
Discovery and Characterization of Actively Replicating DNA and Retro-Transcribing Viruses in Lower Vertebrate Hosts Based on RNA Sequencing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Metatranscriptomics and Transcriptomics Datasets
2.2. Discovery of DNA and Retro-Transcribing Viruses
2.3. Virus Genome Characterization and Abundance Estimations
2.4. Phylogenetic Analyses
2.5. Virus–Host Codivergence Analyses
2.6. Data Availability
3. Results
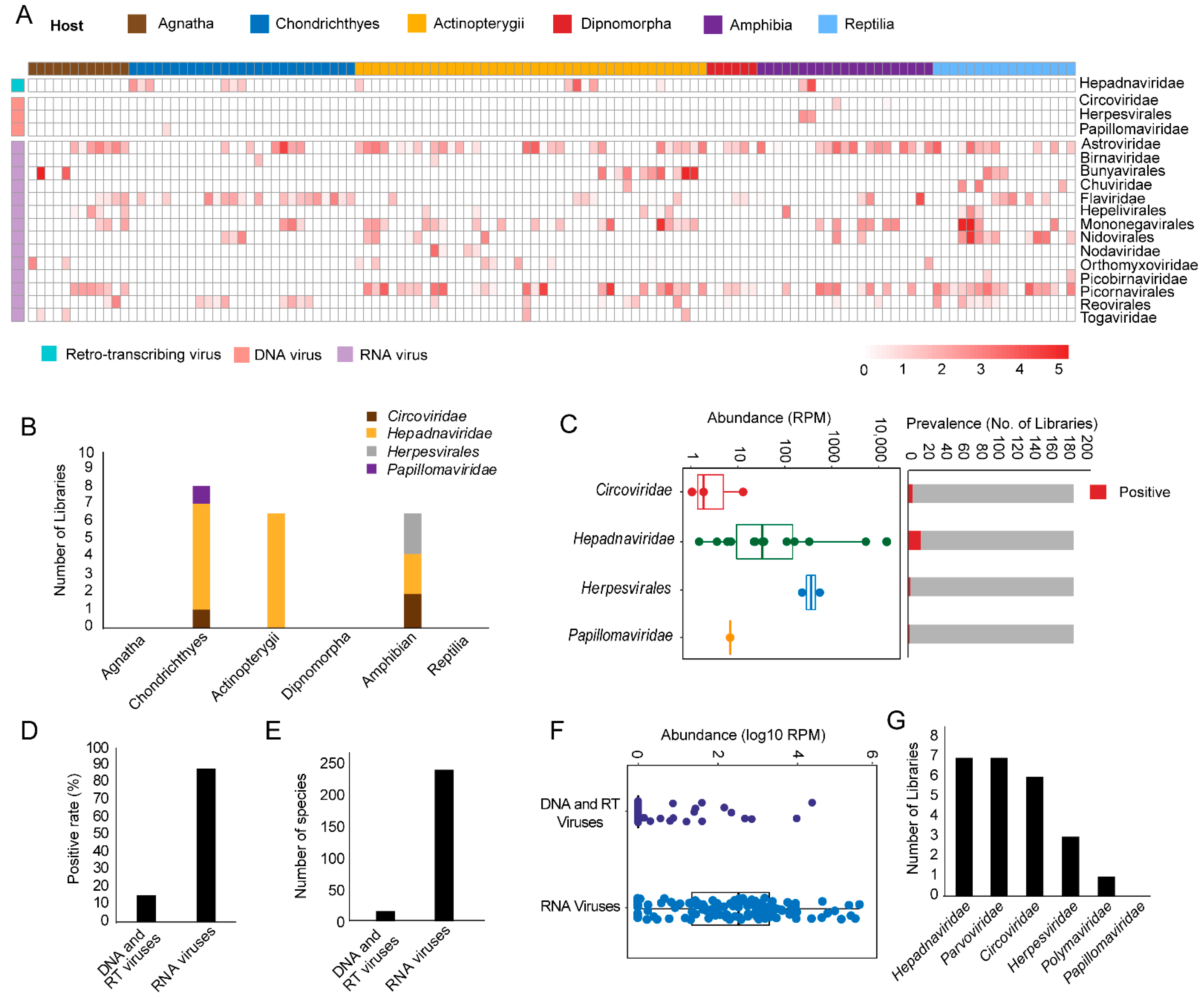
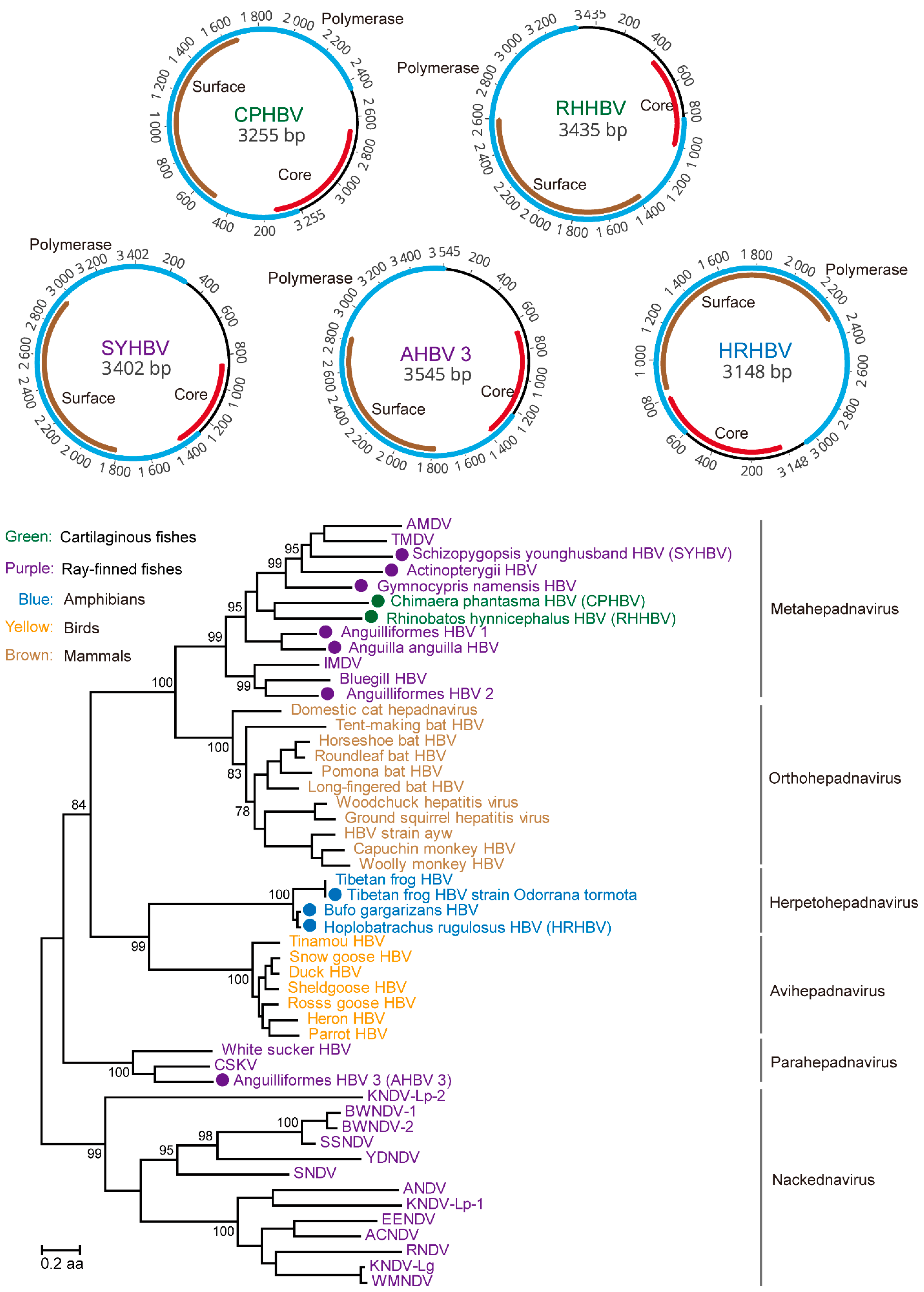
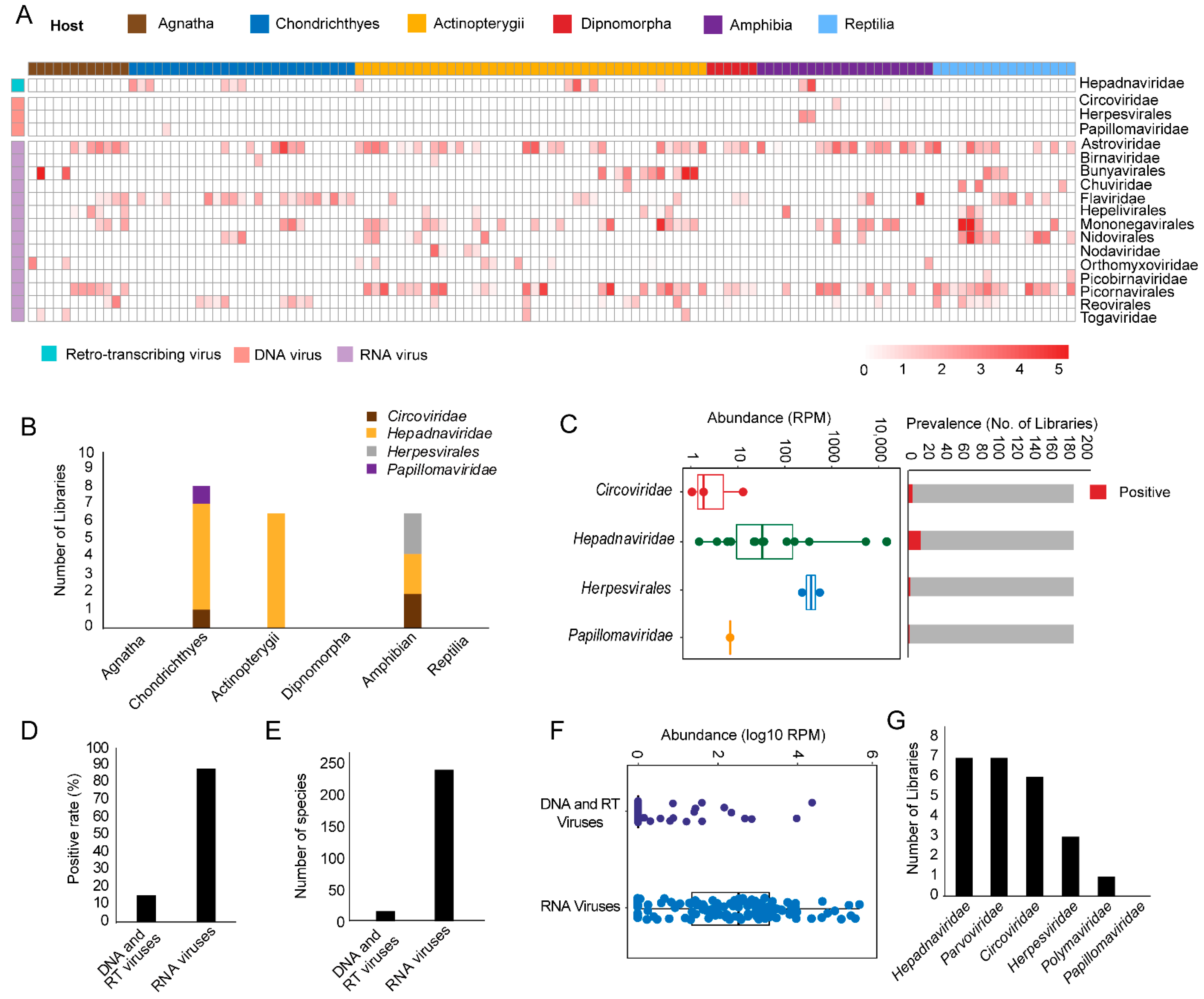
3.1. Discovery and Characterization of DNA and RT Viruses Based on Metatranscriptomics and Transcriptomics Data
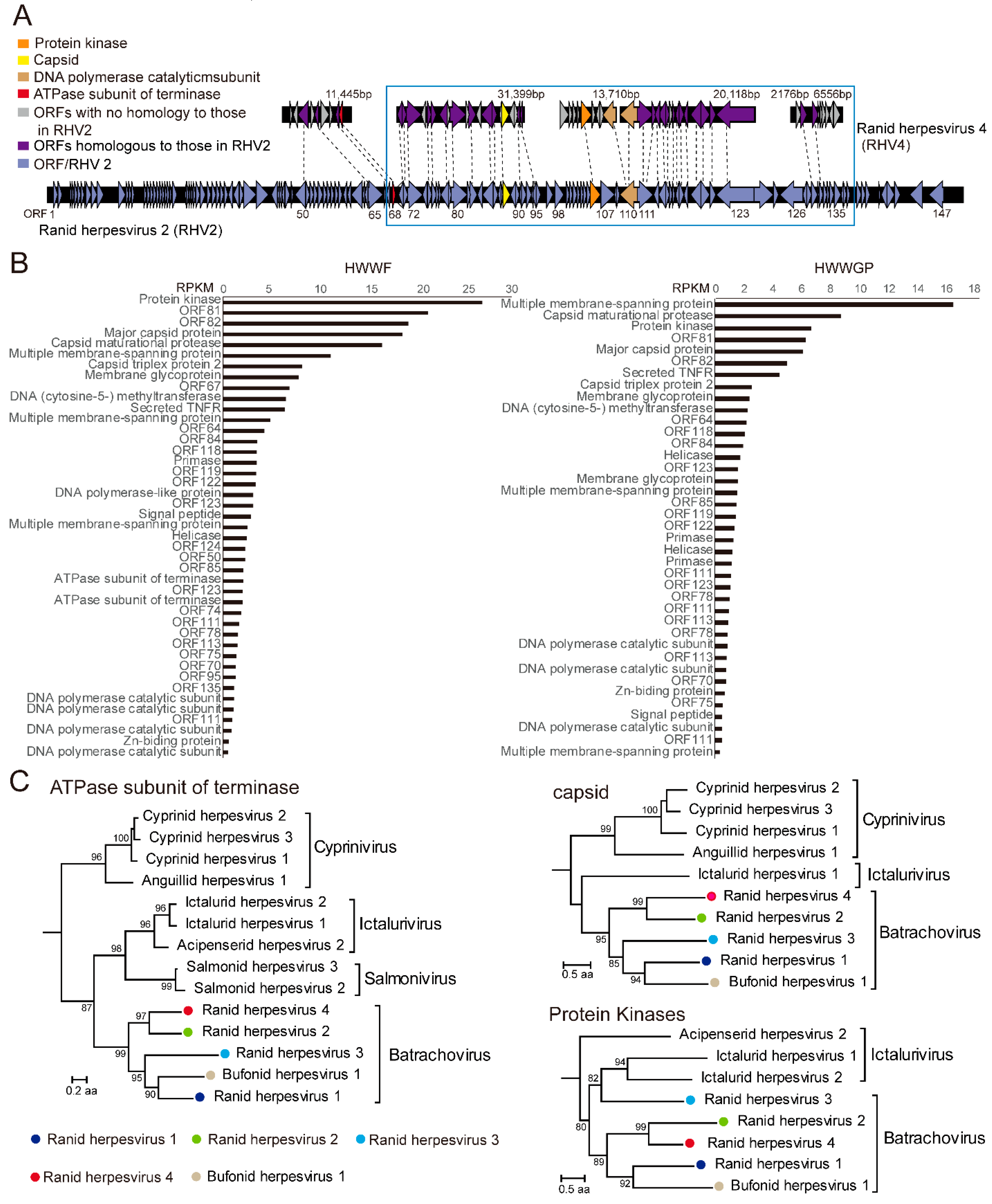
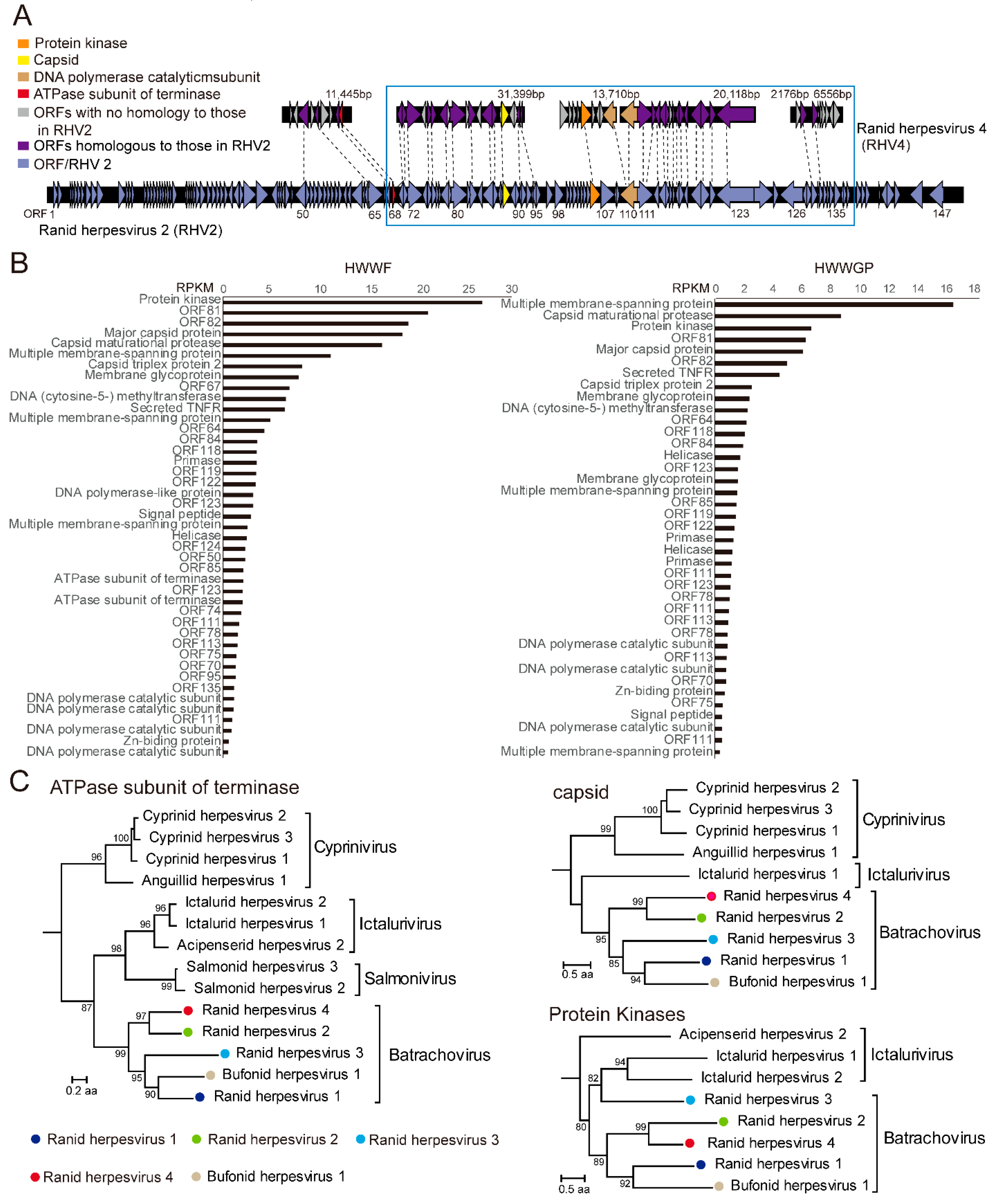
3.2. A Candidate Member of Alloherpesvirus Discovered in Rana Rugulosa
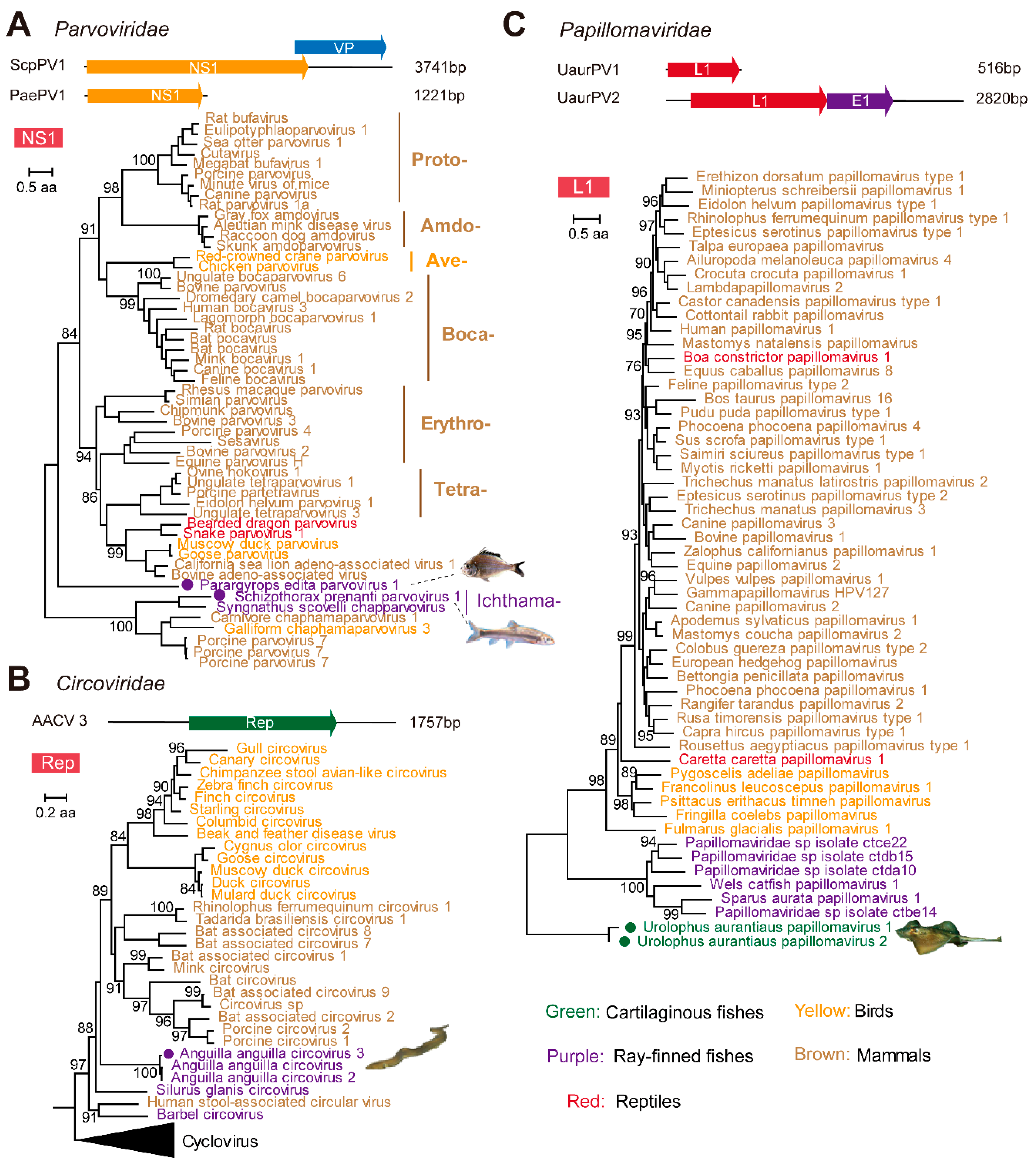
3.3. Identification and Characterization of Small DNA Viruses
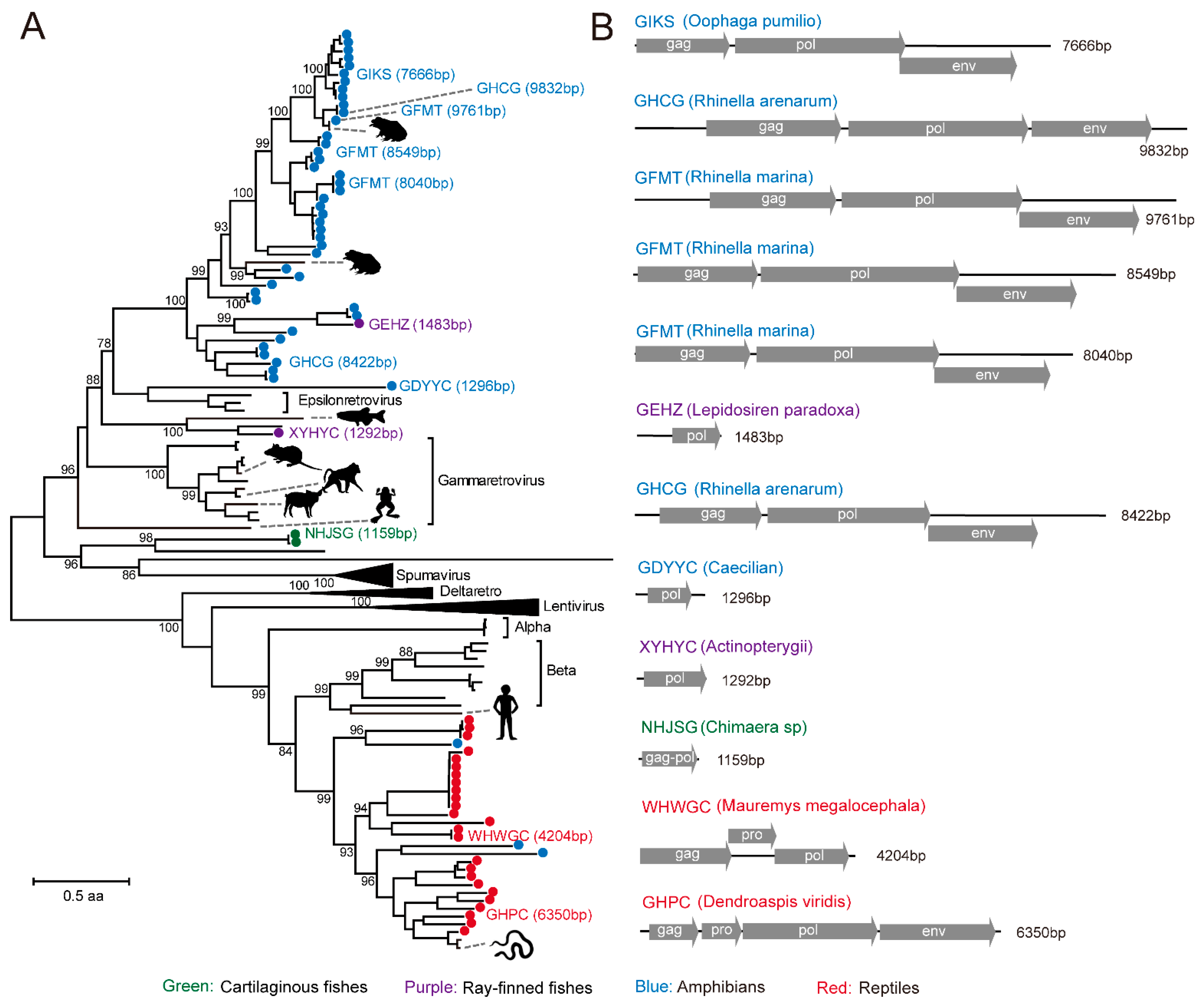
3.4. Identification of RT Viruses and Endogenous Virus Element of RT Viruses
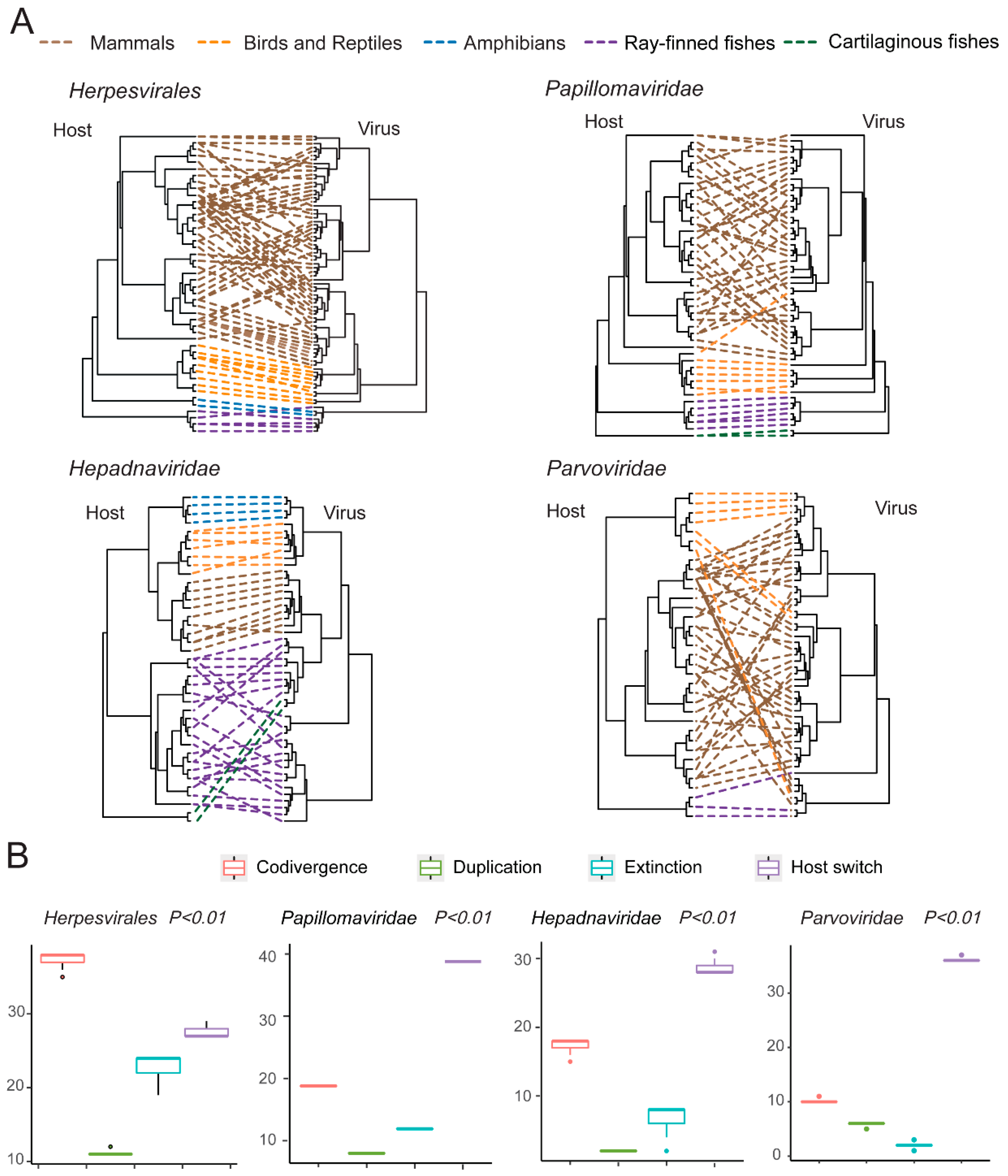
3.5. Testing the Codivergence Relationship between the DNA Viruses and Their Vertebrate Hosts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lander, G.C. The Structure of an Infectious P22 Virion Shows the Signal for Headful DNA Packaging. Science 2006, 312, 1791–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolduc, B.; Shaughnessy, D.P.; Wolf, Y.I.; Koonin, E.V.; Roberto, F.F.; Young, M. Identification of Novel Positive-Strand RNA Viruses by Metagenomic Analysis of Archaea-Dominated Yellowstone Hot Springs. J. Virol. 2012, 86, 5562–5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, S.; Chung, B.Y.; Bass, D.; Moureau, G.; Tang, S.; McAlister, E.; Culverwell, C.L.; Glücksman, E.; Wang, H.; Brown, T.D.; et al. Novel virus discovery and genome reconstruction from field RNA samples reveals highly divergent viruses in dipteran hosts. PLoS ONE 2013, 8, e80720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 2015, 4, e05378. