
Modeling of CCR5 Recognition by HIV-1 gp120: How the Viral Protein Exploits the Conformational Plasticity of the Coreceptor

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Homology Modeling
2.2. Molecular Dynamics Simulation
2.3. Definition of Protein Domains
2.4. Trajectory Analysis
2.4.1. Deviation of the Atomic Coordinates of the Main Chain of Proteins
- RMSDt—the RMSD of the frame t
- min—the minimized structure, taken as reference
- x—the position of the atom n
- N—the total number of atoms in the set.
2.4.2. Fluctuation of Atomic Coordinates Per Residue
- RMSFp,r—the RMSF for the residue r of the protein p
- avg—the averaged structure
- x—the position of the atom
- T—the total number of structures
2.4.3. Clustering of Structures
2.4.4. Projection of CCR5 Extracellular and Intracellular Helix Extremities
2.4.5. Matrix of Structural Similarities
- t1 and t2—the frame of the simulated systems 1 and 2, respectively
- d—the analyzed CCR5 domain
- N—the total number of atoms in d
- x—the position of the atom n
2.4.6. Frequency Mapping of Non-Covalent Intermolecular Interactions
2.4.7. Principal Component Analysis
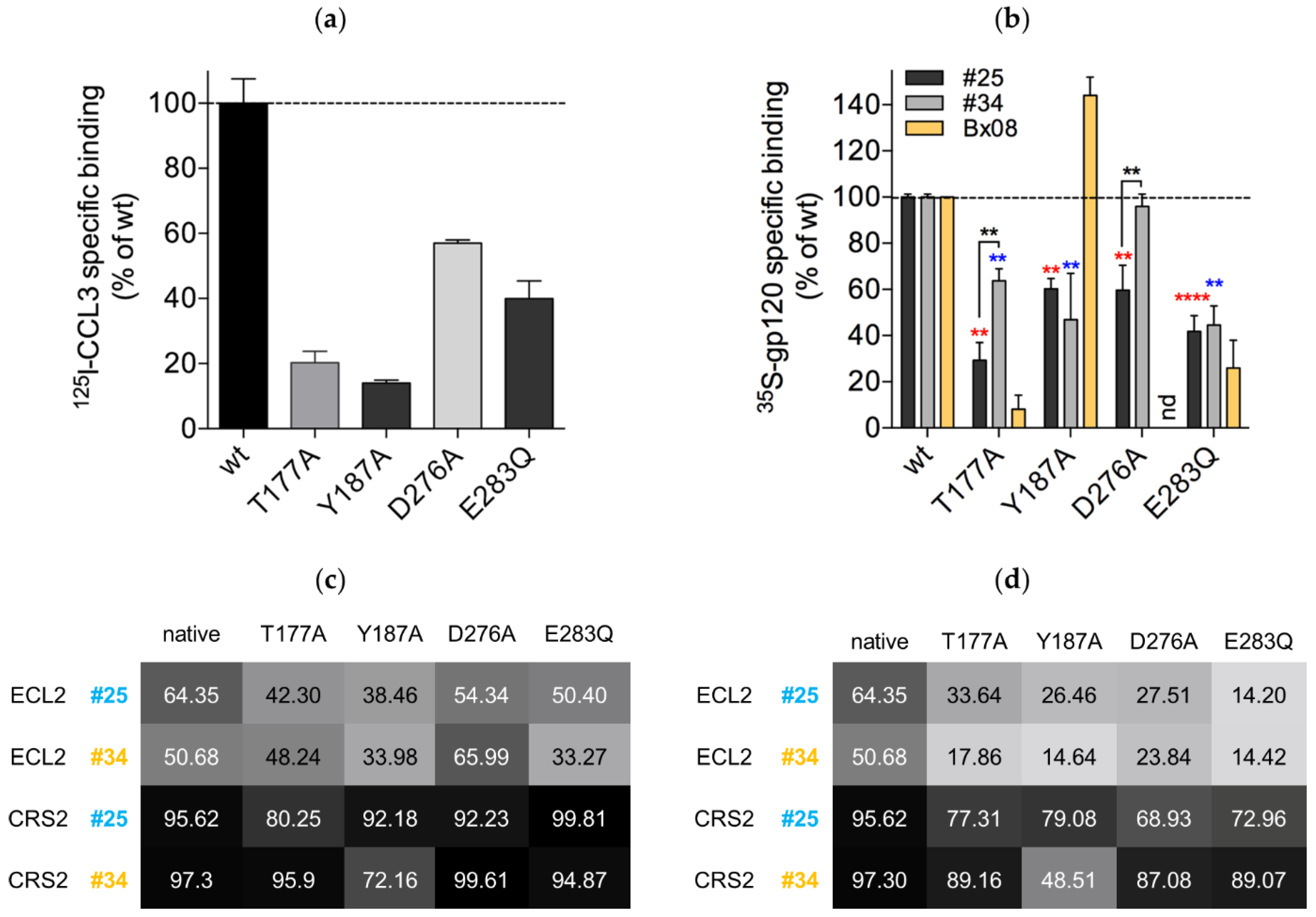
2.5. Binding Experiments to Wild-Type CCR5 and CCR5 Mutants
3. Results
3.1. General Description of the Modeling and Molecular Dynamics Simulations
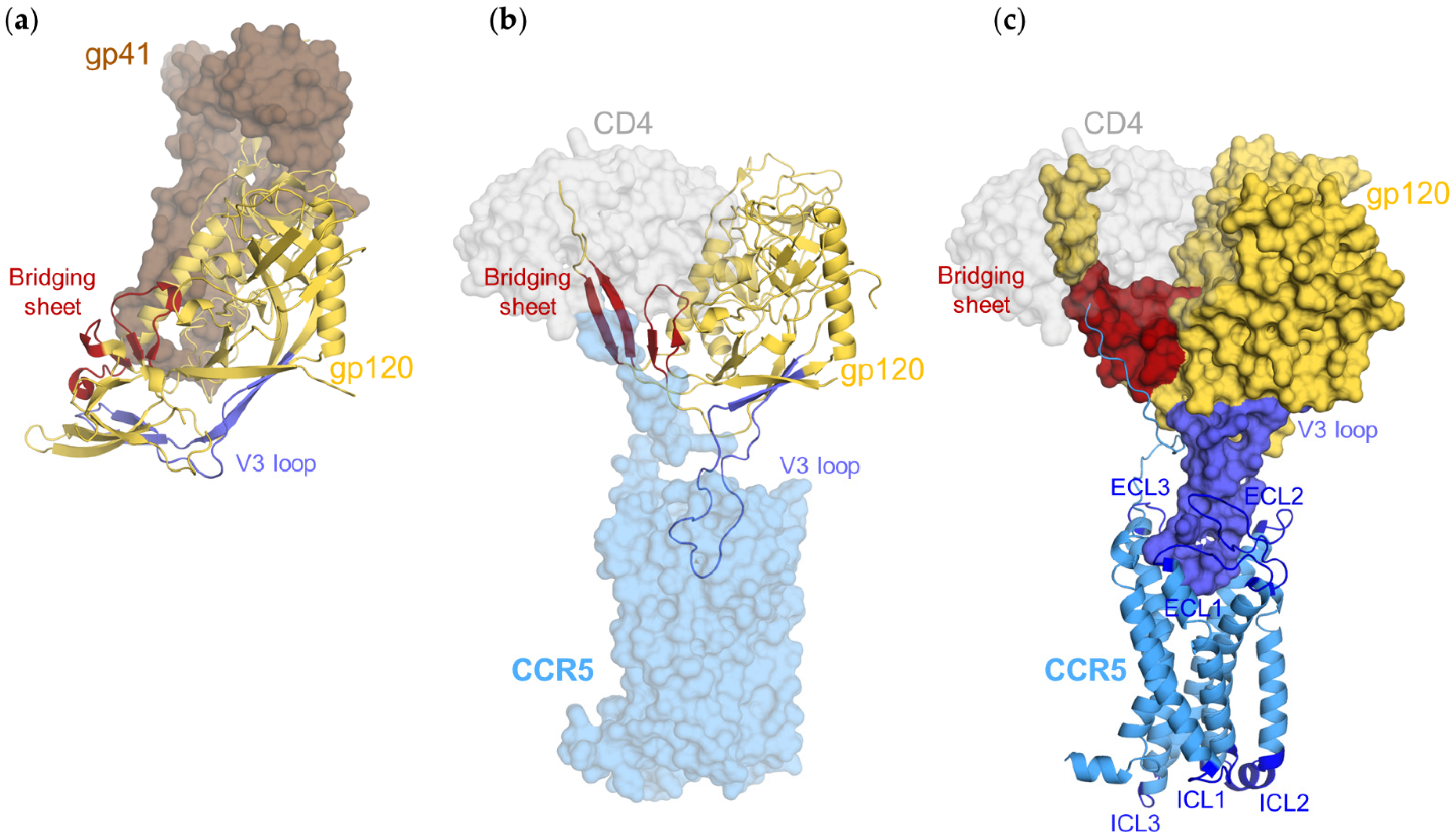
3.2. CD4–gp120–CCR5 Is a Flexible Complex
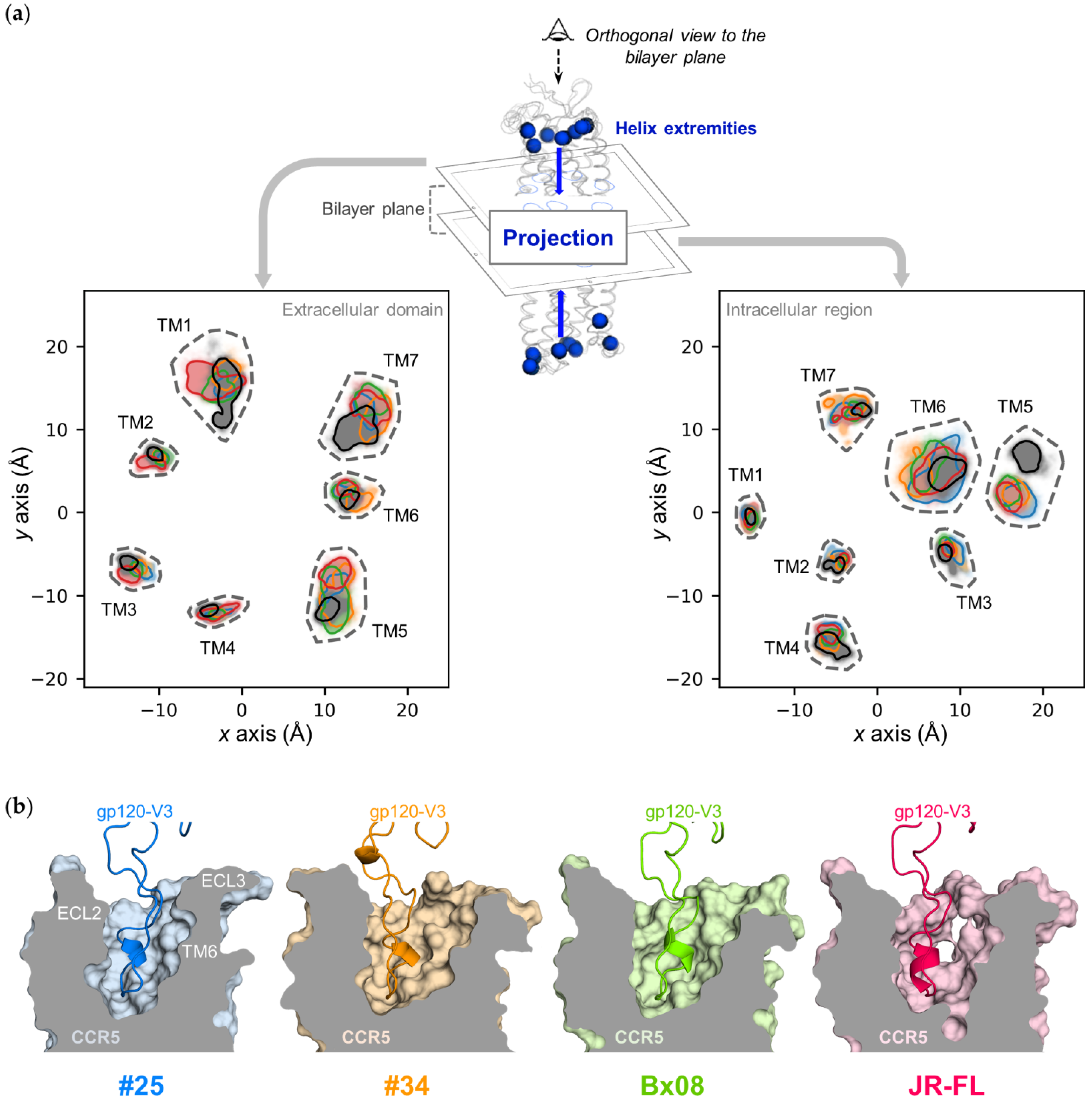
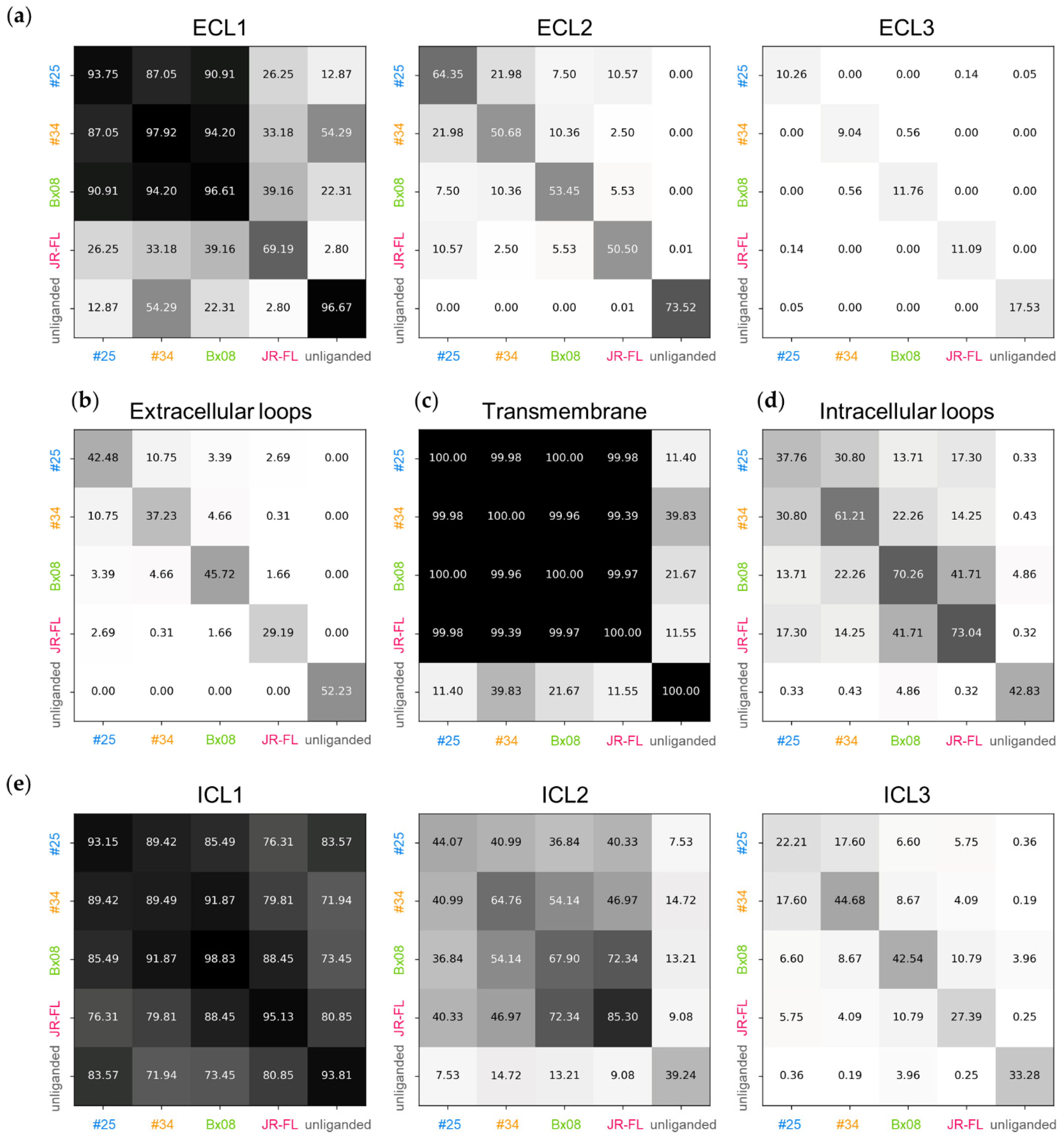
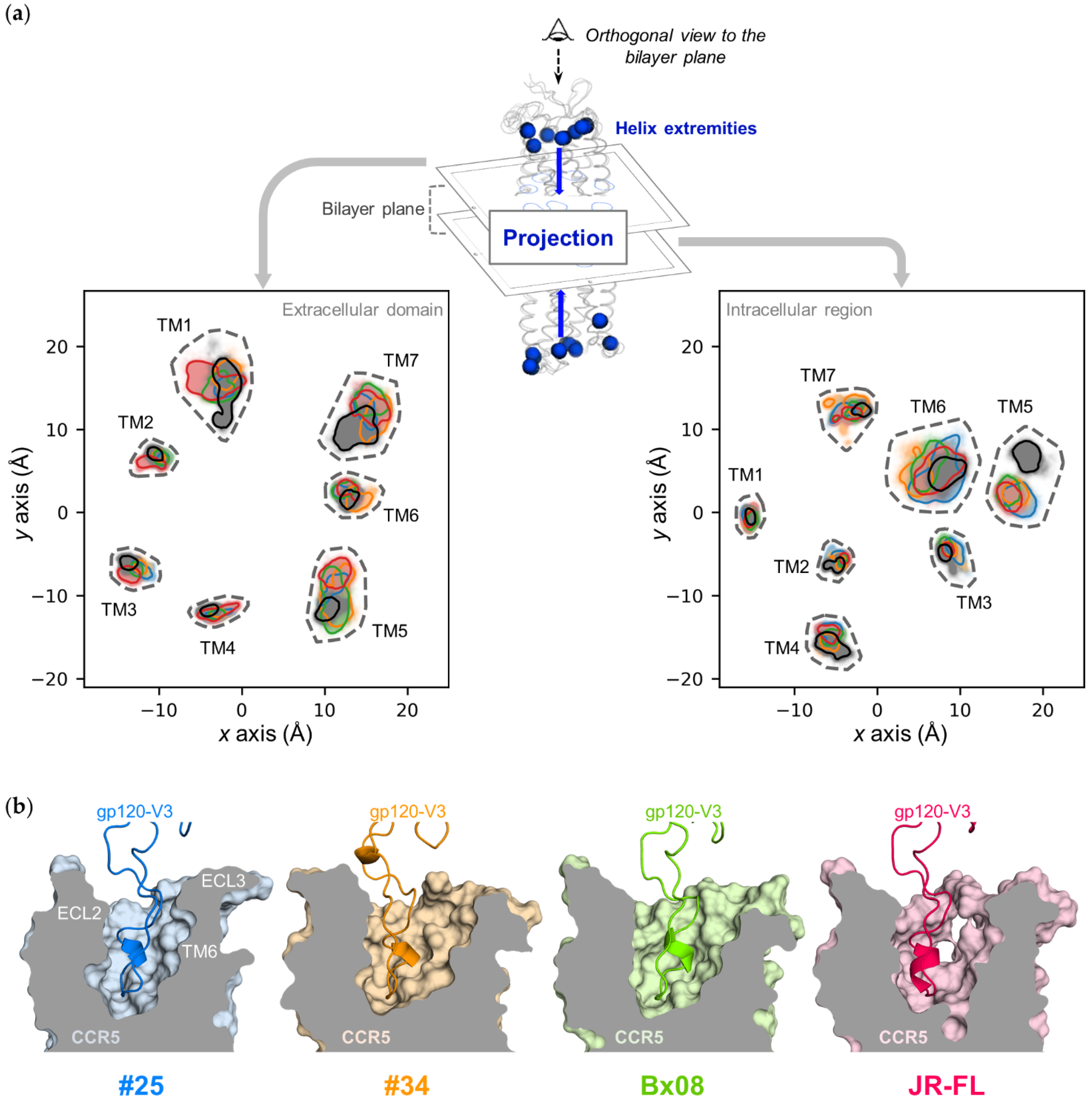
3.3. Gp120s Differentially Shape the Extracellular Side of CCR5
3.4. Gp120s Differentially Shape the Intracellular Side of CCR5
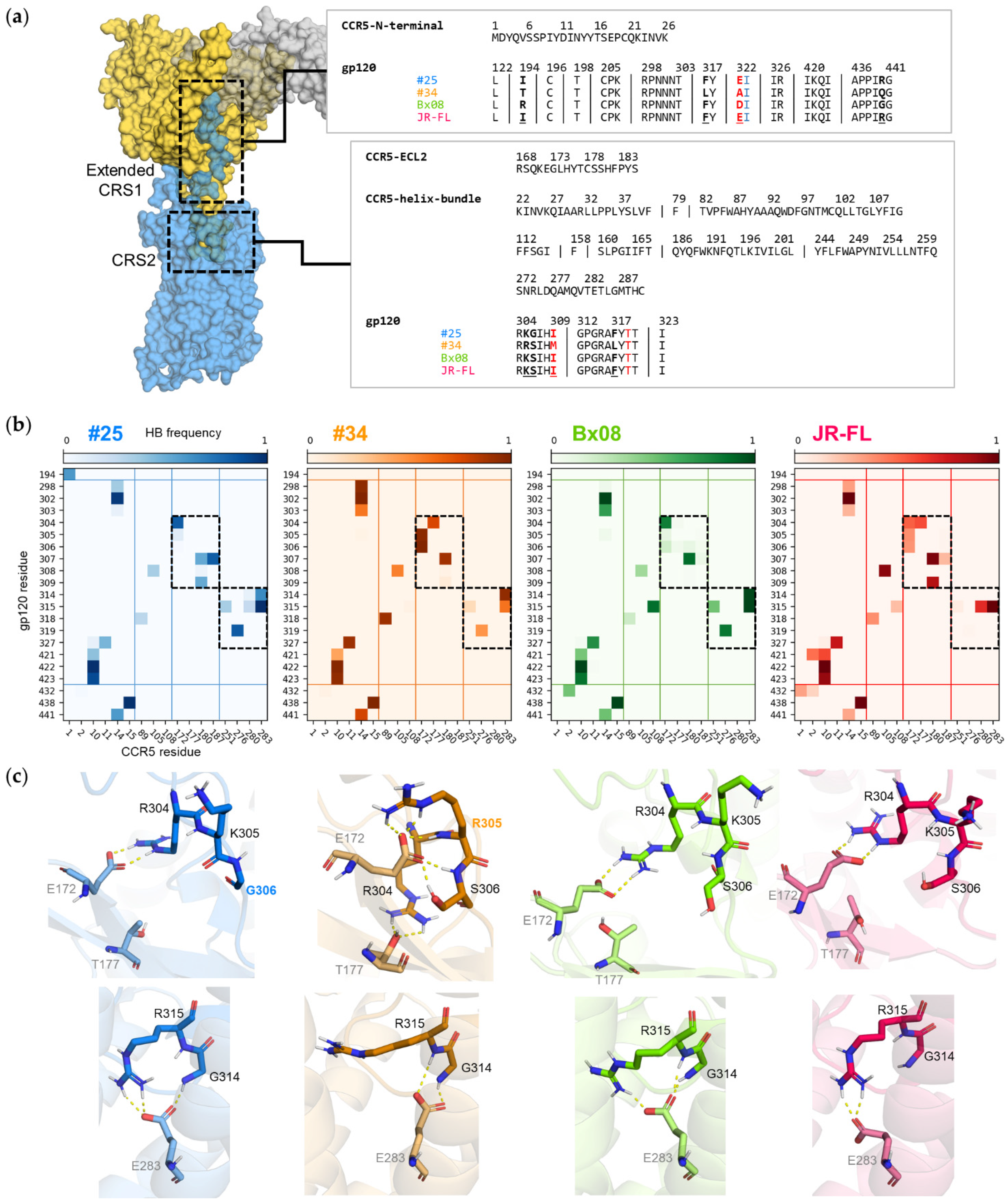
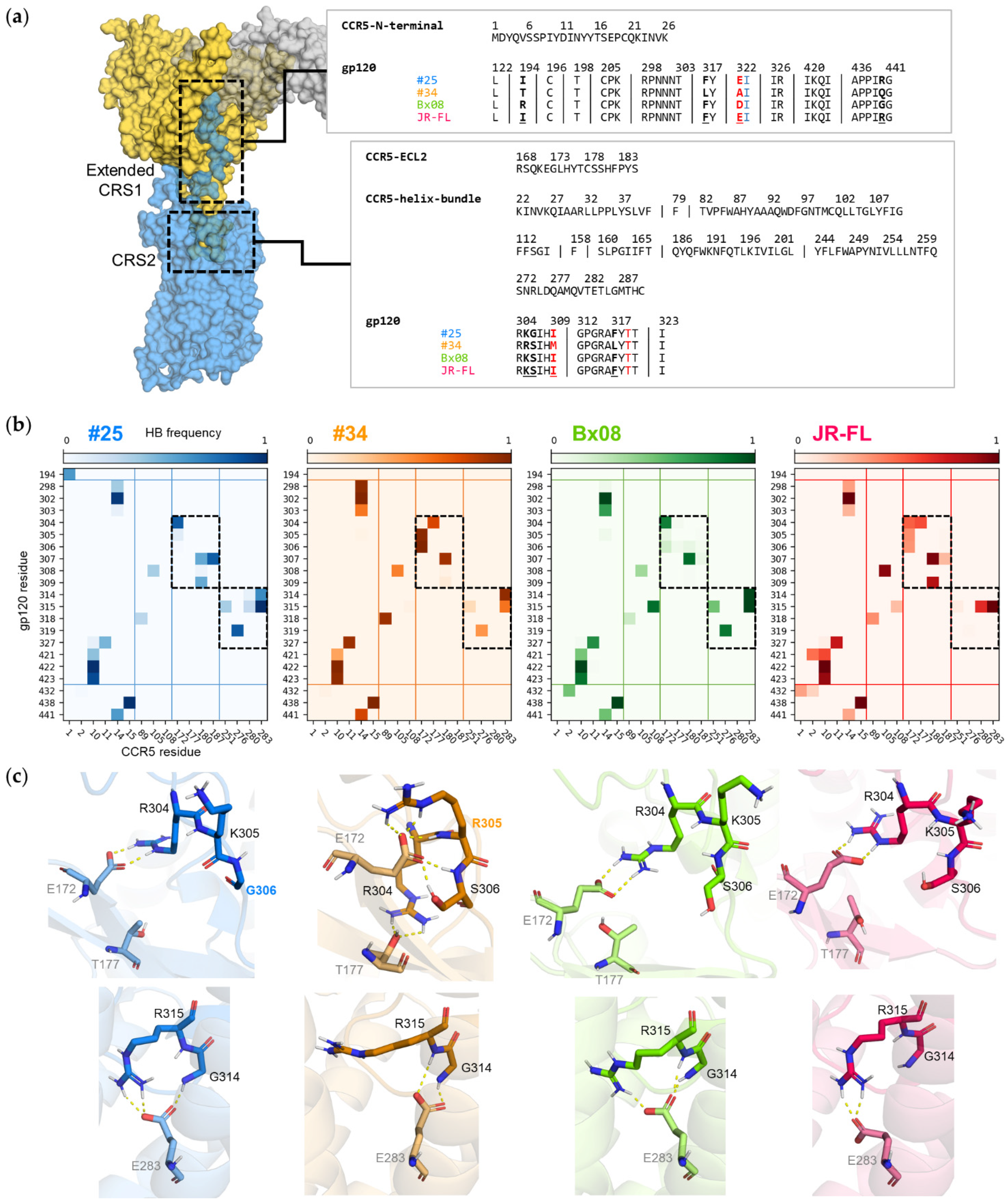
3.5. Gp120s Show Similar yet Different Binding Modes to CCR5
3.6. Mutations in CRS2 Differentially Influence the Binding of Distinct gp120s to CCR5
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, B. Molecular Mechanism of HIV-1 Entry. Trends Microbiol. 2019, 27, 878–891. [Google Scholar] [CrossRef]
- Wang, Q.; Finzi, A.; Sodroski, J. The Conformational States of the HIV-1 Envelope Glycoproteins. Trends Microbiol. 2020, 28, 655–667. [Google Scholar] [CrossRef]
- Li, Z.; Li, W.; Lu, M.; Bess, J.; Chao, C.W.; Gorman, J.; Terry, D.S.; Zhang, B.; Zhou, T.; Blanchard, S.C.; et al. Subnanometer Structures of HIV-1 Envelope Trimers on Aldrithiol-2-Inactivated Virus Particles. Nat. Struct. Mol. Biol. 2020, 27, 726–734. [Google Scholar] [CrossRef]
- Liu, J.; Bartesaghi, A.; Borgnia, M.J.; Sapiro, G.; Subramaniam, S. Molecular Architecture of Native HIV-1 Gp120 Trimers. Nature 2008, 455, 109–113. [Google Scholar] [CrossRef]
- Lu, M.; Ma, X.; Castillo-Menendez, L.R.; Gorman, J.; Alsahafi, N.; Ermel, U.; Terry, D.S.; Chambers, M.; Peng, D.; Zhang, B.; et al. Associating HIV-1 Envelope Glycoprotein Structures with States on the Virus Observed by SmFRET. Nature 2019, 568, 415–419. [Google Scholar] [CrossRef]
