
Evolutionary Relationships of Ljungan Virus Variants Circulating in Multi-Host Systems across Europe

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
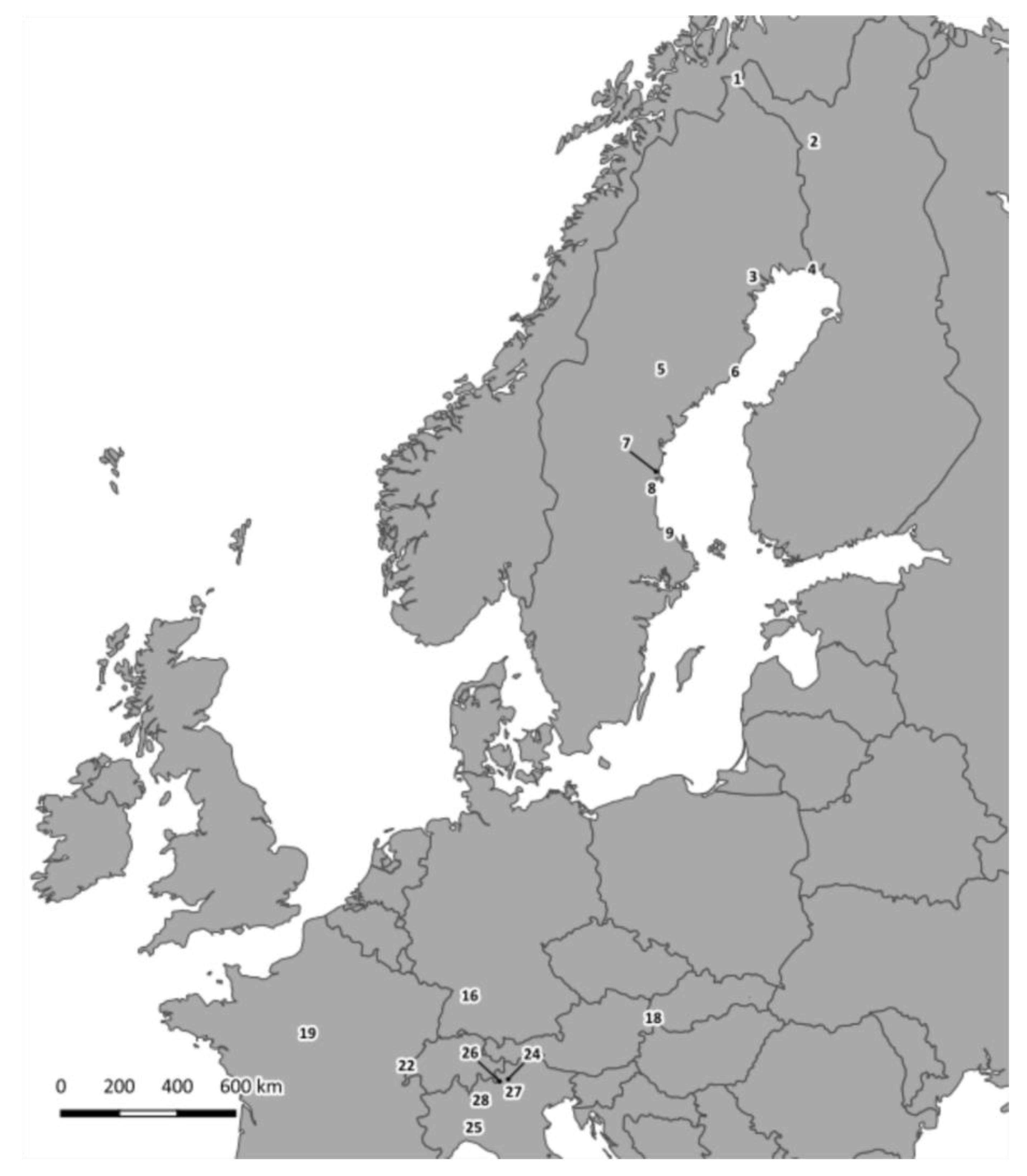
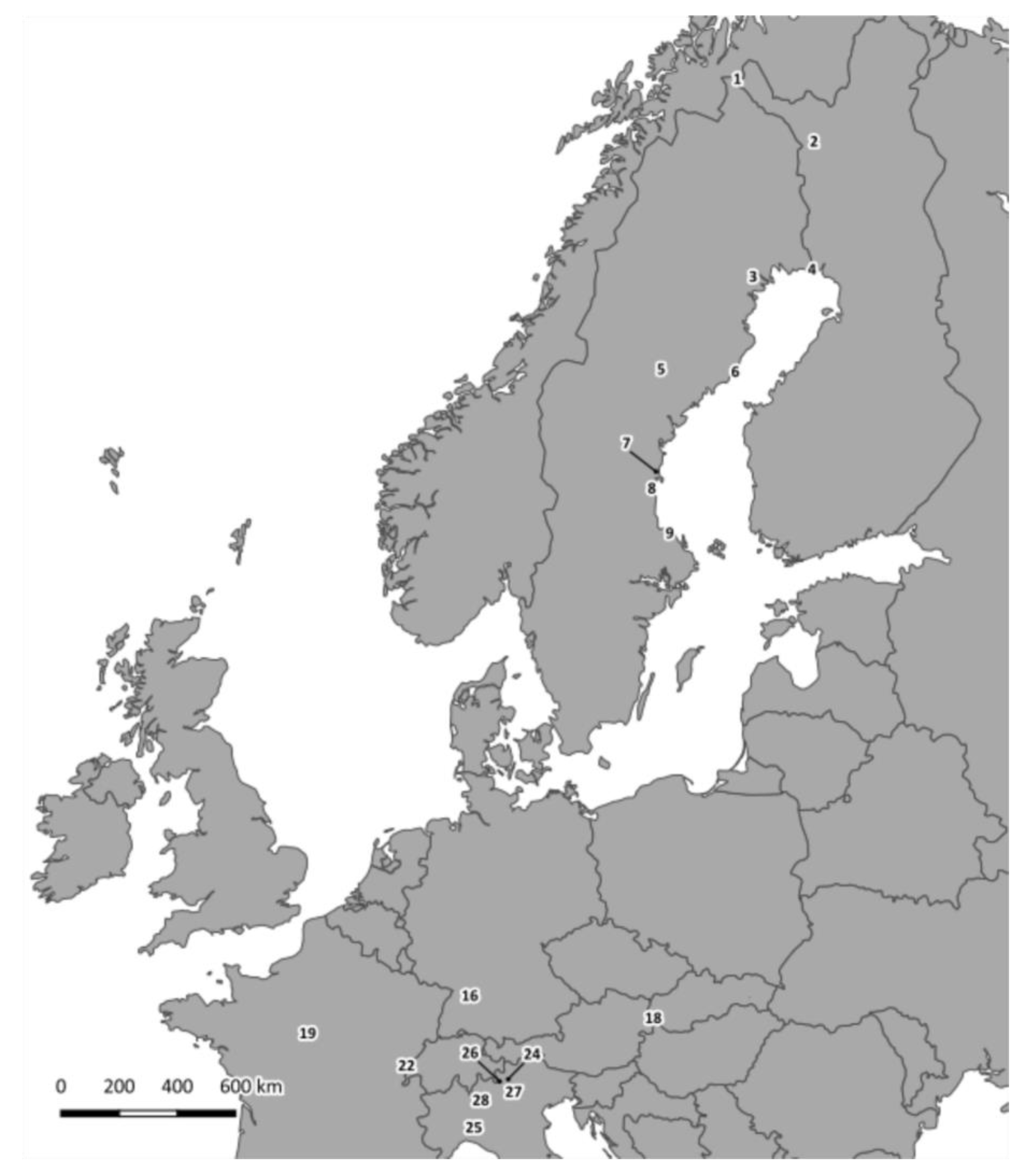
2.1. Sample Collection
2.2. RNA Extraction, Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and Sequencing
2.3. Genotyping Using the VP1 Region
2.4. Analysis of Potential Selection on VP1 Region
2.5. Phylogenetic Analysis of the 3Dpol Region
2.6. Network Analysis Using the 5′-UTR
3. Results
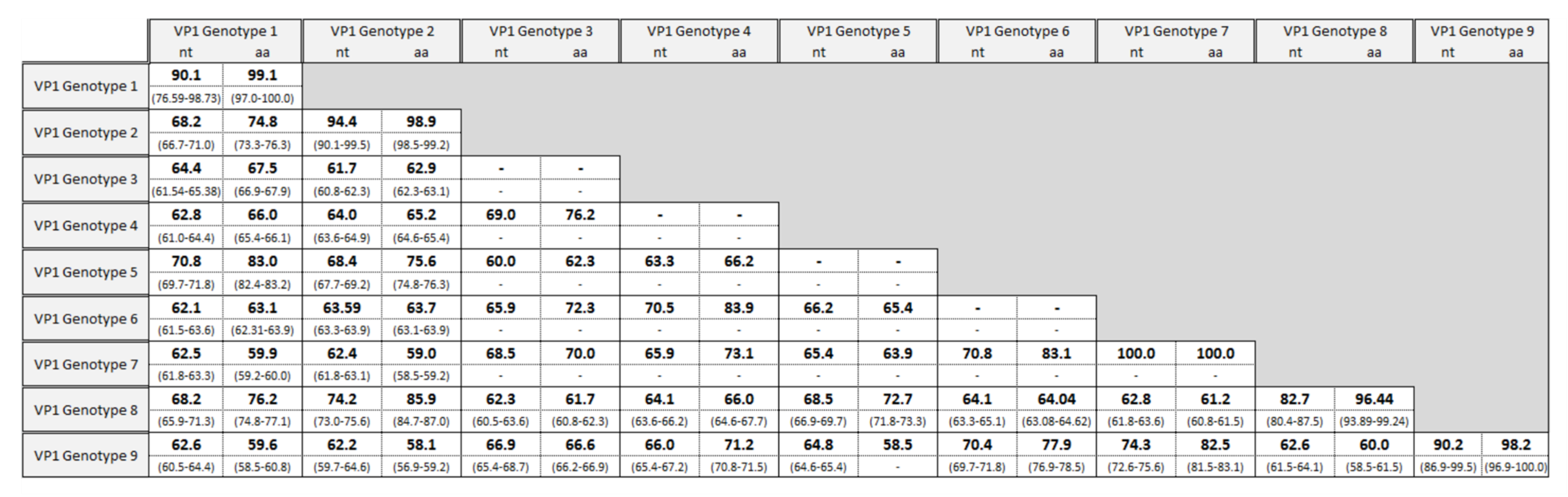
3.1. Genotyping Using the VP1 Region
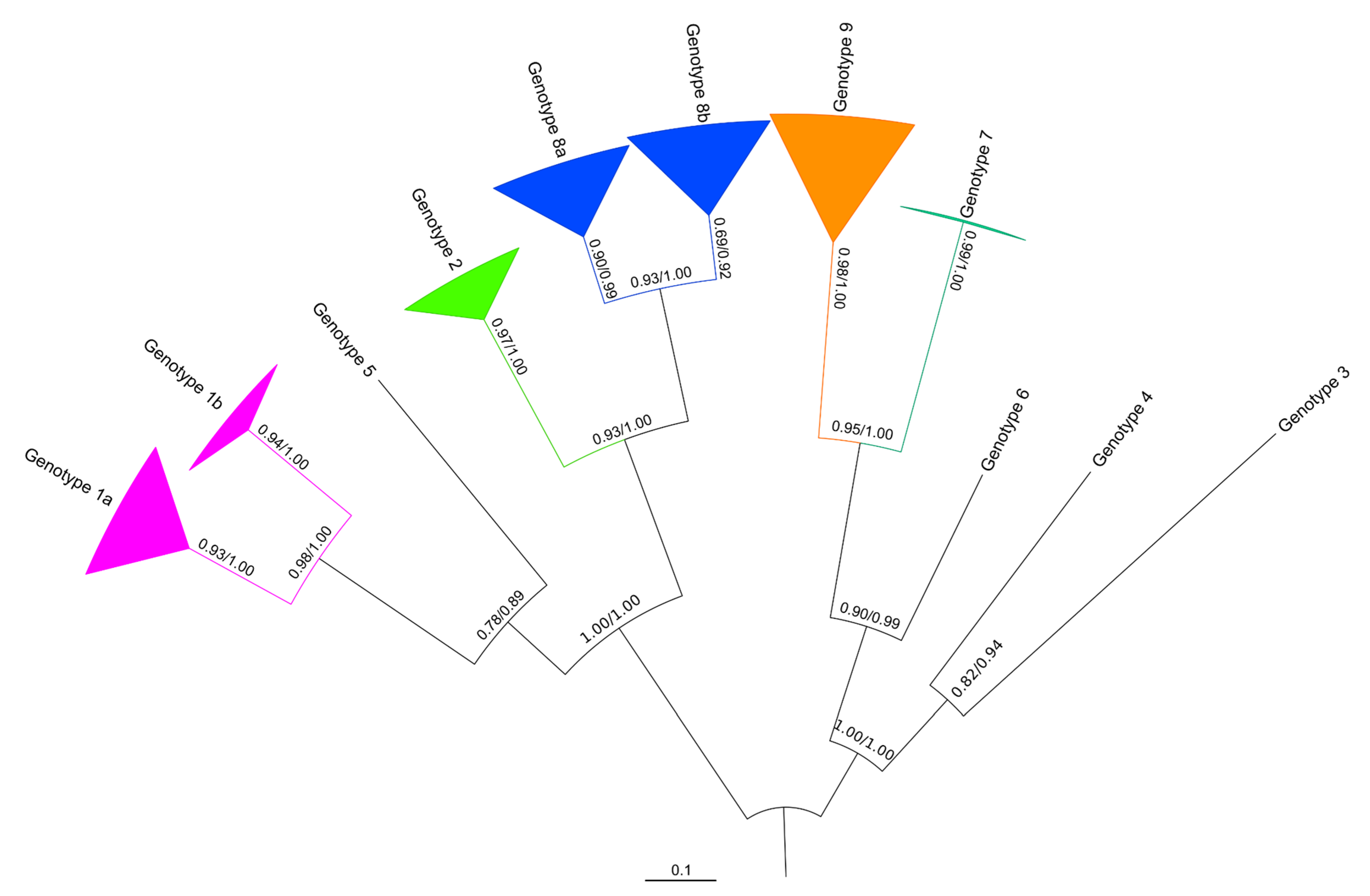
3.2. Phylogeny of the VP1 Region
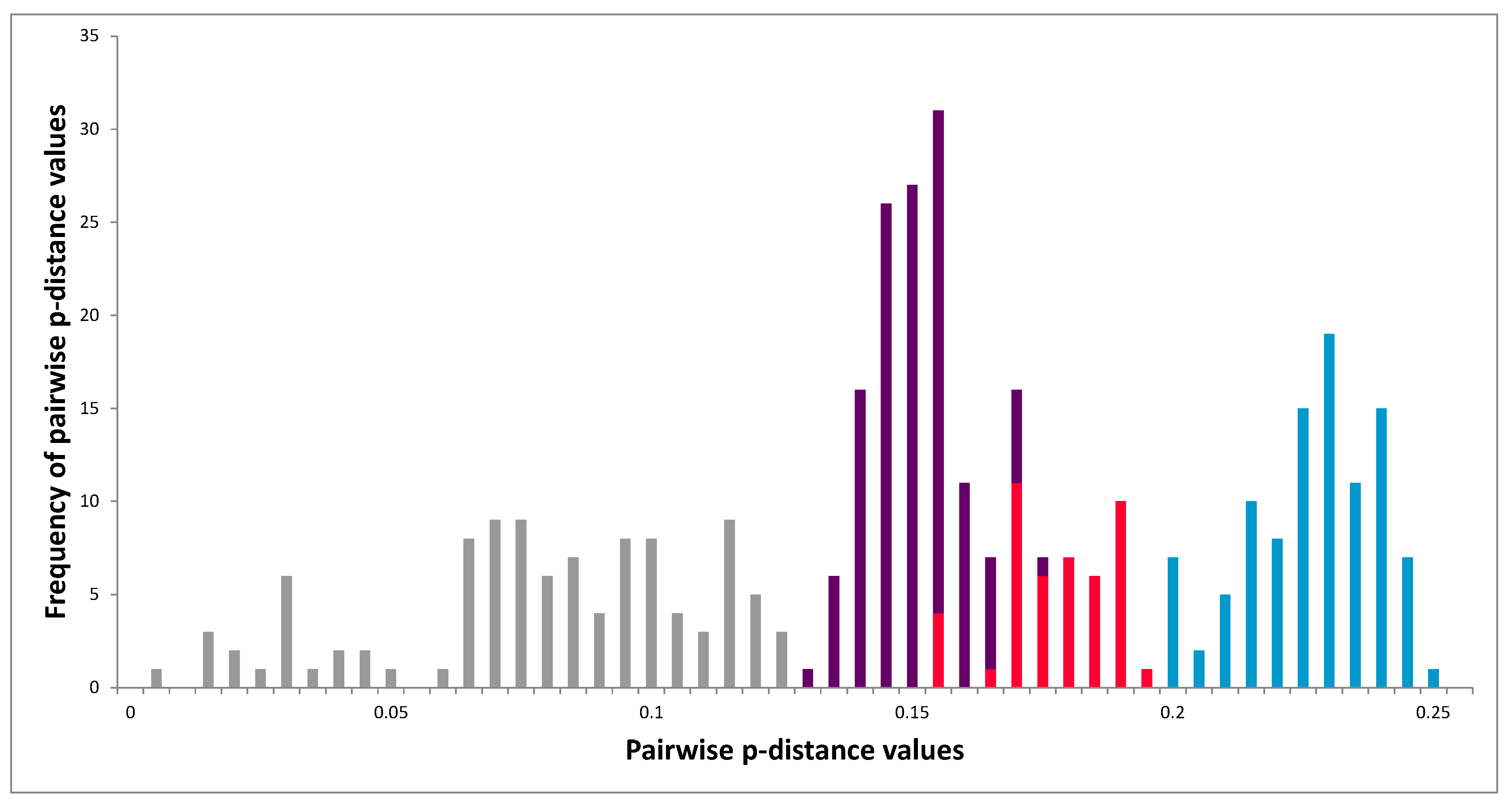
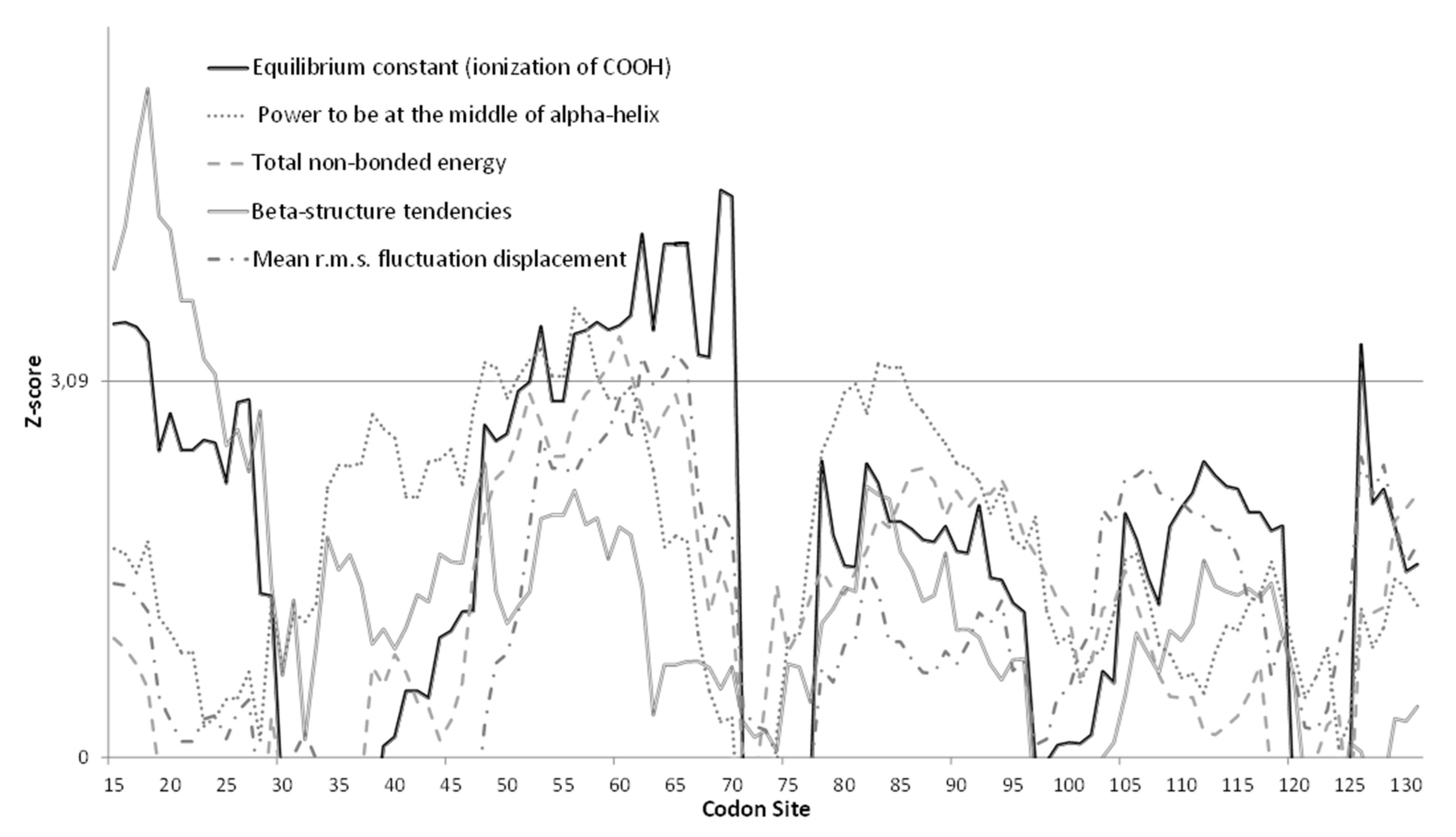
3.3. Molecular Analysis of VP1 Sequences
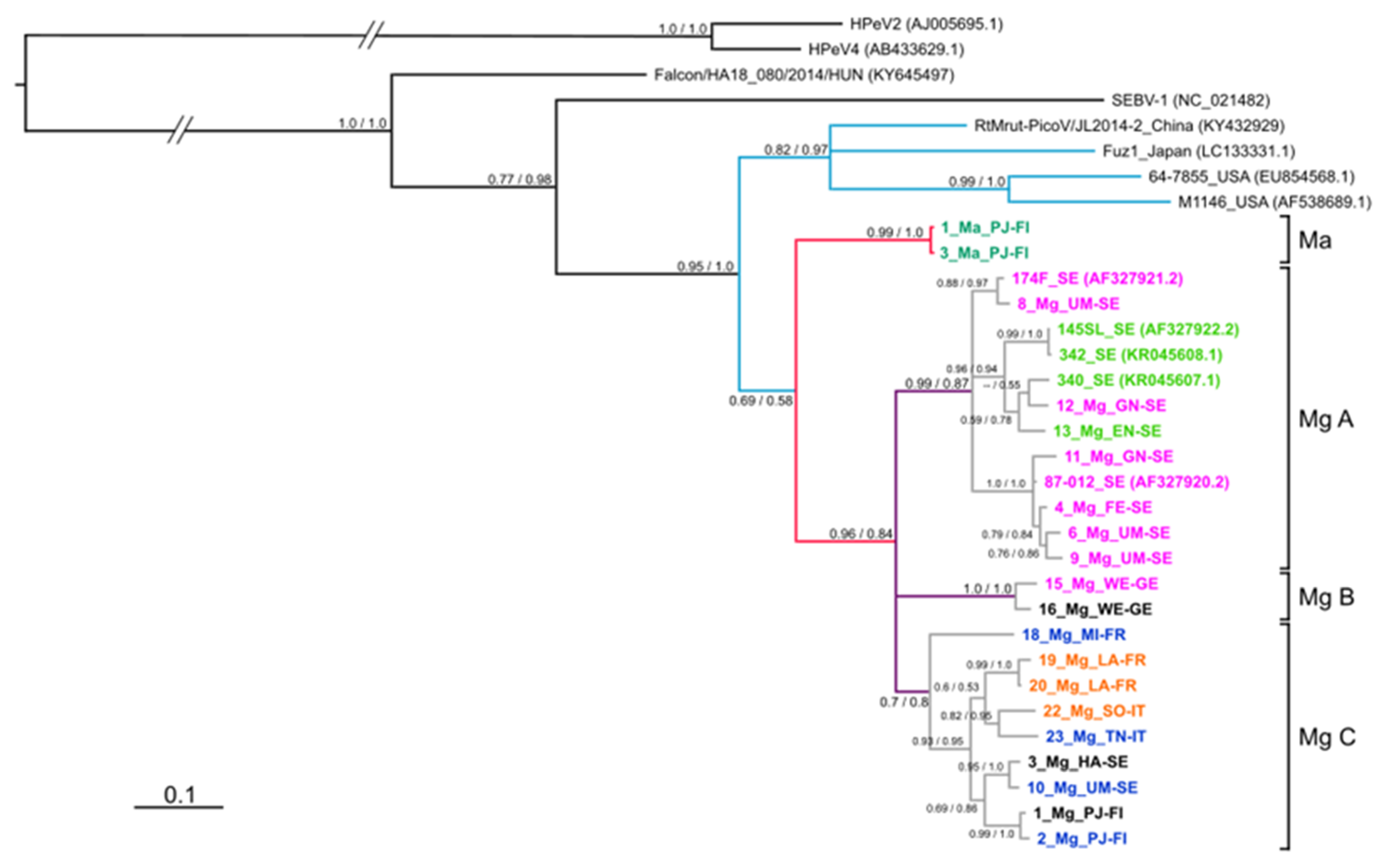
3.4. Phylogeny of 3Dpol Region
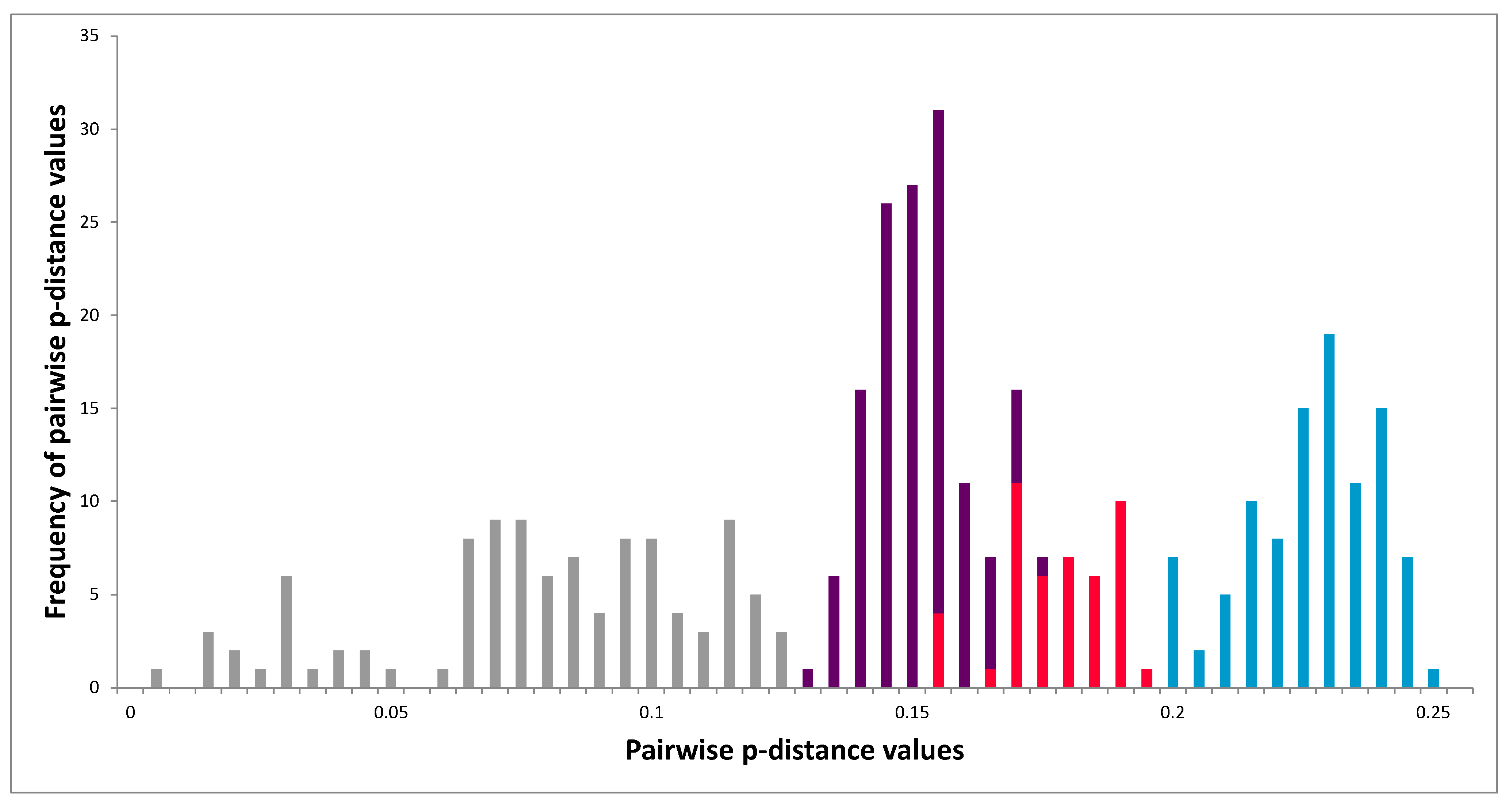
3.5. Molecular Analysis of 3Dpol Region
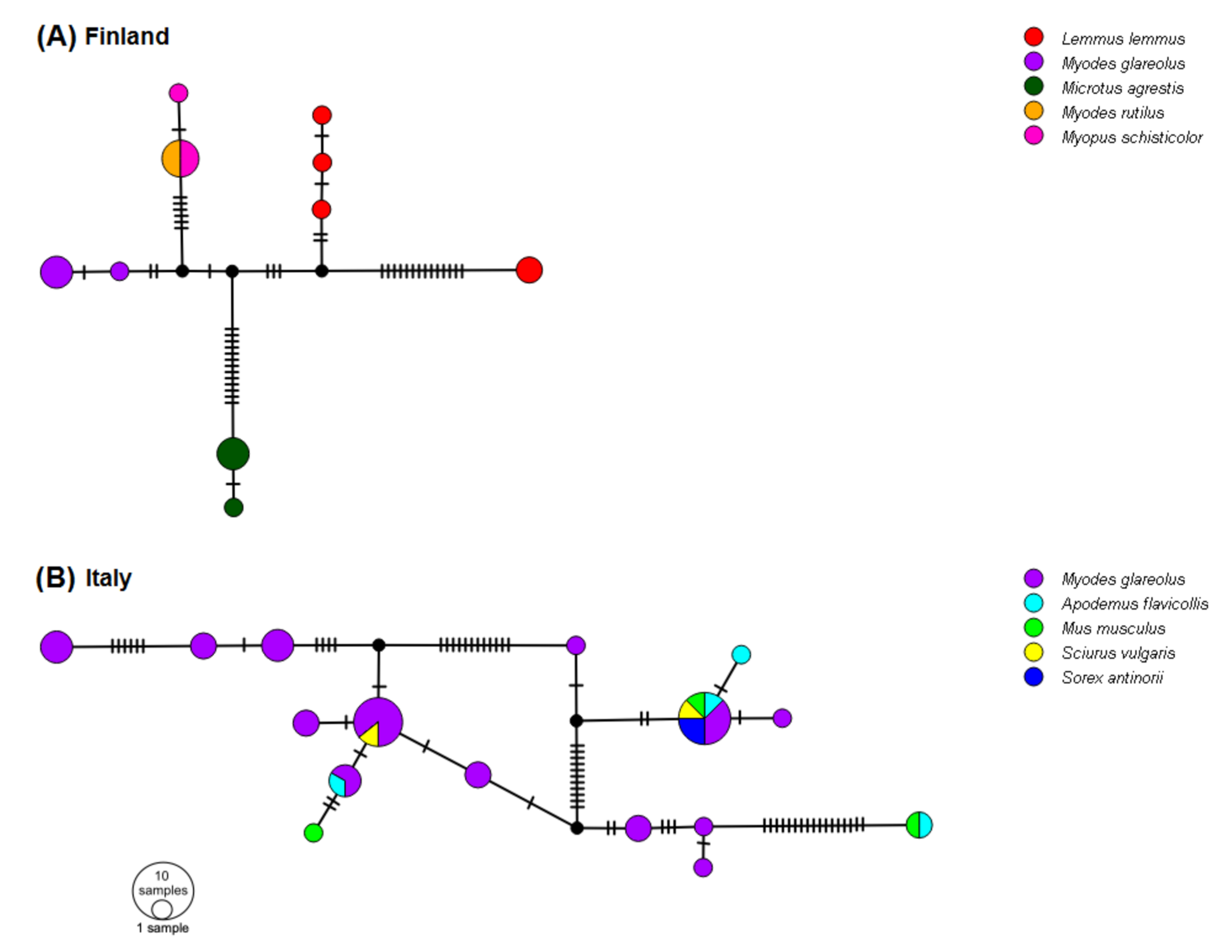
3.6. Networks of 5′-UTR Haplotypes
4. Discussion
4.1. LV Phylogeny and Evolution
4.2. Host Specificity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mills, J.N.; Fulhorst, C.F. Small Mammal-Associated Zoonoses. Vector-Borne Zoonotic Dis. 2010, 10, 547. [Google Scholar] [CrossRef]
- Han, B.A.; Schmidt, J.P.; Bowden, S.E.; Drake, J. Rodent reservoirs of future zoonotic diseases. Proc. Natl. Acad. Sci. USA 2015, 112, 7039–7044. [Google Scholar] [CrossRef] [Green Version]
- Hansen, A.; Xiang, J.; Liu, Q.; Tong, M.X.; Sun, Y.; Liu, X.; Chen, K.; Cameron, S.; Hanson-Easey, S.; Han, G.-S.; et al. Experts’ Perceptions on China’s Capacity to Manage Emerging and Re-emerging Zoonotic Diseases in an Era of Climate Change. Zoonoses Public Health 2016, 201664, 527–536. [Google Scholar] [CrossRef]
- Lankau, E.W.; Sinclair, J.R.; Schroeder, B.A.; Galland, G.G.; Marano, N. Public Health Implications of Changing Rodent Im-portation Patterns United States, 1999–2013. Transbound Emerg. Dis. 2017, 64, 528–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, K.; Dressel, K.M.; Niedrig, M.; Mertens, M.; Schüle, S.A.; Groschup, M.H. Public Health and Vector-Borne Diseases A New Concept for Risk Governance. Zoonoses Public Health 2013, 60, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Nii-Trebi, N.I. Emerging and Neglected Infectious Diseases: Insights, Advances, and Challenges. Biomed. Res. Int. 2017, 2017, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Reuter, G.; Boros, Á.; Tóth, Z.; Gia Phan, T.; Delwart, E.; Pankovics, P. A highly divergent picornavirus in an amphibian, the smooth newt (Lissotriton vulgaris). J. Gen. Virol. 2015, 96, 2607–2613. [Google Scholar] [CrossRef] [Green Version]
- Barbknecht, M.; Sepsenwol, E.S.; Leis, M.; Tuttle-Lau, M.; Gaikowski, N.J.; Knowles, B.; Lasee, B.; Hoffman, M.A. Characteri-zation of a new picornavirus isolated from the freshwater fish Lepomis macrochirus. J. Gen. Virol. 2014, 95, 601–613. [Google Scholar] [CrossRef] [Green Version]