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Shi, M.; Zhang, Y.Z.; Holmes, E.C. Meta-transcriptomics and The Evolutionary Biology of RNA Viruses. Virus Res. 2017, 243, 83–90. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Chen, X.; Tian, J.H.; Chen, L.J.; Li, K.; Wang, W.; Eden, J.S.; Shen, J.J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef]
- Roediger, B.; Lee, Q.; Tikoo, S.; Cobbin, J.C.A.; Henderson, J.M.; Jormakka, M.; O’Rourke, M.B.; Padula, M.P.; Pinello, N.; Henry, M.; et al. An Atypical Parvovirus Drives Chronic Tubulointerstitial Nephropathy and Kidney Fibrosis. Cell 2018, 175, 530–543.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, A.F.; Shi, M.; Eden, J.S.; Zhang, Y.Z.; Holmes, E.C. Diversity and Evolution of Novel Invertebrate DNA Viruses Revealed by Meta-Transcriptomics. Viruses 2019, 11, 1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrai, M.; Brussel, K.V.; Shi, M.; Li, C.X.; Chang, W.S.; Munday, J.S.; Voss, K.; McLuckie, A.; Taylor, D.; Laws, A.; et al. Identification of a Novel papillomavirus Associated with Squamous Cell Carcinoma in a Domestic Cat. Viruses 2020, 12, 124. [Google Scholar] [CrossRef] [Green Version]
- Aghazadeh, M.; Shi, M.; Pesavento, P.A.; Durham, A.C.; Polley, T.; Donahoe, S.L.; Troyer, R.M.; Barrs, V.R.; Holmes, E.C.; Beatty, J.A. Transcriptome Analysis and In Situ Hybridization for FcaGHV1 in Feline Lymphoma. Viruses 2018, 10, 464. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Lin, X.D.; Vasilakis, N.; Tian, J.H.; Li, C.X.; Chen, L.J.; Eastwood, G.; Diao, X.N.; Chen, M.H.; Chen, X.; et al. Divergent Viruses Discovered in Arthropods and Vertebrates Revise the Evolutionary History of the Flaviviridae and Related Viruses. J. Virol. 2015, 90, 659–669. [Google Scholar] [CrossRef] [Green Version]