- Pan, J.; Peng, H.; Chen, B.; Harrison, S.C. Cryo-EM Structure of Full-Length HIV-1 Env Bound with the Fab of Antibody PG16. J. Mol. Biol. 2020, 432, 1158–1168. [Google Scholar] [CrossRef]
- Ozorowski, G.; Pallesen, J.; de Val, N.; Lyumkis, D.; Cottrell, C.A.; Torres, J.L.; Copps, J.; Stanfield, R.L.; Cupo, A.; Pugach, P.; et al. Open and Closed Structures Reveal Allostery and Pliability in the HIV-1 Envelope Spike. Nature 2017, 547, 360–363. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Barnes, C.O.; Yang, Z.; Nussenzweig, M.C.; Bjorkman, P.J. Partially Open HIV-1 Envelope Structures Exhibit Conformational Changes Relevant for Coreceptor Binding and Fusion. Cell Host Microbe 2018, 24, 579–592.e4. [Google Scholar] [CrossRef] [Green Version]
- Munro, J.B.; Gorman, J.; Ma, X.; Zhou, Z.; Arthos, J.; Burton, D.R.; Koff, W.C.; Courter, J.R.; Smith, A.B.; Kwong, P.D.; et al. Conformational Dynamics of Single HIV-1 Envelope Trimers on the Surface of Native Virions. Science 2014, 346, 759–763. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Guo, Y.-C.; Zhang, X.-L.; Deng, L.; Sang, P.; Yang, L.-Q.; Liu, S.-Q. CD4-Binding Obstacles in Conformational Transitions and Allosteric Communications of HIV Gp120. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183217. [Google Scholar] [CrossRef]
- Maeda, K.; Das, D.; Ogata-Aoki, H.; Nakata, H.; Miyakawa, T.; Tojo, Y.; Norman, R.; Takaoka, Y.; Ding, J.; Arnold, G.F.; et al. Structural and Molecular Interactions of CCR5 Inhibitors with CCR5. J. Biol. Chem. 2006, 281, 12688–12698. [Google Scholar] [CrossRef] [Green Version]
- Tam, K.; Schultz, M.; Reyes-Robles, T.; Vanwalscappel, B.; Horton, J.; Alonzo, F.; Wu, B.; Landau, N.R.; Torres, V.J. Staphylococcus aureus Leukocidin LukED and HIV-1 Gp120 Target Different Sequence Determinants on CCR5. mBio 2016, 7, e02024-16. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Fan, S.; Sun, Z. Structural and Functional Characterization of the Human CCR5 Receptor in Complex with HIV Gp120 Envelope Glycoprotein and CD4 Receptor by Molecular Modeling Studies. J. Mol. Model. 2003, 9, 329–336. [Google Scholar] [CrossRef]
- Napier, K.B.; Wang, Z.; Peiper, S.C.; Trent, J.O. CCR5 Interactions with the Variable 3 Loop of Gp120. J. Mol. Model. 2007, 13, 29–41. [Google Scholar] [CrossRef]
- Tamamis, P.; Floudas, C.A. Molecular Recognition of CCR5 by an HIV-1 Gp120 V3 Loop. PLoS ONE 2014, 9, e95767. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Han, G.W.; Abagyan, R.; Wu, B.; Stevens, R.C.; Cherezov, V.; Kufareva, I.; Handel, T.M. Structure of CC Chemokine Receptor 5 with a Potent Chemokine Antagonist Reveals Mechanisms of Chemokine Recognition and Molecular Mimicry by HIV. Immunity 2017, 46, 1005–1017.e5. [Google Scholar] [CrossRef] [Green Version]
- Shaik, M.M.; Peng, H.; Lu, J.; Rits-Volloch, S.; Xu, C.; Liao, M.; Chen, B. Structural Basis of Coreceptor Recognition by HIV-1 Envelope Spike. Nature 2019, 565, 318. [Google Scholar] [CrossRef]