- Mayo, M.A.; Pringle, C.R. Virus taxonomy 1997. J. Gen. Virol. 1998, 79, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Rogers, N. Evolution of Picornaviruses: Impacts of Recombination and Selection. Ph.D. Thesis, Brigham Young University, Provo, UT, USA, 2008. [Google Scholar]
- Lewis-Rogers, N.; Crandall, K.A. Evolution of Picornaviridae: An examination of phylogenetic relationships and cophylogeny. Mol. Phylogenetics Evol. 2010, 54, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Niklasson, B.; Hörnfeldt, B.; Lundman, B. Could myocarditis, insulin-dependent diabetes mellitus, and Guillain–Barré syn-drome be caused by one or more infectious agents carried by rodents? Emerg. Infect. Dis. 1998, 4, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Niklasson, B.; Samsioe, A.; Papadogiannakis, N.; Kawecki, A.; Hörnfeldt, B.; Saade, G.R.; Klitz, W. Association of zoonotic Ljungan virus with intrauterine fetal deaths. Birth Defects Res. Part A Clin. Mol. Teratol. 2007, 79, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Niklasson, B.; Samsioe, A.; Papadogiannakis, N.; Gustafsson, S.; Klitz, W. Zoonotic Ljungan virus associated with central nervous system malformations in terminated pregnancy. Birth Defects Res. Part A Clin. Mol. Teratol. 2009, 85, 542–545. [Google Scholar] [CrossRef]
- Krous, H.F.; Langlois, N.E. Ljungan virus: A commentary on its association with fetal and infant morbidity and mortality in animals and humans. Birth Defects Res. Part A Clin. Mol. Teratol. 2010, 88, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Tapia, G.; Cinek, O.; Rasmussen, T.; Grinde, B.; Rønningen, K.S. No Ljungan Virus RNA in Stool Samples from the Norwegian Environmental Triggers of Type 1 Diabetes (MIDIA) Cohort Study. Diabetes Care 2010, 33, 1069–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Wang, F.; Huang, J.; Xin, H. Evaluation of the association of zoonotic Ljungan virus with perinatal deaths and fetal malformation. Birth Defects Res. Part C Embryo Today Rev. 2015, 105, 81–85. [Google Scholar] [CrossRef]
- Jääskeläinen, A.J.; Nurminen, N.; Kolehmainen, P.; Smura, T.; Tauriainen, S.; Toppari, J.; Ilonen, J.; Veijola, R.; Knip, M.; Hyöty, H.; et al. No Association between Ljungan Virus Seropositivity and the Beta-cell Damaging Process in the Finnish Type 1 Di-abetes Prediction and Prevention Study Cohort. Pediatr. Infect. Dis. J. 2019, 38, 314–316. [Google Scholar] [CrossRef] [PubMed]
- Lundstig, A.; McDonald, S.L.; Maziarz, M.; Weldon, W.C.; Vaziri-Sani, F.; Lernmark, Å.; Nilsson, A.-L. Neutralizing Ljungan virus antibodies in children with newly diagnosed type 1 diabetes. J. Gen. Virol. 2021, 102, 001602. [Google Scholar] [CrossRef] [PubMed]