- Lauber, C.; Seitz, S.; Mattei, S.; Suh, A.; Beck, J.; Herstein, J.; Börold, J.; Salzburger, W.; Kaderali, L.; Briggs, J.A.G.; et al. Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses. Cell Host Microbe 2017, 22, 387–399.e6. [Google Scholar] [CrossRef] [Green Version]
- Pénzes, J.J.; Pham, H.T.; Benkö, M.; Tijssen, P. Novel parvoviruses in reptiles and genome sequence of a lizard parvovirus shed light on Dependoparvovirus genus evolution. J. Gen. Virol. 2015, 96, 2769. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Fu, Y.; Xie, J.; Cheng, J.; Ghabrial, S.A.; Li, G.; Peng, Y.; Yi, X.; Jiang, D. Widespread Endogenization of Densoviruses and Parvoviruses in Animal and Human Genomes. J. Virol. 2011, 85, 9863–9876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Zheng, J.; Jiang, Y. A new iridovirus isolated from soft-shelled turtle. Virus Res. 1999, 63, 147–151. [Google Scholar] [CrossRef]
- Yan, Y.; Cui, H.; Jiang, S.; Huang, Y.; Huang, X.; Wei, S.; Xu, W.; Qin, Q. Identification of a novel marine fish virus, Singapore grouper iridovirus-encoded microRNAs expressed in grouper cells by Solexa sequencing. PLoS ONE 2011, 6, e19148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- 1Grayfer, L.; De Jesus Andino, F.; Robert, J. The Amphibian (Xenopus laevis) Type I Interferon Response to Frog Virus 3: New Insight into Ranavirus Pathogenicity. J. Virol. 2014, 88, 5766–5777. [Google Scholar] [CrossRef] [Green Version]
- Jakob, N.J.; Kehm, R.; Gelderblom, H.R. A novel fish herpesvirus of Osmerus eperlanus. Virus Genes 2010, 41, 81–85. [Google Scholar] [CrossRef]
- Davison, A.J.; Cunningham, C.; Sauerbier, W.; McKinnell, R.G. Genome sequences of two frog herpesviruses. J. Gen. Virol. 2006, 87, 3509–3514. [Google Scholar] [CrossRef]
- Sim, R.R.; Norton, T.M.; Bronson, E.; Allender, M.C.; Stedman, N.; Childress, A.L.; Wellehan, J.F.X. Identification of a novel herpesvirus in captive Eastern box turtles (Terrapene carolina carolina). Vet. Microbiol. 2015, 175, 218–223. [Google Scholar] [CrossRef]