- Zhou, Q.; Yang, D.; Wu, M.; Guo, Y.; Guo, W.; Zhong, L.; Cai, X.; Dai, A.; Jang, W.; Shakhnovich, E.I.; et al. Common Activation Mechanism of Class A GPCRs. eLife 2019, 8, e50279. [Google Scholar] [CrossRef]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J.; et al. Structure of the CCR5 Chemokine Receptor–HIV Entry Inhibitor Maraviroc Complex. Science 2013, 341, 1387–1390. [Google Scholar] [CrossRef] [Green Version]
- Colin, P.; Bénureau, Y.; Staropoli, I.; Wang, Y.; Gonzalez, N.; Alcami, J.; Hartley, O.; Brelot, A.; Arenzana-Seisdedos, F.; Lagane, B. HIV-1 Exploits CCR5 Conformational Heterogeneity to Escape Inhibition by Chemokines. Proc. Natl. Acad. Sci. USA 2013, 110, 9475–9480. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, C.A. Chapter Eight—Receptor Conformation and Constitutive Activity in CCR5 Chemokine Receptor Function and HIV Infection; Pharmacology & Therapeutics of Constitutively Active Receptors. In Advances in Pharmacology; Tao, Y.-X., Ed.; Academic Press: Cambridge, MA, USA, 2014; Volume 70, pp. 215–263. [Google Scholar]
- Wu, Y.; Yoder, A. Chemokine Coreceptor Signaling in HIV-1 Infection and Pathogenesis. PLOS Pathog. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornò, M.D.; Donninelli, G.; Varano, B.; Sacco, L.D.; Masotti, A.; Gessani, S. HIV-1 Gp120 Activates the STAT3/Interleukin-6 Axis in Primary Human Monocyte-Derived Dendritic Cells. J. Virol. 2014, 88, 11045–11055. [Google Scholar] [CrossRef] [Green Version]
- Fantuzzi, L.; Spadaro, F.; Purificato, C.; Cecchetti, S.; Podo, F.; Belardelli, F.; Gessani, S.; Ramoni, C. Phosphatidylcholine-Specific Phospholipase C Activation Is Required for CCR5-Dependent, NF-KB–Driven CCL2 Secretion Elicited in Response to HIV-1 Gp120 in Human Primary Macrophages. Blood 2008, 111, 3355–3363. [Google Scholar] [CrossRef]
- Liu, Q.-H.; Williams, D.A.; McManus, C.; Baribaud, F.; Doms, R.W.; Schols, D.; Clercq, E.D.; Kotlikoff, M.I.; Collman, R.G.; Freedman, B.D. HIV-1 Gp120 and Chemokines Activate Ion Channels in Primary Macrophages through CCR5 and CXCR4 Stimulation. Proc. Natl. Acad. Sci. USA 2000, 97, 4832–4837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melar, M.; Ott, D.E.; Hope, T.J. Physiological Levels of Virion-Associated Human Immunodeficiency Virus Type 1 Envelope Induce Coreceptor-Dependent Calcium Flux. J. Virol. 2007, 81, 1773–1785. [Google Scholar] [CrossRef] [Green Version]
- Missé, D.; Esteve, P.-O.; Renneboog, B.; Vidal, M.; Cerutti, M.; St Pierre, Y.; Yssel, H.; Parmentier, M.; Veas, F. HIV-1 Glycoprotein 120 Induces the MMP-9 Cytopathogenic Factor Production That Is Abolished by Inhibition of the P38 Mitogen-Activated Protein Kinase Signaling Pathway. Blood 2001, 98, 541–547. [Google Scholar] [CrossRef] [Green Version]
- Tomkowicz, B.; Lee, C.; Ravyn, V.; Cheung, R.; Ptasznik, A.; Collman, R.G. The Src Kinase Lyn Is Required for CCR5 Signaling in Response to MIP-1β and R5 HIV-1 Gp120 in Human Macrophages. Blood 2006, 108, 1145–1150. [Google Scholar] [CrossRef]