- Niklasson, B.; Kinnunen, L.; Hörnfeldt, B.; Hörling, J.; Benemar, C.; Hedlund, K.O.; Matskova, L.; Hyypiä, T.; Winberg, G. A New Picornavirus Isolated from Bank Voles (Clethrionomys glareolus). Virology 1999, 255, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Jääskeläinen, A.J.; Kolehmainen, P.; Voutilainen, L.; Hauffe, H.C.; Kallio-Kokko, H.; Lappalainen, M.; Tolf, C.; Lindberg, A.M.; Henttonen, H.; Vaheri, A.; et al. Evidence of Ljungan virus specific antibodies in humans and rodents, Finland. J. Med. Virol. 2013, 85, 2001–2008. [Google Scholar] [CrossRef]
- Jääskeläinen, A.J.; Voutilainen, L.; Lehmusto, R.; Henttonen, H.; Lappalainen, M.; Kallio-Kokko, H.; Vaheri, A.; Vapalahti, O. Serological survey in the Finnish human population implies human-to-human transmission of Ljungan virus or antigenically related viruses. Epidemiol. Infect. 2015, 144, 1278–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fevola, C.; Forbes, K.M.; Mäkelä, S.; Putkuri, N.; Hauffe, H.C.; Kallio-Kokko, H.; Mustonen, J.; Jääskeläinen, A.J.; Vaheri, A. Lymphocytic choriomeningitis, Ljungan and orthopoxvirus seroconversions in patients hospitalized due to acute Puumala hantavirus infection. J. Clin. Virol. 2016, 84, 48–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fevola, C.; Kuivanen, S.; Smura, T.; Vaheri, A.; Kallio-Kokko, H.; Hauffe, H.C.; Vapalahti, O.; Jääskeläinen, A.J. Seropreva-lence of lymphocytic choriomeningitis virus and Ljungan virus in Finnish patients with suspected neurological infections. J. Med. Virol. 2018, 90, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Fevola, C.; Rossi, C.; Rosso, F.; Girardi, M.; Rosà, R.; Manica, M.; Delucchi, L.; Rocchini, D.; Garzon-Lopez, C.X.; Arnoldi, D.; et al. Geographical Distribution of Ljungan Virus in Small Mammals in Europe. Vector-Borne Zoonotic Dis. 2020, 20, 692–782. [Google Scholar] [CrossRef] [PubMed]
- Niklasson, B.; Höornfeldt, B.; Nyholm, E.; Niedrig, M.; Donoso-Mantke, P.; Gelderblom, H.R.; Lermark, A. Type 1 diabetes in Swedish bank voles: Signs of disease in both colonized and wild cyclic populations at peak density. Ann. N. Y. Acad. Sci. 2003, 1005, 170–175. [Google Scholar] [CrossRef]
- Niklasson, B.; Heller, K.E.; Schønecker, B.; Bildsøe, M.; Daniels, T.; Hampe, C.; Widlund, P.; Simonson, W.T.; Schaefer, J.B.; Rutledge, E.; et al. Development of Type 1 Diabetes in Wild Bank Voles Associated with Islet Autoantibodies and the Novel Ljungan Virus. Exp. Diabesity Res. 2003, 4, 35–44. [Google Scholar] [CrossRef]
- Niklasson, B.; Nyholm, E.; Feinstein, R.E.; Samsioe, A.; Hörnfeldt, B. Diabetes and myocarditis in voles and lemmings at cyclic peak densities—induced by Ljungan virus? Oecologia 2006, 150, 1–7. [Google Scholar] [CrossRef]
- Samsioe, A.; Feinstein, R.; Saade, G.; Sjöholm, Å.; Hörnfeldt, B.; Fundele, R.; Klitz, W.; Niklasson, B. Intrauterine death, fetal malformation, and delayed pregnancy in Ljungan virus-infected mice. Birth Defects Res. Part B Dev. Reprod. Toxicol. 2006, 77, 251–256. [Google Scholar] [CrossRef]
