
Immunisation Using Novel DNA Vaccine Encoding Virus Membrane Fusion Complex and Chemokine Genes Shows High Protection from HSV-2

Abstract
1. Introduction
2. Materials and Methods
2.1. Vaccines
2.2. Animals and Virus
2.3. Immunisation
2.4. Virus Challenge
2.5. Neutralising Antibody Assay
2.6. Quantification of Virus DNA by PCR
2.7. DNA Cell Transfection
2.8. RNA Purification
2.9. Reverse Transcriptase Polymerase Chain Reaction Amplification, RT-PCR
2.10. Cell Fusion Assay and Imaging
2.11. Statistics
3. Results
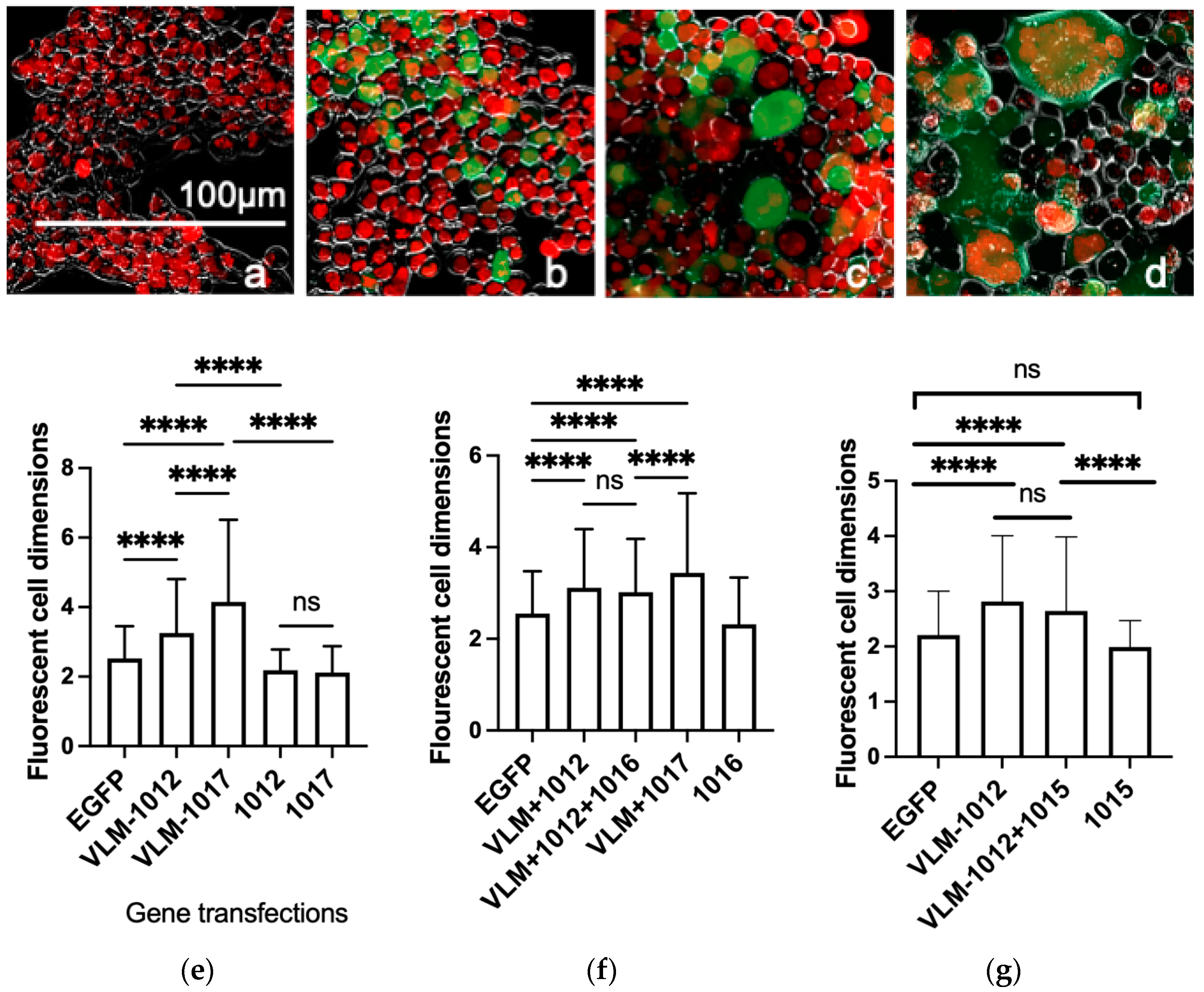
3.1. Functional In Vitro Gene Expression of the Virus Entry Complex Modulates Cell Fusion
3.2. Gene Immunisation In Vivo Induces Efficient HSV-2 Neutralising Antibodies
3.3. Gene Immunisation Highly Protective against HSV-2 Acute Disease and Infection
3.4. Gene Immunisation Protective against Establishment HSV-2 Latency
3.5. Gene Immunisation Protective against Recurrent Disease
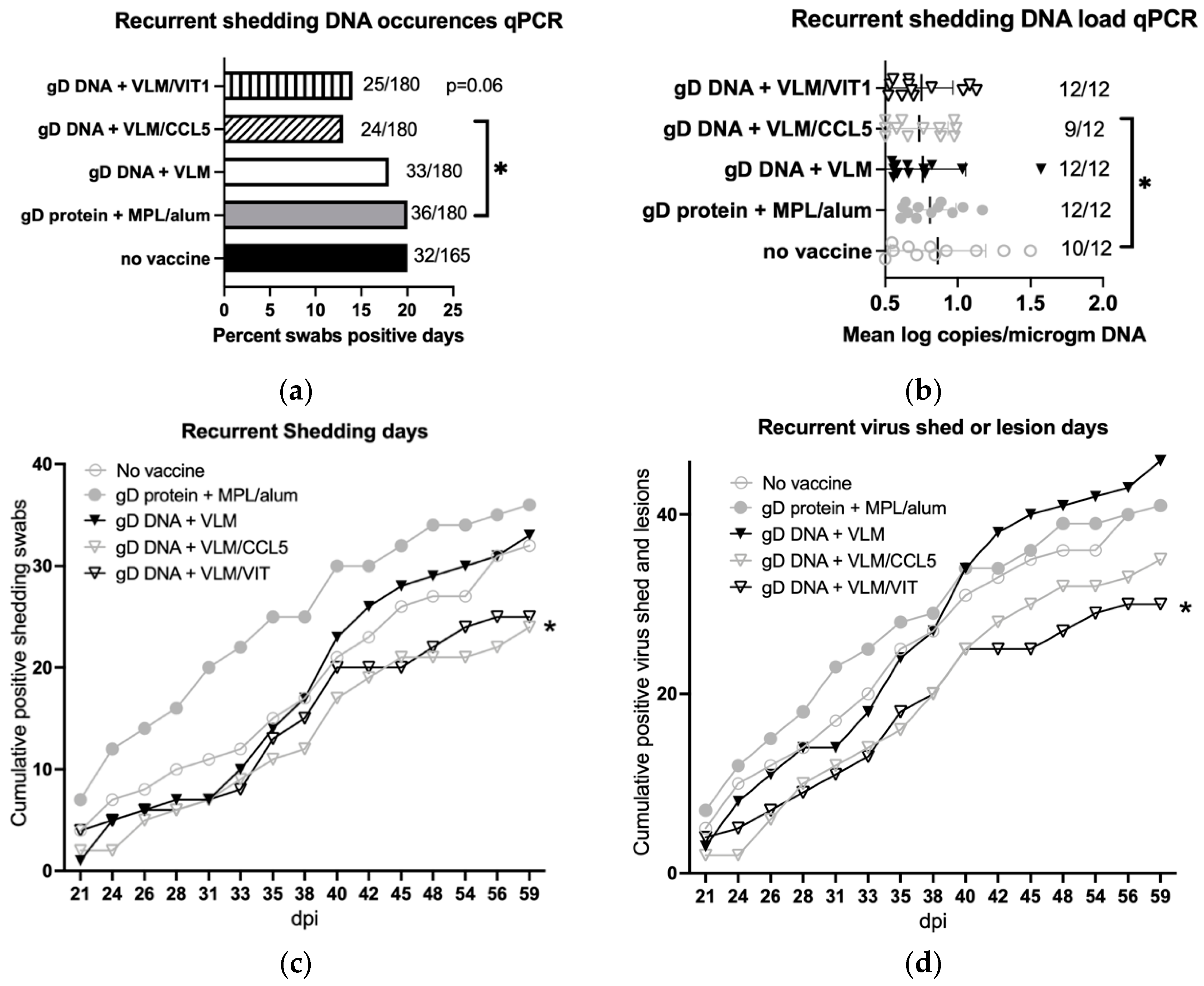
3.6. Gene Immunisation Reduces Recurrent Virus Shedding and Lesions
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- James, C.; Harfouche, M.; Welton, N.J.; Turner, K.M.; Abu-Raddad, L.J.; Gottlieb, S.L.; Looker, K.J. Herpes simplex virus: Global infection prevalence and incidence estimates, 2016. Bull. World Health Organ. 2020, 98, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Truong, N.R.; Smith, J.B.; Sandgren, K.J.; Cunningham, A.L. Mechanisms of Immune Control of Mucosal HSV Infection: A Guide to Rational Vaccine Design. Front. Immunol. 2019, 10, 373. [Google Scholar] [CrossRef] [PubMed]
- Melvin, A.J.; Mohan, K.M.; Vora, S.B.; Selke, S.; Sullivan, E.; Wald, A. Neonatal Herpes Simplex Virus Infection: Epidemiology and Outcomes in the Modern Era. J. Pediatr. Infect. Dis. Soc. 2022, 11, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Silhol, R.; Coupland, H.; Baggaley, R.F.; Miller, L.; Staadegaard, L.; Gottlieb, S.L.; Stannah, J.; Turner, K.M.E.; Vickerman, P.; Hayes, R.; et al. What Is the Burden of Heterosexually Acquired HIV Due to HSV-2? Global and Regional Model-Based Estimates of the Proportion and Number of HIV Infections Attributable to HSV-2 Infection. J. Acquir. Immune Defic. Syndr. 2021, 88, 19–30. [Google Scholar] [CrossRef]
- Henze, L.; Buhl, C.; Sandherr, M.; Cornely, O.A.; Heinz, W.J.; Khodamoradi, Y.; Kiderlen, T.R.; Koehler, P.; Seidler, A.; Sprute, R.; et al. Management of herpesvirus reactivations in patients with solid tumours and hematologic malignancies: Update of the Guidelines of the Infectious Diseases Working Party (AGIHO) of the German Society for Hematology and Medical Oncology (DGHO) on herpes simplex virus type 1, herpes simplex virus type 2, and varicella zoster virus. Ann. Hematol. 2022, 101, 491–511. [Google Scholar] [CrossRef]