- Colin, P.; Zhou, Z.; Staropoli, I.; Garcia-Perez, J.; Gasser, R.; Armani-Tourret, M.; Benureau, Y.; Gonzalez, N.; Jin, J.; Connell, B.J.; et al. CCR5 Structural Plasticity Shapes HIV-1 Phenotypic Properties. PLOS Pathog. 2018, 14, e1007432. [Google Scholar] [CrossRef] [Green Version]
- Berro, R.; Klasse, P.J.; Lascano, D.; Flegler, A.; Nagashima, K.A.; Sanders, R.W.; Sakmar, T.P.; Hope, T.J.; Moore, J.P. Multiple CCR5 Conformations on the Cell Surface Are Used Differentially by Human Immunodeficiency Viruses Resistant or Sensitive to CCR5 Inhibitors. J. Virol. 2011, 85, 8227–8240. [Google Scholar] [CrossRef] [Green Version]
- Flegler, A.J.; Cianci, G.C.; Hope, T.J. CCR5 Conformations Are Dynamic and Modulated by Localization, Trafficking and G Protein Association. PLoS ONE 2014, 9, e89056. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.M.; Kasprowicz, R.; Hartley, O.; Signoret, N. CCR5 Susceptibility to Ligand-Mediated down-Modulation Differs between Human T Lymphocytes and Myeloid Cells. J. Leukoc. Biol. 2015, 98, 59–71. [Google Scholar] [CrossRef]
- Lee, B.; Sharron, M.; Blanpain, C.; Doranz, B.J.; Vakili, J.; Setoh, P.; Berg, E.; Liu, G.; Guy, H.R.; Durell, S.R.; et al. Epitope Mapping of CCR5 Reveals Multiple Conformational States and Distinct but Overlapping Structures Involved in Chemokine and Coreceptor Function. J. Biol. Chem. 1999, 274, 9617–9626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannert, N.; Craig, S.; Farzan, M.; Sogah, D.; Santo, N.V.; Choe, H.; Sodroski, J. Sialylated O-Glycans and Sulfated Tyrosines in the NH2-Terminal Domain of CC Chemokine Receptor 5 Contribute to High Affinity Binding of Chemokines. J. Exp. Med. 2001, 194, 1661–1674. [Google Scholar] [CrossRef] [Green Version]
- Scurci, I.; Akondi, K.B.; Pinheiro, I.; Paolini-Bertrand, M.; Borgeat, A.; Cerini, F.; Hartley, O. CCR5 Tyrosine Sulfation Heterogeneity Generates Cell Surface Receptor Subpopulations with Different Ligand Binding Properties. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129753. [Google Scholar] [CrossRef]
- Blanpain, C.; Vanderwinden, J.-M.; Cihak, J.; Wittamer, V.; Le Poul, E.; Issafras, H.; Stangassinger, M.; Vassart, G.; Marullo, S.; Schloōndorff, D.; et al. Multiple Active States and Oligomerization of CCR5 Revealed by Functional Properties of Monoclonal Antibodies. Mol. Biol. Cell 2002, 13, 723–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.H.; Taub, D. Cholesterol Is Essential for Macrophage Inflammatory Protein 1β Binding and Conformational Integrity of CC Chemokine Receptor 5. Blood 2002, 99, 4298–4306. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Colin, P.; Staropoli, I.; Lima-Fernandes, E.; Ferret, C.; Demir, A.; Rogée, S.; Hartley, O.; Randriamampita, C.; Scott, M.G.H.; et al. Targeting Spare CC Chemokine Receptor 5 (CCR5) as a Principle to Inhibit HIV-1 Entry. J. Biol. Chem. 2014, 289, 19042–19052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Momboisse, F.; Boncompain, G.; Koensgen, F.; Zhou, Z.; Cordeiro, N.; Arenzana-Seisdedos, F.; Perez, F.; Lagane, B.; Kellenberger, E.; et al. CCR5 Adopts Three Homodimeric Conformations That Control Cell Surface Delivery. Sci. Signal. 