- Hauffe, H.C.; Fevola, C.; Rossi, C.; Rizzoli, A.; Niemimaa, J.; Henttonen, H. Is rodent-borne Ljungan virus responsible for mortality in migrating Norwegian lemmings (Lemmus lemmus)? In INTERACT: International Network for Terrestrial Research and Monitoring in the Arctic. Stories of Arctic Science; Callaghan, T.V., Savela, H., Eds.; DCE Danish Centre for Environment and Energy, Aarhus University: Aarhus, Denmark, 2015; pp. 120–121. [Google Scholar]
- Tolf, C.; Gullberg, M.; Johansson, E.S.; Tesh, R.B.; Andersson, B.; Lindberg, A.M. Molecular characterization of a novel Ljungan virus (Parechovirus; Picornaviridae) reveals a fourth genotype and indicates ancestral recombination. J. Gen. Virol. 2009, 90, 843–853. [Google Scholar] [CrossRef]
- Pounder, K.; Watts, P.C.; Niklasson, B.; Kallio, E.R.; Marston, D.A.; Fooks, A.R.; Begon, M.; McElhinney, L.M. Genome characterisation of two Ljungan virus isolates from wild bank voles (Myodes glareolus) in Sweden. Infect. Genet. Evol. 2015, 36, 156–164. [Google Scholar] [CrossRef]
- Johansson, E.S.; Niklasson, B.; Tesh, R.B.; Shafren, D.R.; Da Rosa, A.P.A.T.; Lindberg, A.M. Molecular characterization of M1146, an American isolate of Ljungan virus (LV) reveals the presence of a new LV genotype. J. Gen. Virol. 2003, 84, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Mitake, H.; Fujii, Y.; Nagai, M.; Ito, N.; Okadera, K.; Okada, K.; Sakoda, Y. Isolation of a novel Ljungan virus from wild birds in Japan. J. Gen. Virol. 2016, 97, 1818–1822. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lu, L.; Du, J.; Yang, L.; Ren, X.; Liu, B.; Jiang, J.; Yang, J.; Dong, J.; Sun, L.; et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 2018, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Forbes, K.M.; Voutilainen, L.; Jääskeläinen, A.; Sironen, T.; Kinnunen, P.M.; Stuart, P.; Vapalahti, O.; Henttonen, H.; Huitu, O. Serological Survey of Rodent-Borne Viruses in Finnish Field Voles. Vector-Borne Zoonotic Dis. 2014, 14, 278–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvert, J.; Chieochansin, T.; Benschop, K.S.; Leitch, E.M.; Drexler, J.F.; Grywna, K.; da Costa Ribeiro, H., Jr.; Drosten, C.; Harvala, H.; Poovorawan, Y.; et al. Recombination dynamics of human Parechoviruses: Investigation of type-specific differ-ences in frequency and epidemiological correlates. J. Gen. Virol. 2010, 91, 1229–1238. [Google Scholar] [CrossRef]
- Hyypiä, T.; Auvinen, P.; Maaronen, M. Polymerase Chain Reaction for Human Picornaviruses. J. Gen. Virol. 1989, 70, 3261–3268. [Google Scholar] [CrossRef]
- Mantke, O.D.; Kallies, R.; Niklasson, B.; Nitsche, A.; Niedrig, M. A new quantitative real-time reverse transcriptase PCR assay and melting curve analysis for detection and genotyping of Ljungan virus strains. J. Virol. Methods 2007, 141, 71–77. [Google Scholar] [CrossRef]
