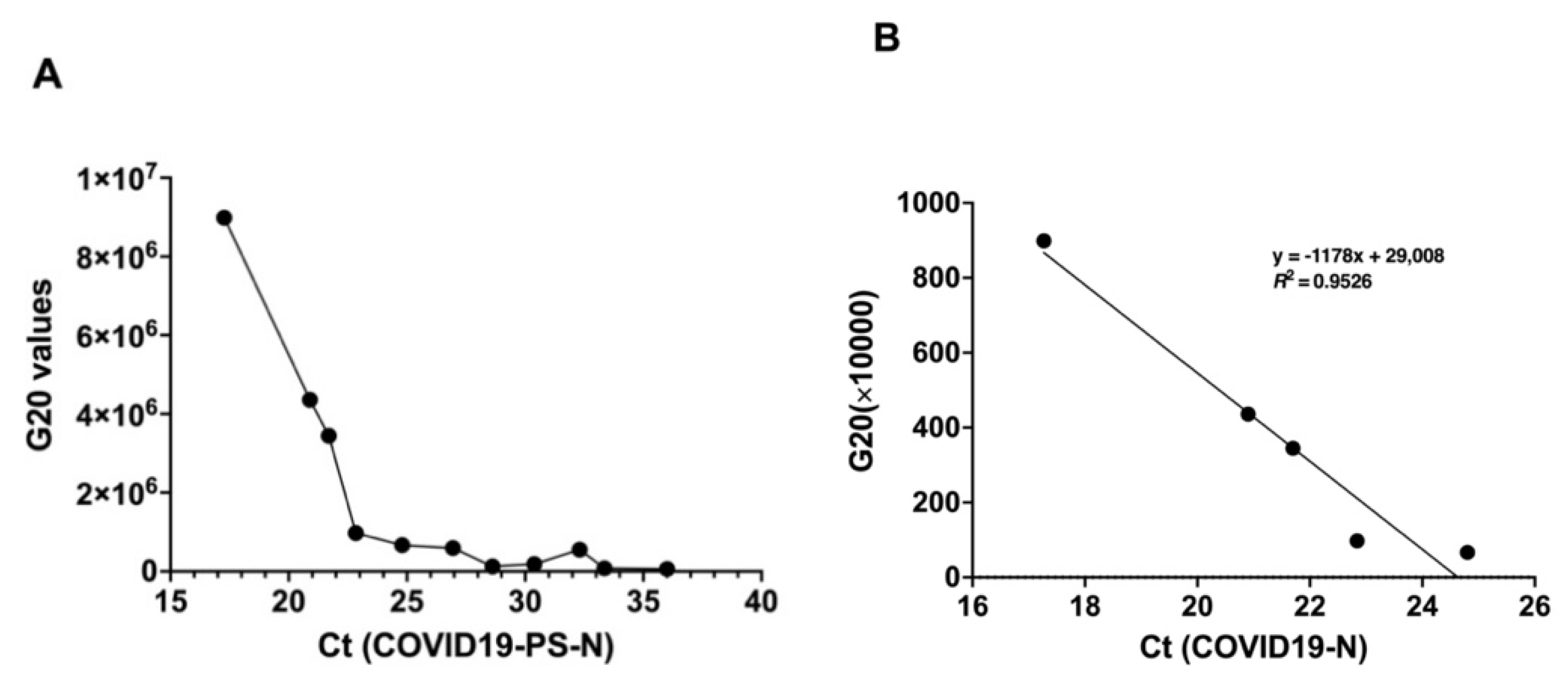
2.5. Limit of Detection (LOD) of GenBody™ COVID-19 Ag Test
To determine the LOD of the developed kit, cultured and inactivated SARS-CoV-2 virus obtained from Zeptomatrix Inc. (Buffalo, NY, USA) and the recombinant SARS-CoV-2 NP from the previous study [
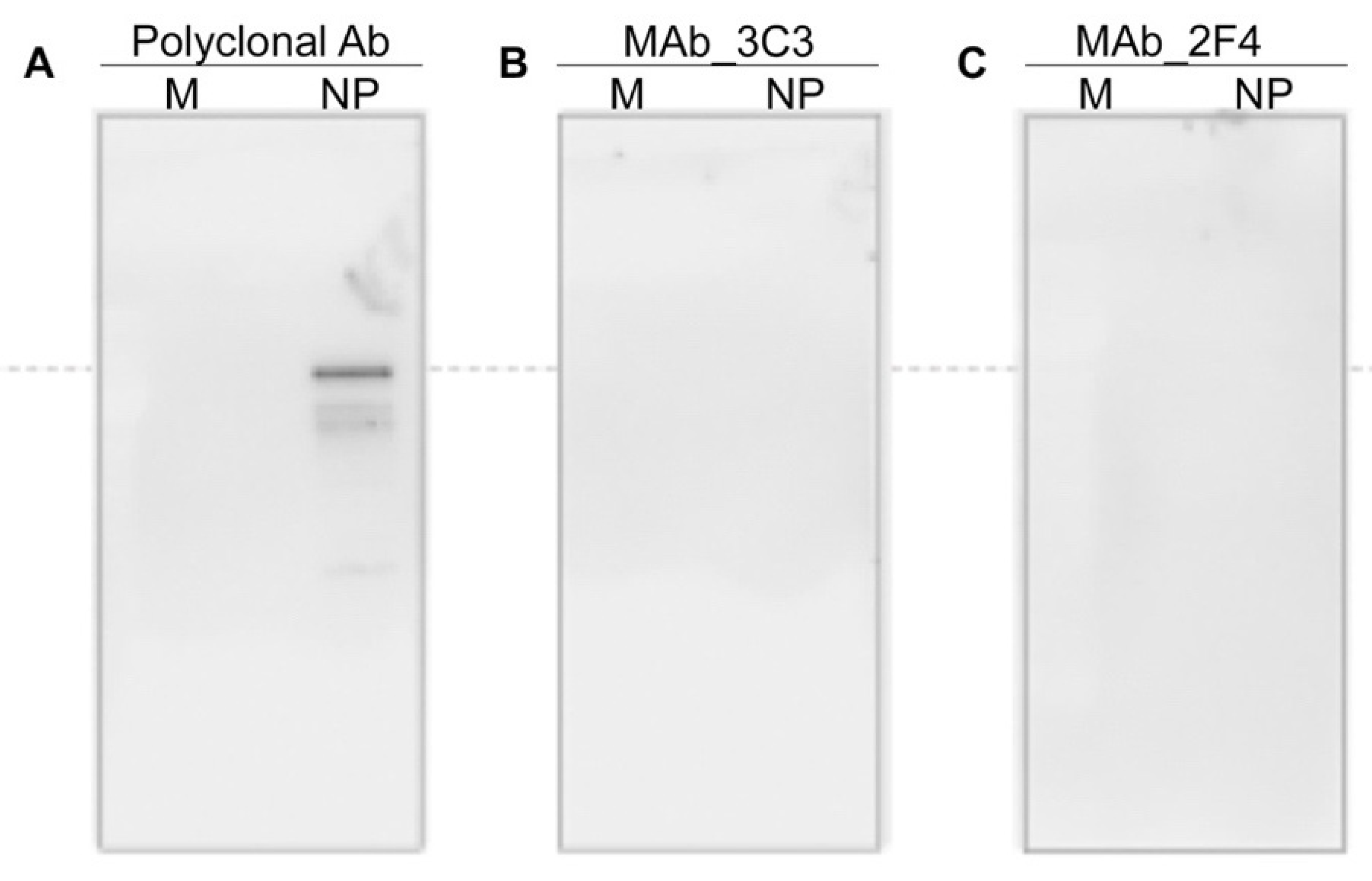
12], along with anti-NP MAbs 3C3 and 2F4, were used. An IC Reader (GenBody Confiscope G20) was used to quantitate the intensities of the test lines. The G20 cut off value was set at 100,000 and samples with a G20 value > 100,000 were considered positive. The analytical sensitivity of the GenBody™ COVID-19 Ag test was evaluated by the comparison of TCID
50 values/concentration of recombinant SARS-CoV-2 NP antigen with the corresponding G20 values. The lowest TCID
50 value/concentration of recombinant NP antigen at which the G20 value was assessed to be positive was designated as the LOD of the GenBody™ COVID-19 Ag test.