2018, 11, eaal2869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward Realistic Biological Membrane Simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [Green Version]
- Ponder, J.W.; Case, D.A. Force Fields for Protein Simulations. In Advances in Protein Chemistry; Protein Simulations; Academic Press: Cambridge, MA, USA, 2003; Volume 66, pp. 27–85. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR Structure Models and Ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef] [Green Version]
- Marcou, G.; Rognan, D. Optimizing Fragment and Scaffold Docking by Use of Molecular Interaction Fingerprints. J. Chem. Inf. Model. 2007, 47, 195–207. [Google Scholar] [CrossRef]
- Gasser, R.; Hamoudi, M.; Pellicciotta, M.; Zhou, Z.; Visdeloup, C.; Colin, P.; Braibant, M.; Lagane, B.; Negroni, M. Buffering Deleterious Polymorphisms in Highly Constrained Parts of HIV-1 Envelope by Flexible Regions. Retrovirology 2016, 13, 50. [Google Scholar] [CrossRef] [Green Version]
- Lagane, B.; Ballet, S.; Planchenault, T.; Balabanian, K.; Poul, E.L.; Blanpain, C.; Percherancier, Y.; Staropoli, I.; Vassart, G.; Oppermann, M.; et al. Mutation of the DRY Motif Reveals Different Structural Requirements for the CC Chemokine Receptor 5-Mediated Signaling and Receptor Endocytosis. Mol. Pharmacol. 2005, 67, 1966–1976. [Google Scholar] [CrossRef]
- Garcia-Perez, J.; Rueda, P.; Staropoli, I.; Kellenberger, E.; Alcami, J.; Arenzana-Seisdedos, F.; Lagane, B. New Insights into the Mechanisms Whereby Low Molecular Weight CCR5 Ligands Inhibit HIV-1 Infection. J. Biol. Chem. 2011, 286, 4978–4990. [Google Scholar] [CrossRef] [Green Version]
- Benureau, Y.; Colin, P.; Staropoli, I.; Gonzalez, N.; Garcia-Perez, J.; Alcami, J.; Arenzana-Seisdedos, F.; Lagane, B. Guidelines for Cloning, Expression, Purification and Functional Characterization of Primary HIV-1 Envelope Glycoproteins. J. Virol. Methods 2016, 236, 184–195. [Google Scholar] [CrossRef]
- Garcia-Perez, J.; Staropoli, I.; Azoulay, S.; Heinrich, J.-T.; Cascajero, A.; Colin, P.; Lortat-Jacob, H.; Arenzana-Seisdedos, F.; Alcami, J.; Kellenberger, E.; et al. A Single-Residue Change in the HIV-1 V3 Loop Associated with Maraviroc Resistance Impairs CCR5 Binding Affinity While Increasing Replicative Capacity. Retrovirology 2015, 12, 50. [Google Scholar] [CrossRef] [Green Version]