- Benschop, K.; Molenkamp, R.; van der Ham, A.; Wolthers, K.; Beld, M. Rapid detection of human parechoviruses in clinical samples by real-time PCR. J. Clin. Virol. 2008, 41, 69–74. [Google Scholar] [CrossRef]
- Holtz, L.R.; Finkbeiner, S.R.; Kirkwood, C.D.; Wang, D. Identification of a novel picornavirus related to cosaviruses in a child with acute diarrhea. Virol. J. 2008, 5, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Fevola, C.; Rossi, C.; Rosà, R.; Nordström, Å.; Ecke, F.; Magnusson, M.; Miller, A.L.; Niemimaa, J.; Olsson, G.E.; Jääskeläinen, A.J.; et al. Distribution and Seasonal Variation of Ljungan Virus in Bank Voles (Myodes glareolus) in Fennoscandia. J. Wildl. Dis. 2017, 53, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Flemister, M.R.; Brown, B.A.; Pallansch, M.A. Typing of Human Enteroviruses by Partial Sequencing of VP1. J. Clin. Microbiol. 1999, 37, 1288–1293. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogeny. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar] [CrossRef] [Green Version]
- Pond, S.; Frost, S.D.W. Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Pond, S.L.K. Detecting Individual Sites Subject to Episodic Diversifying Selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delport, W.; Poon, A.; Frost, S.D.W.; Pond, S.L.K. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef] [Green Version]
- Woolley, S.; Johnson, J.; Smith, M.J.; Crandall, K.A.; McClellan, D.A. TreeSAAP: Selection on Amino Acid Properties using phylogenetic trees. Bioinformatics 2003, 19, 671–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.-H. Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J. Mol. Evol. 1993, 36, 96–99. [Google Scholar] [CrossRef]
- McClellan, D.A.; Palfreyman, E.J.; Smith, M.J.; Moss, J.L.; Christensen, R.G.; Sailsbery, J.K. Physicochemical Evolution and Molecular Adaptation of the Cetacean and Artiodactyl Cytochrome b Proteins. Mol. Biol. Evol. 2005, 22, 437–455. [Google Scholar] [CrossRef] [Green Version]
- Clement, M.J.; Snell, Q.; Walker, P.; Posada, D.; Crandall, K.A. TCS: Estimating gene genealogies. In Proceedings of the 16th International Parallel and Distributed Processing Symposium, Lauderdale, FL, USA, 15–19 April 2002; Volume 3, p. 184. [Google Scholar]
- Tapparel, C.; Junier, T.; Gerlach, D.; Van Belle, S.; Turin, L.; Cordey, S.; Mühlemann, K.; Regamey, N.; Aubert, J.-D.; Soccal, P.M.; et al. New Respiratory Enterovirus and Recombinant Rhinoviruses among Circulating Picornaviruses. Emerg. Infect. Dis. 2009, 15, 719–726. [Google Scholar] [CrossRef]