- Smith, C.R.; Pogany, L.; Auguste, U.; Steben, M.; Lau, T. Does suppressive antiviral therapy for herpes simplex virus prevent transmission in an HIV-positive population? A systematic review. Can. Commun. Dis. Rep. 2016, 42, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Agyemang, E.; Magaret, A.S.; Selke, S.; Johnston, C.; Corey, L.; Wald, A. Herpes Simplex Virus Shedding Rate: Surrogate Outcome for Genital Herpes Recurrence Frequency and Lesion Rates, and Phase 2 Clinical Trials End Point for Evaluating Efficacy of Antivirals. J. Infect. Dis. 2018, 218, 1691–1699. [Google Scholar] [CrossRef]
- Belshe, R.B.; Leone, P.A.; Bernstein, D.I.; Wald, A.; Levin, M.J.; Stapleton, J.T.; Gorfinkel, I.; Morrow, R.L.; Ewell, M.G.; Stokes-Riner, A.; et al. Efficacy results of a trial of a herpes simplex vaccine. N. Engl. J. Med. 2012, 366, 34–43. [Google Scholar] [CrossRef]
- Corey, L.; Langenberg, A.G.; Ashley, R.; Sekulovich, R.E.; Izu, A.E.; Douglas, J.M., Jr.; Handsfield, H.H.; Warren, T.; Marr, L.; Tyring, S.; et al. Recombinant glycoprotein vaccine for the prevention of genital HSV-2 infection: Two randomized controlled trials. Chiron HSV Vaccine Study Group. JAMA 1999, 282, 331–340. [Google Scholar] [CrossRef]
- Stanberry, L.R.; Spruance, S.L.; Cunningham, A.L.; Bernstein, D.I.; Mindel, A.; Sacks, S.; Tyring, S.; Aoki, F.Y.; Slaoui, M.; Denis, M.; et al. Glycoprotein-D-adjuvant vaccine to prevent genital herpes. N. Engl. J. Med. 2002, 347, 1652–1661. [Google Scholar] [CrossRef]
- Belshe, R.B.; Blevins, T.P.; Yu, Y.; Nethington, A.E.; Bellamy, A.; Bryant, C.; Morrison, L.A. Neutralizing Antibody Kinetics and Immune Protection Against HSV-1 Genital Disease in Vaccinated Women. J. Infect. Dis. 2022, jiac067. [Google Scholar] [CrossRef] [PubMed]
- Belshe, R.B.; Heineman, T.C.; Bernstein, D.I.; Bellamy, A.R.; Ewell, M.; van der Most, R.; Deal, C.D. Correlate of immune protection against HSV-1 genital disease in vaccinated women. J. Infect. Dis. 2014, 209, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, B.; Grunewald, K. Herpesvirus membrane fusion—A team effort. Curr. Opin. Struct. Biol. 2020, 62, 112–120. [Google Scholar] [CrossRef]
- Hilterbrand, A.T.; Heldwein, E.E. Go go gadget glycoprotein!: HSV-1 draws on its sizeable glycoprotein tool kit to customize its diverse entry routes. PLoS Pathog. 2019, 15, e1007660. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.A. A Comparison of Plasmid DNA and mRNA as Vaccine Technologies. Vaccines 2019, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Gianni, T.; Leoni, V.; Chesnokova, L.S.; Hutt-Fletcher, L.M.; Campadelli-Fiume, G. alphavbeta3-integrin is a major sensor and activator of innate immunity to herpes simplex virus-1. Proc. Natl. Acad. Sci. USA 2012, 109, 19792–19797. [Google Scholar] [CrossRef]