- Farzan, M.; Mirzabekov, T.; Kolchinsky, P.; Wyatt, R.; Cayabyab, M.; Gerard, N.P.; Gerard, C.; Sodroski, J.; Choe, H. Tyrosine Sulfation of the Amino Terminus of CCR5 Facilitates HIV-1 Entry. Cell 1999, 96, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Tang, M.; Zhang, M.-Y.; Majeed, S.; Montabana, E.; Stanfield, R.L.; Dimitrov, D.S.; Korber, B.; Sodroski, J.; Wilson, I.A.; et al. Structure of a V3-Containing HIV-1 Gp120 Core. Science 2005, 310, 1025–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, J.L.; Hu, Q.; Navenot, J.-M.; Peiper, S.C. Role of CD4 Hinge Region in GP120 Utilization by Immunoglobulin Domain 1. Biochem. Biophys. Res. Commun. 2002, 292, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, M.; Bettadapura, R.; Bajaj, C. Computational Refinement and Validation Protocol for Proteins with Large Variable Regions Applied to Model HIV Env Spike in CD4 and 17b Bound State. Structure 2015, 23, 1138–1149. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Perez, J.; Rueda, P.; Alcami, J.; Rognan, D.; Arenzana-Seisdedos, F.; Lagane, B.; Kellenberger, E. Allosteric Model of Maraviroc Binding to CC Chemokine Receptor 5 (CCR5). J. Biol. Chem. 2011, 286, 33409–33421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanpain, C.; Doranz, B.J.; Bondue, A.; Govaerts, C.; De Leener, A.; Vassart, G.; Doms, R.W.; Proudfoot, A.; Parmentier, M. The Core Domain of Chemokines Binds CCR5 Extracellular Domains While Their Amino Terminus Interacts with the Transmembrane Helix Bundle. J. Biol. Chem. 2003, 278, 5179–5187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genoud, S.; Kajumo, F.; Guo, Y.; Thompson, D.; Dragic, T. CCR5-Mediated Human Immunodeficiency Virus Entry Depends on an Amino-Terminal Gp120-Binding Site and on the Conformational Integrity of All Four Extracellular Domains. J. Virol. 1999, 73, 1645–1648. [Google Scholar] [CrossRef] [Green Version]
- Anastassopoulou, C.G.; Ketas, T.J.; Klasse, P.J.; Moore, J.P. Resistance to CCR5 Inhibitors Caused by Sequence Changes in the Fusion Peptide of HIV-1 Gp41. Proc. Natl. Acad. Sci. USA 2009, 106, 5318–5323. [Google Scholar] [CrossRef] [Green Version]
- Abrol, R.; Trzaskowski, B.; Goddard, W.A.; Nesterov, A.; Olave, I.; Irons, C. Ligand- and Mutation-Induced Conformational Selection in the CCR5 Chemokine G Protein-Coupled Receptor. Proc. Natl. Acad. Sci. USA 2014, 111, 13040–13045. [Google Scholar] [CrossRef] [Green Version]
- Bostock, M.J.; Solt, A.S.; Nietlispach, D. The Role of NMR Spectroscopy in Mapping the Conformational Landscape of GPCRs. Curr. Opin. Struct. Biol. 2019, 57, 145–156. [Google Scholar] [CrossRef]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein–Coupled Receptor Activation. Annu. Rev. Biochem. 2014, 87, 897–919. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, M.; Touma, A.M.; Dysthe, M.; Sadler, F.; Sivaramakrishnan, S.; Vaidehi, N. Conformational Plasticity of the Intracellular Cavity of GPCR−G-Protein Complexes Leads to G-Protein Promiscuity and Selectivity. Proc. Natl. Acad. Sci. USA 2019, 116, 11956–11965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleetwood, O.; Carlsson, J.; Delemotte, L. Identification of Ligand-Specific G Protein-Coupled Receptor States and Prediction of Downstream Efficacy via Data-Driven Modeling. eLife 2021, 10, e60715. [Google Scholar] [CrossRef]