- Olijve, L.; Jennings, L.; Walls, T. Human Parechovirus: An Increasingly Recognized Cause of Sepsis-Like Illness in Young Infants. Clin. Microbiol. Rev. 2018, 31, e00047-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensen, T.; Belsham, G.J. Identification of a short, highly conserved, motif required for picornavirus capsid precursor processing at distal sites. PLoS Pathog. 2019, 15, e1007509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kok, C.C.; McMINN, P.C. Picornavirus RNA-dependent RNA polymerase. Int. J. Biochem. Cell Biol. 2009, 41, 498–502. [Google Scholar] [CrossRef]
- Holmes, E. The Evolutionary Genetics of Emerging Viruses. Annu. Rev. Ecol. Evol. Syst. 2009, 40, 353–372. [Google Scholar] [CrossRef]
- Usherwood, E.J.; Nash, A.A. Lymphocyte recognition of picornaviruses. J. Gen. Virol. 1995, 76, 499–508. [Google Scholar] [CrossRef]
- Benschop, K.S.M.; Williams, Ç.H.; Wolthers, K.C.; Stanway, G.; Simmonds, P. Widespread recombination within human parechoviruses: Analysis of temporal dynamics and constraints. J. Gen. Virol. 2008, 89, 1030–1035. [Google Scholar] [CrossRef]
- Lukashev, A.N. Recombination among picornaviruses. Rev. Med. Virol. 2010, 20, 327–337. [Google Scholar] [CrossRef]
- Drexler, J.F.; Grywna, K.; Lukashev, A.; Stöcker, A.; Almeida, P.S.; Wieseler, J.; Ribeiro, T.C.; Petersen, N.; da Costa Ribeiro, H., Jr.; Belalov, I.; et al. Full genome sequence analysis of parechoviruses from Brazil reveals geographical patterns in the evo-lution of non-structural genes and intratypic recombination in the capsid region. J. Gen. Virol. 2011, 92, 564–571. [Google Scholar] [CrossRef]
- Thoi, T.C.; Than, V.T.; Kim, W. Whole genomic characterization of a Korean human parechovirus type 1 (HPeV1) identifies recombination events. J. Med. Virol. 2014, 86, 2084–2091. [Google Scholar] [CrossRef]
- Zhao, X.; Shi, Y.; Xia, Y. Genome analysis revealed novel genotypes and recombination of the human parechoviruses prevalent in children in Eastern China. Gut Pathog. 2016, 8, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Alexandersen, S.; Nelson, T.M.; Hodge, J.; Druce, J. Evolutionary and network analysis of virus sequences from infants infected with an Australian recombinant strain of human parechovirus type 3. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Williams, Ç.H.; Panayiotou, M.; Girling, G.D.; Peard, C.I.; Oikarinen, S.; Hyoty, H.; Stanway, G. Evolution and conservation in human parechovirus genomes. J. Gen. Virol. 2009, 90, 1702–1712. [Google Scholar] [CrossRef]
- Zoll, J.; Galama, J.M.D.; van Kuppeveld, F.J.M. Identification of Potential Recombination Breakpoints in Human Parechoviruses. J. Virol. 2009, 83, 3379–3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benschop, K.S.M.; De Vries, M.; Minnaar, R.P.; Stanway, G.; Van Der Hoek, L.; Wolthers, K.C.; Simmonds, P. Comprehensive full-length sequence analyses of human parechoviruses: Diversity and recombination. J. Gen. Virol. 2009, 91, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Hauffe, H.C.; Niklasson, B.; Olsson, T.; Bianchi, A.; Rizzoli, A.; Klitz, W. Ljungan Virus Detected in Bank Voles (Myodes glareolus) and Yellow-Necked Mice (Apodemus flavicollis) from Northern Italy. J. Wildl. Dis. 2010, 46, 262–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salisbury, A.-M.; Begon, M.; Dove, W.; Niklasson, B.; Stewart, J.P. Ljungan virus is endemic in rodents in the UK. Arch. Virol. 2013, 159, 547–551. [Google Scholar] [CrossRef]