2.6. Clinical Specimens
In the clinical evaluation study conducted at Yeungnam University Medical Centre, South Korea, tests were performed according to the instructions for use of the GenBody™ COVID-19 Ag test (COVAG025) with residual Nasopharyngeal swabs transferred in VTM, ESwapTM 482C (COAPN Diagnostic Inc., Carlsbad, CA, USA)/T-SWAB TRANSPORTTM CTM (Noble Biosciences Inc., Gyeonggi-do, Korea), from 30 positive and 100 negative specimens confirmed by the EUA-approved real-time RT-PCR assay Allplex™ 2019-nCoV (Seegene Inc., Seoul, Korea)
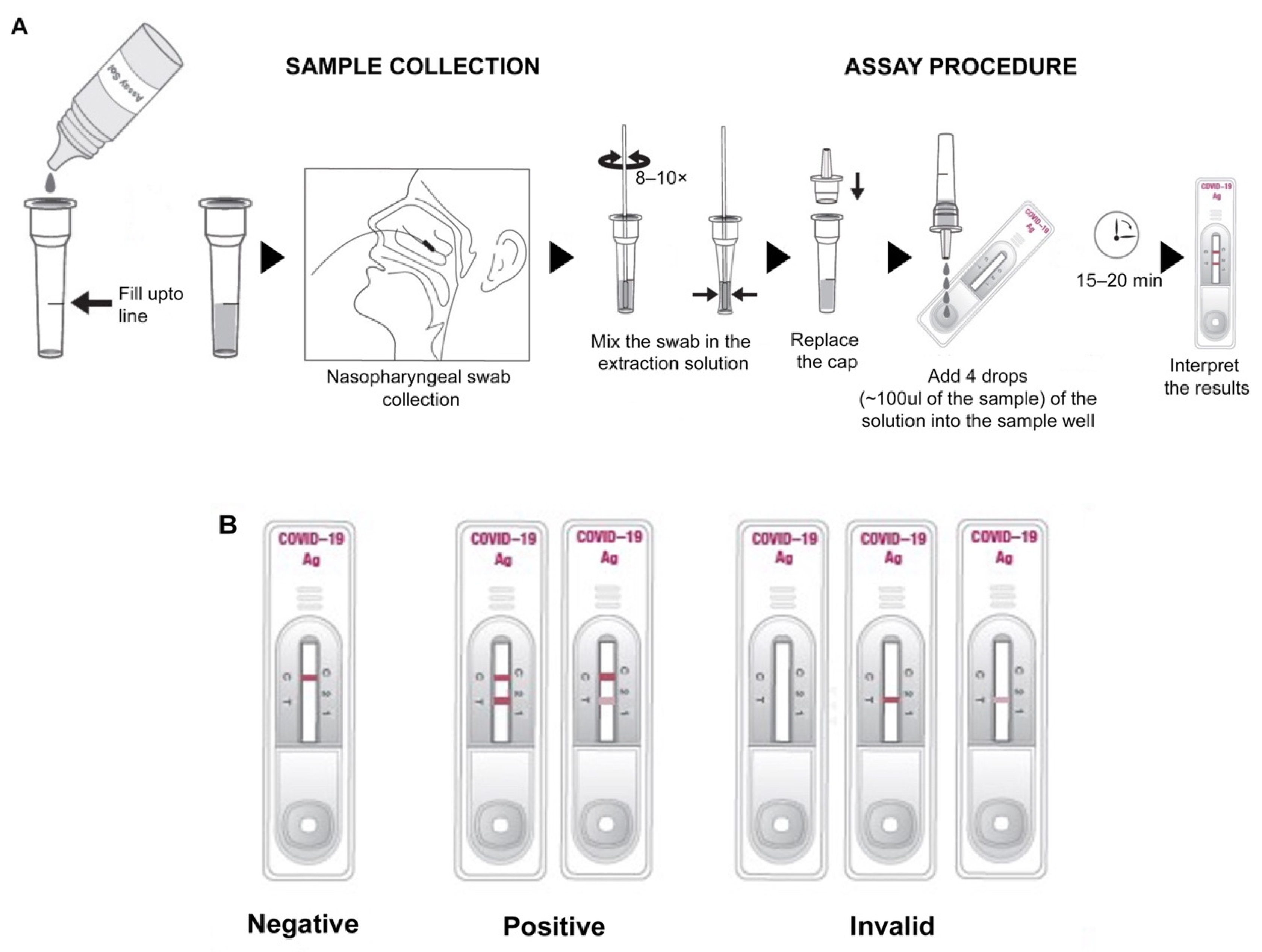
For a prospective clinical evaluation study at Rao’s pathlab, India, a second set of clinical samples, including nasopharyngeal swabs from symptomatic and asymptomatic individuals from multiple centers, were collected (
Figure 2A) and transferred in VTM, followed by analysis within 24 h with the GenBody™ COVAG025 and compared simultaneously with the EUA-approved EURORealTime SARS-CoV-2.
In both the cases, sample collection was performed according to the specimen collection guidelines of the Centers for Disease Control and Prevention (CDC). For nasopharyngeal sample collection, a sterile swab was carefully inserted into the nostril that presented the most secretion upon visual inspection. It was gently rotated, pushing the swab further, until resistance was met at the level of the turbinate (less than one inch into the nostril), followed by repeated rotation against the nasal wall (
Figure 2A). The nasopharyngeal swab was then swirled several times in the vial containing VTM/extraction solution. The extraction solution containing the specimen could be stored at room temperature for up to 1 h or at 2–8 °C (36–46 °F) for up to 12 h.
2.7. Clinical Evaluation of COVAG025
Ten microliters of specimen were loaded into the sample well of the device and three drops (~100 μL) of buffer solution were subsequently loaded into the sample well (
Figure 2A). Results were interpreted within 15 min. The appearance of the control and test lines was assessed after 15 min. An IC Reader (GenBody
TM Confiscope G20, Chungcheongnam-do, Korea) was used to quantitate the intensities of the test lines. The G20 cut off value was set at 100,000 and samples with a G20 value > 100,000 were considered positive. Tests were considered valid if a color appeared at the control line (
Figure 2B). If a red color appeared at the test line, the specimen was supposed to contain SARS-CoV-2 Ag.