- Springael, J.-Y.; de Poorter, C.; Deupi, X.; Van Durme, J.; Pardo, L.; Parmentier, M. The Activation Mechanism of Chemokine Receptor CCR5 Involves Common Structural Changes but a Different Network of Interhelical Interactions Relative to Rhodopsin. Cell. Signal. 2007, 19, 1446–1456. [Google Scholar] [CrossRef] [PubMed]
- Arthos, J.; Rubbert, A.; Rabin, R.L.; Cicala, C.; Machado, E.; Wildt, K.; Hanbach, M.; Steenbeke, T.D.; Swofford, R.; Farber, J.M.; et al. CCR5 Signal Transduction in Macrophages by Human Immunodeficiency Virus and Simian Immunodeficiency Virus Envelopes. J. Virol. 2000, 74, 6418–6424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicala, C.; Arthos, J.; Martinelli, E.; Censoplano, N.; Cruz, C.C.; Chung, E.; Selig, S.M.; Ryk, D.V.; Yang, J.; Jagannatha, S.; et al. R5 and X4 HIV Envelopes Induce Distinct Gene Expression Profiles in Primary Peripheral Blood Mononuclear Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 3746–3751. [Google Scholar] [CrossRef] [Green Version]
- Cicala, C.; Arthos, J.; Selig, S.M.; Dennis, G.; Hosack, D.A.; Ryk, D.V.; Spangler, M.L.; Steenbeke, T.D.; Khazanie, P.; Gupta, N.; et al. HIV Envelope Induces a Cascade of Cell Signals in Non-Proliferating Target Cells That Favor Virus Replication. Proc. Natl. Acad. Sci. USA 2002, 99, 9380–9385. [Google Scholar] [CrossRef] [Green Version]
- Del Corno, M.; Liu, Q.-H.; Schols, D.; de Clercq, E.; Gessani, S.; Freedman, B.D.; Collman, R.G. HIV-1 Gp120 and Chemokine Activation of Pyk2 and Mitogen-Activated Protein Kinases in Primary Macrophages Mediated by Calcium-Dependent, Pertussis Toxin–Insensitive Chemokine Receptor Signaling. Blood 2001, 98, 2909–2916. [Google Scholar] [CrossRef] [Green Version]
- Weissman, D.; Rabin, R.L.; Arthos, J.; Rubbert, A.; Dybul, M.; Swofford, R.; Venkatesan, S.; Farber, J.M.; Fauci, A.S. Macrophage-Tropic HIV and SIV Envelope Proteins Induce a Signal through the CCR5 Chemokine Receptor. Nature 1997, 389, 981–985. [Google Scholar] [CrossRef]
- Araújo, L.A.L.; Almeida, S.E.M. HIV-1 Diversity in the Envelope Glycoproteins: Implications for Viral Entry Inhibition. Viruses 2013, 5, 595–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemelaar, J. Implications of HIV Diversity for the HIV-1 Pandemic. J. Infect. 2013, 66, 391–400. [Google Scholar] [CrossRef]
- Lynch, R.M.; Shen, T.; Gnanakaran, S.; Derdeyn, C.A. Appreciating HIV Type 1 Diversity: Subtype Differences in Env. AIDS Res. Hum. Retroviruses 2009, 25, 237–248. [Google Scholar] [CrossRef]