- Hansen, T.F.; Stenseth, N.C.; Henttonen, H.; Tast, J. Interspecific and intraspecific competition as causes of direct and delayed density dependence in a fluctuating vole population. Proc. Natl. Acad. Sci. USA 1999, 96, 986–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longdon, B.; Hadfield, J.D.; Webster, C.L.; Obbard, D.J.; Jiggins, F.M. Host phylogeny determines viral persistence and repli-cation in novel hosts. PLoS Pathog. 2011, 7, e1002260. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.; Yan, Q.; He, S.; Zhuang, S.; Niu, J.; Xia, N. Specific primer amplification of the VP1 region directed by 5′ UTR sequence analysis: Enterovirus testing and identification in clinical samples from hand-foot-and-mouth disease patients. J. Virol. Methods 2013, 193, 463–469. [Google Scholar] [CrossRef]
- Hörling, J.; Lundkvist, Å.; Jaarola, M.; Plyusnin, A.; Tegelström, H.; Persson, K.; Lehväslaiho, H.; Hörnfeldt, B.; Vaheri, A.; Niklasson, B. Distribution and genetic heterogeneity of Puumala virus in Sweden. J. Gen. Virol. 1996, 77, 2555–2562. [Google Scholar] [CrossRef]
- Jaarola, M.; Tegelströom, H.; Fredga, K. Colonization history in Fennoscandian rodents. Biol. J. Linn. Soc. 1999, 68, 113–127. [Google Scholar] [CrossRef]
- Plyusnin, A.; Vapalahti, O.; Lehväslaiho, H.; Apekina, N.; Mikhailova, T.; Gavrilovskaya, I.; Laakkonen, J.; Niemimaa, J.; Henttonen, H.; Brummer-Korvenkontio, M.; et al. Genetic variation of wild Puumala viruses within the serotype, local rodent populations and individual animal. Virus Res. 1995, 38, 25–41. [Google Scholar] [CrossRef]
- Dekonenko, A.; Yakimenko, V.; Ivanov, A.; Morozov, V.; Nikitin, P.; Khasanova, S.; Dzagurova, T.; Tkachenko, E.; Schmaljohn, C. Genetic similarity of Puumala viruses found in Finland and western Siberia and of the mitochondrial DNA of their rodent hosts suggests a common evolutionary origin. Infect. Genet. Evol. 2003, 3, 245–257. [Google Scholar] [CrossRef]
- Brünner, H.; Lugon-Moulin, N.; Balloux, F.; Fumagalli, L.; Hausser, J. A taxonomical re-evaluation of the Valais chromosome race of the common shrew Sorex araneus (Insectivora: Soricidae). Acta Theriol. 2002, 47, 245–275. [Google Scholar] [CrossRef]
- Colangelo, P.; Aloise, G.; Franchini, P.; Annesi, F.; Amori, G. Mitochondrial DNA reveals hidden diversity and an ancestral lineage of the bank vole in the Italian peninsula. J. Zool. 2011, 287, 41–52. [Google Scholar] [CrossRef]