- Davison, A.J.; Wright, K.M.; Harrach, B. DNA sequence of frog adenovirus. J. Gen. Virol. 2000, 81 Pt 10, 2431–2439. [Google Scholar] [CrossRef]
- Kovács, G.M.; LaPatra, S.E.; D’Halluin, J.C.; Benko, M. Phylogenetic analysis of the hexon and protease genes of a fish adenovirus isolated from white sturgeon (Acipenser transmontanus) supports the proposal for a new adenovirus genus. Virus Res. 2003, 98, 27–34. [Google Scholar] [CrossRef]
- Farkas, S.L.; Benkő, M.; Élő, P.; Ursu, K.; Dán, Á.; Ahne, W.; Harrach, B. Genomic and phylogenetic analyses of an adenovirus isolated from a corn snake (Elaphe guttata) imply a common origin with members of the proposed new genus Atadenovirus. J. Gen. Virol. 2002, 83 Pt 10, 2403. [Google Scholar] [CrossRef]
- López-Bueno, A.; Mavian, C.; Labella, A.M.; Castro, D.; Borrego, J.J.; Alcami, A.; Alejo, A. Concurrence of Iridovirus, Polyomavirus, and a Unique Member of a New Group of Fish papillomaviruses in Lymphocystis Disease-Affected Gilthead Sea Bream. J. Virol. 2016, 90, 8768–8779. [Google Scholar] [CrossRef] [Green Version]
- Herbst, L.H.; Lenz, J.; Doorslaer, K.V.; Chen, Z.; Stacy, B.A.; Wellehan, J.F.X.; Manire, C.A.; Burk, R.D. Genomic characterization of two novel reptilian papillomaviruses, Chelonia mydas papillomavirus 1 and Caretta caretta papillomavirus 1. Virology 2009, 383, 131–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, C.M.; Chen, M.M.; Wang, C.S.; Liu, P.C.; Nan, F.H. Isolation of a novel polyomavirus, related to Japanese eel endothelial cell-infecting virus, from marbled eels, Anguilla marmorata (Quoy & Gaimard). J. Fish Dis. 2016, 39, 889–897. [Google Scholar] [PubMed]
- Cooper, J.E.; Gschmeissner, S.; Holt, P.E. Viral particles in a papilloma from a green lizard (Lacerta viridis). Lab. Anim. 1982, 16, 12–13. [Google Scholar] [CrossRef] [Green Version]