- Campadelli-Fiume, G.; Collins-McMillen, D.; Gianni, T.; Yurochko, A.D. Integrins as Herpesvirus Receptors and Mediators of the Host Signalosome. Annu. Rev. Virol. 2016, 3, 215–236. [Google Scholar] [CrossRef]
- Holm, C.K.; Jensen, S.B.; Jakobsen, M.R.; Cheshenko, N.; Horan, K.A.; Moeller, H.B.; Gonzalez-Dosal, R.; Rasmussen, S.B.; Christensen, M.H.; Yarovinsky, T.O.; et al. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat. Immunol. 2012, 13, 737–743. [Google Scholar] [CrossRef]
- Reed, S.G.; Carter, D.; Casper, C.; Duthie, M.S.; Fox, C.B. Correlates of GLA family adjuvants’ activities. Semin. Immunol. 2018, 39, 22–29. [Google Scholar] [CrossRef]
- Sin, J.; Kim, J.J.; Pachuk, C.; Satishchandran, C.; Weiner, D.B. DNA vaccines encoding interleukin-8 and RANTES enhance antigen-specific Th1-type CD4(+) T-cell-mediated protective immunity against herpes simplex virus type 2 in vivo. J. Virol. 2000, 74, 11173–11180. [Google Scholar] [CrossRef]
- Yan, Y.; Hu, K.; Fu, M.; Deng, X.; Luo, S.; Tong, L.; Guan, X.; He, S.; Li, C.; Jin, W.; et al. CCL19 and CCL28 Assist Herpes Simplex Virus 2 Glycoprotein D To Induce Protective Systemic Immunity against Genital Viral Challenge. mSphere 2021, 6, e00058-21. [Google Scholar] [CrossRef] [PubMed]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Peligero-Cruz, C.; Givony, T.; Sebe-Pedros, A.; Dobes, J.; Kadouri, N.; Nevo, S.; Roncato, F.; Alon, R.; Goldfarb, Y.; Abramson, J. IL18 signaling promotes homing of mature Tregs into the thymus. Elife 2020, 9, e58213. [Google Scholar] [CrossRef] [PubMed]
- Schlecker, E.; Stojanovic, A.; Eisen, C.; Quack, C.; Falk, C.S.; Umansky, V.; Cerwenka, A. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J. Immunol. 2012, 189, 5602–5611. [Google Scholar] [CrossRef] [PubMed]
- Snelgrove, S.L.; Abeynaike, L.D.; Thevalingam, S.; Deane, J.A.; Hickey, M.J. Regulatory T Cell Transmigration and Intravascular Migration Undergo Mechanistically Distinct Regulation at Different Phases of the Inflammatory Response. J. Immunol. 2019, 203, 2850–2861. [Google Scholar] [CrossRef]
- Villarreal, D.O.; L’Huillier, A.; Armington, S.; Mottershead, C.; Filippova, E.V.; Coder, B.D.; Petit, R.G.; Princiotta, M.F. Targeting CCR8 Induces Protective Antitumor Immunity and Enhances Vaccine-Induced Responses in Colon Cancer. Cancer Res. 2018, 78, 5340–5348. [Google Scholar] [CrossRef]