4. Discussion
The containment of rapidly surging SARS-CoV-2 cases requires faster and accurate diagnostics. Limitations of the gold standard nucleic acid amplification tests (NAATs) [
17] pave the way for quicker and accurate antigen/antibody-mediated rapid diagnostic tests (RDTs). The high demand for rapid and accurate diagnostics led to the production of a large number of RDTs, but most of them still await clinical evaluation. We developed a novel RDT, GenBody
TM COVID-19 Ag test COVAG025, with a LOD of 6.93 × 10
1 for SARS-CoV-2 inactivated virus and 0.39 µg/mL for recombinant SARS-CoV-2 NP. We describe here the results of two diverse clinical evaluation studies of COVAG025 at two different locations in comparison to EUA-approved real-time RT-PCR assays following the norms and guidelines on the review and approval of in vitro diagnostic devices for COVID 19, as recommended by the South Korean Ministry of Food and Drug Safety. The overall diagnostic sensitivity/specificity of the retrospective and prospective studies was 90/98% and 94/100%, respectively. This was in compliance with WHO guidelines for the evaluation of Ag-RDTs, which recommends a minimum of 80% sensitivity and 97% specificity for Ag-RDTs. Our data was compliant with other independent clinical evaluations [
18,
19] with low rates of false positivity.
Antigen-based RDTs for SARS-CoV-2 are reported to target multiple antigens, including the SARS-CoV-2 spike or nucleocapsid protein. Recent studies support the efficacy of the nucleocapsid protein as a detection target in these types of antigen-based assays [
8,
20]. Reports involving SARS-related viruses demonstrate the secretion of high levels of the nucleocapsid protein relative to the other viral proteins [
21]. Thus, the GenBody
TM COVID-19 Ag test COVAG025, utilizing NP as the target antigen, was assumed to be successful in detecting SARS-CoV-2.
One of the unique strengths of this study is the clinical evaluation of both retrospective residual samples as well as prospective unknown samples from the point of collection. The diagnostic sensitivity and specificity were both elevated in the diagnosis of unknown samples at the point of collection, rather than residual samples stored in VTM. The number of false positives also decreased in the diagnosis of unknown samples at the collection site, elevating the positive prediction value to 100%.
Ag-RDT has been reported to have high specificity, but its use in clinical diagnosis is sometimes questioned because of its low sensitivity. Recent studies showed Ag-RDT is unable to detect SARS-CoV-2 at a Ct value of more than 19 [
22]. Other reports have revealed the diagnostic sensitivity of Ag-RDT to be 95% at Ct values < 25 but declines drastically to 20–40% when the Ct value is > 25 [
23]. In contrast to all these observations, the GenBody
TM COVID-19 Ag test COVAG025 showed diagnostic sensitivities of 90–100% at Ct levels ≤ 25 in residual samples and unconfirmed fresh samples, which also further persisted at Ct levels > 25. This proved the superior clinical diagnostic ability of the GenBody
TM COVID-19 Ag test COVAG025 in diagnosing SARS-CoV-2.
In asymptomatic individuals, the overall sensitivity of the GenBody
TM COVID-19 Ag test was estimated to be 75% (95% CI: 19.41–99.37%). Despite not displaying any noticeable symptoms, these cases may contribute to the spread of the virus. This study’s results provide substantial evidence that the point-of-care GenBody
TM COVID-19 Ag test can accurately identify SARS-CoV-2 antigens in people with suspected COVID-19, as well as in asymptomatic people with a high viral load. Previous reports have shown that asymptomatic persons have similar viral loads to symptomatic persons [
24,
25]; therefore, this Ag-RDT could be used in this type of population for proper monitoring and isolation.
5. Conclusions
The clinical evaluation of the GenBodyTM COVID-19 Ag test COVAG025 for the rapid detection of SARS-CoV-2 antigen in 130 residual confirmed specimens and 200 nasopharyngeal samples from suspected patients from the site of collection, in comparison with results obtained by using other commercialized RT-PCR assays, exhibited comparable sensitivity and selectivity. Although the sample size was small due to the various constraints of sample collection, the results indicated a trend towards the effectiveness of the GenBodyTM COVID-19 Ag test COVAG025 in the diagnosis of COVID19 based on viral load. Given the speed, low complexity and accuracy of the GenBodyTM COVID-19 Ag test COVAG025, it is predicted to be suitable for the rapid identification of positive patients. Thus, we believe the GenBody™ COVID-19 Ag test has potential for use as a simple and rapid SARS-CoV-2 antigen detection test, especially in high prevalence areas.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}