- Kollinger, G.; Schwab, M.; Anders, F. Virus-like particles induced by bromodeoxyuridine in melanoma and neuroblastoma of Xiphophorus. J. Cancer Res. Clin. Oncol. 1979, 95, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Tarján, Z.; Pénzes, J.J.; Tóth, R.P.; Benkő, M. First detection of circovirus-like sequences in amphibians and novel putative circoviruses in fishes. Acta Vet. Hung. 2014, 62, 134–144. [Google Scholar] [CrossRef] [Green Version]
- McGeoch, D.J.; Rixon, F.J.; Davison, A.J. Topics in herpesvirus genomics and evolution. Virus Res. 2006, 117, 90–104. [Google Scholar] [CrossRef]
- Ssbauer, S.E.; Hne, W.A. Viruses of Lower Vertebrates. J. Vet. Med. Ser. B 2001, 48, 403–475. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O.; Rannala, B. A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Conow, C.; Fielder, D.; Ovadia, Y.; Libeskind-Hadas, R. Jane: A new tool for the cophylogeny reconstruction problem. Algorithms Mol. Biol. 2010, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Origgi, F.C.; Schmidt, B.R.; Lohmann, P.; Otten, P.; Akdesir, E.; Gaschen, V.; Aguilar-Bultet, L.; Wahli, T.; Sattler, U.; Stoffel, M.H. Ranid Herpesvirus 3 and Proliferative Dermatitis in Free-Ranging Wild Common Frogs (Rana Temporaria). Vet. Pathol. 2017, 54, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Doszpoly, A.; Tarján, Z.L.; Glávits, R.; Müller, T.; Benkő, M. Full genome sequence of a novel circo-like virus detected in an adult European eel Anguilla anguilla showing signs of cauliflower disease. Dis. Aquat. Org. 2014, 109, 107–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borzák, R.; Sellyei, B.; Székely, C.; Doszpoly, A. Molecular detection and genome analysis of circoviruses of European eel (Anguilla anguilla) and sichel (Pelecus cultratus). Acta Vet. Hung. 2017, 65, 262. [Google Scholar] [CrossRef] [Green Version]
- Lorincz, M.; Cságola, A.; Farkas, S.L.; Székely, C.; Tuboly, T. First detection and analysis of a fish circovirus. J. Gen. Virol. 2011, 92 Pt 8, 1817–1821. [Google Scholar] [CrossRef]