- Bayry, J.; Tchilian, E.Z.; Davies, M.N.; Forbes, E.K.; Draper, S.J.; Kaveri, S.V.; Hill, A.V.; Kazatchkine, M.D.; Beverley, P.C.; Flower, D.R.; et al. In silico identified CCR4 antagonists target regulatory T cells and exert adjuvant activity in vaccination. Proc. Natl. Acad. Sci. USA 2008, 105, 10221–10226. [Google Scholar] [CrossRef]
- Catusse, J.; Parry, C.M.; Dewin, D.R.; Gompels, U.A. Inhibition of HIV-1 infection by viral chemokine U83A via high-affinity CCR5 interactions that block human chemokine-induced leukocyte chemotaxis and receptor internalization. Blood 2007, 109, 3633–3639. [Google Scholar] [CrossRef]
- Dewin, D.R.; Catusse, J.; Gompels, U.A. Identification and characterization of U83A viral chemokine, a broad and potent beta-chemokine agonist for human CCRs with unique selectivity and inhibition by spliced isoform. J. Immunol. 2006, 176, 544–556. [Google Scholar] [CrossRef]
- Tweedy, J.; Spyrou, M.A.; Pearson, M.; Lassner, D.; Kuhl, U.; Gompels, U.A. Complete Genome Sequence of Germline Chromosomally Integrated Human Herpesvirus 6A and Analyses Integration Sites Define a New Human Endogenous Virus with Potential to Reactivate as an Emerging Infection. Viruses 2016, 8, 19. [Google Scholar] [CrossRef]
- Willis, S.H.; Rux, A.H.; Peng, C.; Whitbeck, J.C.; Nicola, A.V.; Lou, H.; Hou, W.; Salvador, L.; Eisenberg, R.J.; Cohen, G.H. Examination of the kinetics of herpes simplex virus glycoprotein D binding to the herpesvirus entry mediator, using surface plasmon resonance. J. Virol. 1998, 72, 5937–5947. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, J.R.; Crane, R.T. Monophosphoryl lipid A (MPL) formulations for the next generation of vaccines. Methods 1999, 19, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Bourne, N.; Bravo, F.J.; Francotte, M.; Bernstein, D.I.; Myers, M.G.; Slaoui, M.; Stanberry, L.R. Herpes simplex virus (HSV) type 2 glycoprotein D subunit vaccines and protection against genital HSV-1 or HSV-2 disease in guinea pigs. J. Infect. Dis. 2003, 187, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Tweedy, J.G.; Escriva, E.; Topf, M.; Gompels, U.A. Analyses of Tissue Culture Adaptation of Human Herpesvirus-6A by Whole Genome Deep Sequencing Redefines the Reference Sequence and Identifies Virus Entry Complex Changes. Viruses 2017, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Tweedy, J.; Spyrou, M.A.; Hubacek, P.; Kuhl, U.; Lassner, D.; Gompels, U.A. Analyses of germline, chromosomally integrated human herpesvirus 6A and B genomes indicate emergent infection and new inflammatory mediators. J. Gen. Virol. 2015, 96, 370–389. [Google Scholar] [CrossRef] [PubMed]