- Chiocchio, A.; Colangelo, P.; Aloise, G.; Amori, G.; Bertolino, S.; Bisconti, R.; Castiglia, R.; Canestrelli, D. Population genetic structure of the bank vole Myodes glareolus within its glacial refugium in peninsular Italy. J. Zool. Syst. Evol. Res. 2019, 57, 959–969. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Number 2 | Species | Site 3 | Country | VP1 Genotype 4 | 3Dpol Subgroup 5 |
|---|---|---|---|---|---|
| 1-Ma-PJ-FI 6 | Microtus agrestis | PJ | Finland | 7 | Ma |
| 2-Ma-PJ-FI 6 | Microtus agrestis | PJ | Finland | 7 | ND 7 |
| 3-Ma-PJ-FI 6 | Microtus agrestis | PJ | Finland | 7 | Ma |
| 1-Mg-PJ-FI 6 | Myodes glareolus | PJ | Finland | ND | Mg C |
| 2-Mg-PJ-FI 6 | Myodes glareolus | PJ | Finland | 8 | Mg C |
| 3-Mg-HA-SE | Myodes glareolus | HA | Sweden | ND | Mg C |
| 4-Mg-HP-SE | Myodes glareolus | HP | Sweden | 1 | Mg A |
| 5-Mg-FE-SE | Myodes glareolus | FE | Sweden | 1 | ND |
| 6-Mg-UM-SE | Myodes glareolus | UM | Sweden | 1 | Mg A |
| 7-Mg-UM-SE | Myodes glareolus | UM | Sweden | 1 | ND |
| 8-Mg-UM-SE | Myodes glareolus | UM | Sweden | 1 | Mg A |
| 9-Mg-UM-SE | Myodes glareolus | UM | Sweden | 1 | Mg A |
| 10-Mg-UM-SE | Myodes glareolus | UM | Sweden | 8 | Mg C |
| 11-Mg-GN-SE | Myodes glareolus | GN | Sweden | 1 | Mg A |
| 12-Mg-GN-SE | Myodes glareolus | GN | Sweden | 1 | Mg A |
| 13-Mg-EN-SE | Myodes glareolus | EN | Sweden | 2 | Mg A |
| 14-Mg-TI-SE | Myodes glareolus | TI | Sweden | 1 | ND |
| 15-Mg-WE-DE | Myodes glareolus | WE | Germany | 1 | Mg B |
| 16-Mg-WE-DE | Myodes glareolus | WE | Germany | ND | Mg B |
| 17-Mg-FU-SK | Myodes glareolus | FU | Slovakia | 1 | ND |
| 18-Mg-MI-FR | Myodes glareolus | MI | France | 8 | Mg C |
| 19-Mg-LA-FR | Myodes glareolus | LA | France | 9 | Mg C |
| 20-Mg-LA-FR | Myodes glareolus | LA | France | 9 | Mg C |
| 21-Mg-SO-IT 6 | Myodes glareolus | SO | Italy | 9 | ND |
| 22-Mg-SO-IT 6 | Myodes glareolus | SO | Italy | 9 | Mg C |
| 23-Mg-TN-IT 6 | Myodes glareolus | TN | Italy | 8 | Mg C |
| 24-Mg-BS-IT 6 | Myodes glareolus | BS | Italy | 9 | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, C.; Zadra, N.; Fevola, C.; Ecke, F.; Hörnfeldt, B.; Kallies, R.; Kazimirova, M.; Magnusson, M.; Olsson, G.E.; Ulrich, R.G.; et al. Evolutionary Relationships of Ljungan Virus Variants Circulating in Multi-Host Systems across Europe. Viruses 2021, 13, 1317. https://doi.org/10.3390/v13071317
Rossi C, Zadra N, Fevola C, Ecke F, Hörnfeldt B, Kallies R, Kazimirova M, Magnusson M, Olsson GE, Ulrich RG, et al. Evolutionary Relationships of Ljungan Virus Variants Circulating in Multi-Host Systems across Europe. Viruses. 2021; 13(7):1317. https://doi.org/10.3390/v13071317
Chicago/Turabian StyleRossi, Chiara, Nicola Zadra, Cristina Fevola, Frauke Ecke, Birger Hörnfeldt, René Kallies, Maria Kazimirova, Magnus Magnusson, Gert E. Olsson, Rainer G. Ulrich, and et al. 2021. "Evolutionary Relationships of Ljungan Virus Variants Circulating in Multi-Host Systems across Europe" Viruses 13, no. 7: 1317. https://doi.org/10.3390/v13071317