- Lőrincz, M.; Dán, A.; Láng, M.; Csaba, G.; Tóth, A.G.; Székely, C.; Cságola, A.; Tuboly, T. Novel circovirus in European catfish (Silurus glanis). Arch. Virol. 2012, 157, 1173–1176. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, J.H. Family-Hepadnaviridae. In Virus Taxonomy, in Encyclopedia of Virology, 4th ed.; Bamford, D.H., Zuckerman, M., Eds.; Academic Press: Oxford, UK, 2021; pp. 28–37. [Google Scholar]
- Wei, X.; Chen, Y.; Duan, G.; Holmes, E.C.; Cui, J. A reptilian endogenous foamy virus sheds light on the early evolution of retroviruses. Virus Evol. 2019, 5, vez001. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhang, Y.Y.; Wei, X.; Cui, J. Multiple infiltration and cross-species transmission of foamy viruses across Paleozoic to Cenozoic era. J. Virol. 2021. [Google Scholar] [CrossRef]
- Gebremedhn, H.; Deboutte, W.; Schoonvaere, K.; Demaeght, P.; Smet, L.D.; Amssalu, B.; Matthijnssens, J.; Graaf, D.C.D. Metagenomic Approach with the NetoVIR Enrichment Protocol Reveals Virus Diversity within Ethiopian Honey Bees (Apis mellifera simensis). Viruses 2020, 12, 1218. [Google Scholar] [CrossRef]
- Conceição-Neto, N.; Zeller, M.; Lefrère, H.; Bruyn, P.D.; Beller, L.; Deboutte, W.; Yinda, C.K.; Lavigne, R.; Maes, P.; Ranst, M.V.; et al. Modular approach to customise sample preparation procedures for viral metagenomics: A reproducible protocol for virome analysis. Sci. Rep. 2015, 5, 16532. [Google Scholar] [CrossRef] [Green Version]
- Hall, R.J.; Wang, J.; Todd, A.K.; Bissielo, A.B.; Yen, S.; Strydom, H.; Moore, N.E.; Ren, X.; Huang, Q.S.; Carter, P.E.; et al. Evaluation of rapid and simple techniques for the enrichment of viruses prior to metagenomic virus discovery. J. Virol. Methods 2014, 195, 194–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djikeng, A.; Halpin, R.; Kuzmickas, R.; Depasse, J.; Feldblyum, J.; Sengamalay, N.; Afonso, C.; Zhang, X.; Anderson, N.G.; Ghedin, E.; et al. Viral genome sequencing by random priming methods. BMC Genom. 