- Gudnadottir, M.; Helgadottir, H.; Bjarnason, O.; Jonsdottir, K. Virus Isolated from the Brain of a Patient with Multiple Sclerosis. Exp. Neurol. 1964, 9, 85–95. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Stanberry, L.R.; Harrison, C.J.; Kappes, J.C.; Myers, M.G. Antibody response, recurrence patterns and subsequent herpes simplex virus type 2 (HSV-2) re-infection following initial HSV-2 infection of guinea-pigs: Effects of acyclovir. J. Gen. Virol. 1986, 67 Pt 8, 1601–1612. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Tepe, E.R.; Mester, J.C.; Arnold, R.L.; Stanberry, L.R.; Higgins, T. Effects of DNA immunization formulated with bupivacaine in murine and guinea pig models of genital herpes simplex virus infection. Vaccine 1999, 17, 1964–1969. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Farley, N.; Bravo, F.J.; Earwood, J.; McNeal, M.; Fairman, J.; Cardin, R. The adjuvant CLDC increases protection of a herpes simplex type 2 glycoprotein D vaccine in guinea pigs. Vaccine 2010, 28, 3748–3753. [Google Scholar] [CrossRef]
- Bernstein, D.I. Use of the Guinea pig model of genital herpes to evaluate vaccines and antivirals: Review. Antivir. Res. 2020, 180, 104821. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Cardin, R.D.; Smith, G.A.; Pickard, G.E.; Sollars, P.J.; Dixon, D.A.; Pasula, R.; Bravo, F.J. The R2 non-neuroinvasive HSV-1 vaccine affords protection from genital HSV-2 infections in a guinea pig model. NPJ Vaccines 2020, 5, 104. [Google Scholar] [CrossRef] [PubMed]
- Browne, H.; Bruun, B.; Minson, T. Plasma membrane requirements for cell fusion induced by herpes simplex virus type 1 glycoproteins gB, gD, gH and gL. J. Gen. Virol. 2001, 82, 1419–1422. [Google Scholar] [CrossRef] [PubMed]
- Gompels, U.; Minson, A. The properties and sequence of glycoprotein H of herpes simplex virus type 1. Virology 1986, 153, 230–247. [Google Scholar] [CrossRef]
- Cooper, R.S.; Georgieva, E.R.; Borbat, P.P.; Freed, J.H.; Heldwein, E.E. Structural basis for membrane anchoring and fusion regulation of the herpes simplex virus fusogen gB. Nat. Struct. Mol. Biol. 2018, 25, 416–424. [Google Scholar] [CrossRef]
- Vollmer, B.; Prazak, V.; Vasishtan, D.; Jefferys, E.E.; Hernandez-Duran, A.; Vallbracht, M.; Klupp, B.G.; Mettenleiter, T.C.; Backovic, M.; Rey, F.A.; et al. The prefusion structure of herpes simplex virus glycoprotein B. Sci. Adv. 2020, 6, eabc1726. [Google Scholar] [CrossRef]
- Protto, V.; Marcocci, M.E.; Miteva, M.T.; Piacentini, R.; Li Puma, D.D.; Grassi, C.; Palamara, A.T.; De Chiara, G. Role of HSV-1 in Alzheimer’s disease pathogenesis: A challenge for novel preventive/therapeutic strategies. Curr. Opin. Pharm. 2022, 63, 102200. [Google Scholar] [CrossRef]