- Corbisier, J.; Galès, C.; Huszagh, A.; Parmentier, M.; Springael, J.-Y. Biased Signaling at Chemokine Receptors. J. Biol. Chem. 2015, 290, 9542–9554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbisier, J.; Huszagh, A.; Galés, C.; Parmentier, M.; Springael, J.-Y. Partial Agonist and Biased Signaling Properties of the Synthetic Enantiomers J113863/UCB35625 at Chemokine Receptors CCR2 and CCR5. J. Biol. Chem. 2017, 292, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Lorenzen, E.; Ceraudo, E.; Berchiche, Y.A.; Rico, C.A.; Fürstenberg, A.; Sakmar, T.P.; Huber, T. G Protein Subtype–Specific Signaling Bias in a Series of CCR5 Chemokine Analogs. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, S.; Bassoni, D.L.; Campbell, J.J.; Gerard, N.P.; Gerard, C.; Wehrman, T.S. Biased Agonism as a Mechanism for Differential Signaling by Chemokine Receptors. J. Biol. Chem. 2013, 288, 35039–35048. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CCR5 Mutated Residue | Interaction Type | Interacting Residue | gp120#25 | gp120#34 | gp120Bx08 | gp120JR-FL | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Region | Residue | Group | Region | Residue | Group | |||||
| ECL2 | Thr177 | OH | hydrogen bond | V3 | Arg304 | C(NH2)2+ | 0% | 75% | 2% | 57% |
| ECL2 | Glu172 | COO− | 26% | <1% | 66% | 16% | ||||
| TM5 | Tyr187 | OH | hydrogen bond | V3 | Ile307 | NH | 82% | 0% | 8% | 25% |
| ECL2 | Ser180 | CO | 87% | <1% | 3% | 26% | ||||
| Phenyl ring | π-stacking | TM5 | Phe182 | phenyl ring | 15% | <1% | 4% | 4% | ||
| TM7 | Asp276 | COO− | ionic bond | N-ter | Lys22 | NH3+ | 4% | 32% | 60% | 51% |
| hydrogen bond | V3 | Thr319 | OH | 82% | 46% | 82% | 1% | |||
| TM6 | Asn258 | NH2 | 66% | 10% | 8% | 32% | ||||
| N-ter | Lys22 | NH3+ | <1% | 26% | 54% | 48% | ||||
| TM6 | Gln261 | NH2 | 9% | <1% | 2% | 33% | ||||
| TM7 | Glu283 | COO− | ionic bond | V3 | Arg315 | C(NH2)2+ | 100% | 25% | 99% | 100% |
| COO− | hydrogen bond | V3 | Gly314 | NH | 65% | 94% | 98% | 0% | ||
| V3 | Arg315 | C(NH2)2+ | 100% | 25% | ≈100% | ≈100% | ||||
| V3 | Arg315 | NH | <1% | 35% | 97% | 0% | ||||
| TM3 | Tyr108 | OH | 24% | 5% | <1% | 99% | ||||
| TM6 | Tyr251 | OH | 97% | 8% | 75% | 58% | ||||
| TM7 | Gln280 | NH2 | 67% | 40% | <1% | 16% | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacquemard, C.; Koensgen, F.; Colin, P.; Lagane, B.; Kellenberger, E. Modeling of CCR5 Recognition by HIV-1 gp120: How the Viral Protein Exploits the Conformational Plasticity of the Coreceptor. Viruses 2021, 13, 1395. https://doi.org/10.3390/v13071395
Jacquemard C, Koensgen F, Colin P, Lagane B, Kellenberger E. Modeling of CCR5 Recognition by HIV-1 gp120: How the Viral Protein Exploits the Conformational Plasticity of the Coreceptor. Viruses. 2021; 13(7):1395. https://doi.org/10.3390/v13071395
Chicago/Turabian StyleJacquemard, Célien, Florian Koensgen, Philippe Colin, Bernard Lagane, and Esther Kellenberger. 2021. "Modeling of CCR5 Recognition by HIV-1 gp120: How the Viral Protein Exploits the Conformational Plasticity of the Coreceptor" Viruses 13, no. 7: 1395. https://doi.org/10.3390/v13071395
APA StyleJacquemard, C., Koensgen, F., Colin, P., Lagane, B., & Kellenberger, E. (2021). Modeling of CCR5 Recognition by HIV-1 gp120: How the Viral Protein Exploits the Conformational Plasticity of the Coreceptor. Viruses, 13(7), 1395. https://doi.org/10.3390/v13071395