2008, 9, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, R.; Shi, M.; Grueber, C.E.; Holmes, E.C.; Hogg, C.J.; Belov, K.; Barrs, V.R. Fecal Viral Diversity of Captive and Wild Tasmanian Devils Characterized Using Virion-Enriched Metagenomics and Metatranscriptomics. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asplund, M.; Kjartansdóttir, K.R.; Mollerup, S.; Vinner, L.; Fridholm, H.; Herrera, J.A.R.; Friis-Nielsen, J.; Hansen, T.A.; Jensen, R.H.; Nielsen, I.B.; et al. Contaminating viral sequences in high-throughput sequencing viromics: A linkage study of 700 sequencing libraries. Clin. Microbiol. Infect. 2019, 25, 1277–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, E.C. Reagent contamination in viromics: All that glitters is not gold. Clin. Microbiol. Infect. 2019, 25, 1167–1168. [Google Scholar] [CrossRef] [Green Version]
- Katzourakis, A.; Gifford, R.J. Endogenous viral elements in animal genomes. PLoS Genet. 2010, 6, e1001191. [Google Scholar] [CrossRef] [PubMed]
- Origgi, F.C.; Schmidt, B.R.; Lohmann, P.; Otten, P.; Meier, R.K.; Pisano, S.R.R.; Moore-Jones, G.; Tecilla, M.; Sattler, U.; Wahli, T.; et al. Bufonid herpesvirus 1 (BfHV1) associated dermatitis and mortality in free ranging common toads (Bufo bufo) in Switzerland. Sci. Rep. 2018, 8, 14737. [Google Scholar] [CrossRef]
- Lucke, B. A neoplastic disease of the kidney of the frog, Rana pipiens. Am. J. Cancer 1934, 20, 352–379. [Google Scholar] [CrossRef] [Green Version]
- Tweedell, K.S.; Wong, W.Y. Frog kidney tumors induced by herpesvirus cultured in pronephric cells. J. Natl. Cancer Inst. 1974, 52, 621–624. [Google Scholar] [CrossRef]
- Tweedell, K.S. Induced oncogenesis in developing frog kidney cells. Cancer Res. 1967, 27, 2040–2052. [Google Scholar]
- Lucké, B. Carcinoma in the leopard frog: Its probable causation by a virus. J. Exp. Med. 1938, 68, 457–468. [Google Scholar] [CrossRef]