- Lee, S.W.; Markham, P.F.; Coppo, M.J.; Legione, A.R.; Markham, J.F.; Noormohammadi, A.H.; Browning, G.F.; Ficorilli, N.; Hartley, C.A.; Devlin, J.M. Attenuated vaccines can recombine to form virulent field viruses. Science 2012, 337, 188. [Google Scholar] [CrossRef]
- Turner, A.; Bruun, B.; Minson, T.; Browne, H. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 1998, 72, 873–875. [Google Scholar] [CrossRef]
- Gompels, U.A.; Carss, A.L.; Saxby, C.; Hancock, D.C.; Forrester, A.; Minson, A.C. Characterization and sequence analyses of antibody-selected antigenic variants of herpes simplex virus show a conformationally complex epitope on glycoprotein H. J. Virol. 1991, 65, 2393–2401. [Google Scholar] [CrossRef]
- Gompels, U.A.; Minson, A.C. Antigenic properties and cellular localization of herpes simplex virus glycoprotein H synthesized in a mammalian cell expression system. J. Virol. 1989, 63, 4744–4755. [Google Scholar] [CrossRef]
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The structural basis of herpesvirus entry. Nat. Rev. Microbiol. 2021, 19, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schafer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Meng, B.; Abdullahi, A.; Ferreira, I.; Goonawardane, N.; Saito, A.; Kimura, I.; Yamasoba, D.; Gerber, P.P.; Fatihi, S.; Rathore, S.; et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef]
- Rice, S.A. Release of HSV-1 Cell-Free Virions: Mechanisms, Regulation, and Likely Role in Human-Human Transmission. Viruses 2021, 13, 2395. [Google Scholar] [CrossRef] [PubMed]
- Madavaraju, K.; Koganti, R.; Volety, I.; Yadavalli, T.; Shukla, D. Herpes Simplex Virus Cell Entry Mechanisms: An Update. Front. Cell Infect. Microbiol. 2020, 10, 617578. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.I.; Earwood, J.D.; Bravo, F.J.; Cohen, G.H.; Eisenberg, R.J.; Clark, J.R.; Fairman, J.; Cardin, R.D. Effects of herpes simplex virus type 2 glycoprotein vaccines and CLDC adjuvant on genital herpes infection in the guinea pig. Vaccine 2011, 29, 2071–2078. [Google Scholar] [CrossRef][Green Version]
- Grandi, N.; Tramontano, E. Human Endogenous Retroviruses Are Ancient Acquired Elements Still Shaping Innate Immune Responses. Front. Immunol. 2018, 9, 2039. [Google Scholar] [CrossRef]
- Singh, S.; Datta, A.; Schmidtchen, A.; Bhunia, A.; Malmsten, M. Tryptophan end-tagging for promoted lipopolysaccharide interactions and anti-inflammatory effects. Sci. Rep. 2017, 7, 212. [Google Scholar] [CrossRef]
- Yau, W.M.; Wimley, W.C.; Gawrisch, K.; White, S.H. The preference of tryptophan for membrane interfaces. Biochemistry 1998, 37, 14713–14718. [Google Scholar] [CrossRef]