- Naegele, R.F.; Granoff, A.; Darlington, R.W. The presence of the Lucke herpesvirus genome in induced tadpole tumors and its oncogenicity: Koch-Henle postulates fulfilled. Proc. Natl. Acad. Sci. USA 1974, 71, 830–834. [Google Scholar] [CrossRef] [Green Version]
- Rafferty, K.A. The cultivation of inclusion-associated viruses from lucke tumor frogs. Ann. N. Y. Acad. Sci. 1965, 126, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Tisza, M.J.; Pastrana, D.V.; Welch, N.L.; Stewart, B.; Peretti, A.; Starrett, G.J.; Pang, Y.S.; Krishnamurthy, S.R.; Pesavento, P.A.; McDermott, D.H.; et al. Discovery of several thousand highly diverse circular DNA viruses. Elife 2020, 9, e51971. [Google Scholar] [CrossRef] [PubMed]
- Pénzes, J.J.; de Souza, W.M.; Agbandje-McKenna, M.; Gifford, R.J. An Ancient Lineage of Highly Divergent Parvoviruses Infects both Vertebrate and Invertebrate Hosts. Viruses 2019, 11, 525. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.-X.; Wu, W.-C.; Shi, M. Discovery and Characterization of Actively Replicating DNA and Retro-Transcribing Viruses in Lower Vertebrate Hosts Based on RNA Sequencing. Viruses 2021, 13, 1042. https://doi.org/10.3390/v13061042
Chen X-X, Wu W-C, Shi M. Discovery and Characterization of Actively Replicating DNA and Retro-Transcribing Viruses in Lower Vertebrate Hosts Based on RNA Sequencing. Viruses. 2021; 13(6):1042. https://doi.org/10.3390/v13061042
Chicago/Turabian StyleChen, Xin-Xin, Wei-Chen Wu, and Mang Shi. 2021. "Discovery and Characterization of Actively Replicating DNA and Retro-Transcribing Viruses in Lower Vertebrate Hosts Based on RNA Sequencing" Viruses 13, no. 6: 1042. https://doi.org/10.3390/v13061042
APA StyleChen, X.-X., Wu, W.-C., & Shi, M. (2021). Discovery and Characterization of Actively Replicating DNA and Retro-Transcribing Viruses in Lower Vertebrate Hosts Based on RNA Sequencing. Viruses, 13(6), 1042. https://doi.org/10.3390/v13061042