- Catusse, J.; Clark, D.J.; Gompels, U.A. CCR5 signalling, but not DARC or D6 regulatory, chemokine receptors are targeted by herpesvirus U83A chemokine which delays receptor internalisation via diversion to a caveolin-linked pathway. J. Inflamm. (Lond) 2009, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Grochans, S.; Gutowska, I.; Barczak, K.; Baranowska-Bosiacka, I. CC Chemokines in a Tumor: A Review of Pro-Cancer and Anti-Cancer Properties of Receptors CCR5, CCR6, CCR7, CCR8, CCR9, and CCR10 Ligands. Int. J. Mol. Sci. 2020, 21, 619. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Kojder, K.; Siminska, D.; Bohatyrewicz, R.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. CC Chemokines in a Tumor: A Review of Pro-Cancer and Anti-Cancer Properties of the Ligands of Receptors CCR1, CCR2, CCR3, and CCR4. Int. J. Mol. Sci. 2020, 21, 8412. [Google Scholar] [CrossRef] [PubMed]
- Muri, J.; Cecchinato, V.; Cavalli, A.; Shanbhag, A.A.; Matkovic, M.; Biggiogero, M.; Maida, P.A.; Toscano, C.; Ghovehoud, E.; Danelon-Sargenti, G.; et al. Anti-chemokine antibodies after SARS-CoV-2 infection correlate with favorable disease course. bioRxiv 2022. [Google Scholar] [CrossRef]
- Boukhvalova, M.; McKay, J.; Mbaye, A.; Sanford-Crane, H.; Blanco, J.C.; Huber, A.; Herold, B.C. Efficacy of the Herpes Simplex Virus 2 (HSV-2) Glycoprotein D/AS04 Vaccine against Genital HSV-2 and HSV-1 Infection and Disease in the Cotton Rat Sigmodon hispidus Model. J. Virol. 2015, 89, 9825–9840. [Google Scholar] [CrossRef][Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conditions 1 >50% Protection | No Vaccine | gD Protein + MPL/Alum | gD DNA + VLM | gD DNA + VLM/CCL5 | gD DNA + VLM/VIT |
|---|---|---|---|---|---|
| Prevent acute disease | − (1/12) | 75% (9/12) | 100% (12/12) | 100% (12/12) | 75% (9/12) |
| Prevent virus replication | − (4/11) | 75% (9/12) | 92% (11/12) | 92% (11/12) | 83% (10/12) |
| Reduce latency load | − | + | + | + | + |
| Reduce latency detection DRG | − | − | − | − | + |
| Reduce recurrent disease | − (2/9) | 58% (7/12) | − (5/12) | − (6/12) | 58% (7/12) |
| Reduce recurrent virus shed | − | − | − | + | +/− |
| Reduce recurrent virus and disease | − | − | − | − | + |
| Overall | − | + | + | ++ | +++ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gompels, U.A.; Bravo, F.J.; Briggs, S.; Ameri, S.; Cardin, R.D.; Bernstein, D.I. Immunisation Using Novel DNA Vaccine Encoding Virus Membrane Fusion Complex and Chemokine Genes Shows High Protection from HSV-2. Viruses 2022, 14, 2317. https://doi.org/10.3390/v14112317
Gompels UA, Bravo FJ, Briggs S, Ameri S, Cardin RD, Bernstein DI. Immunisation Using Novel DNA Vaccine Encoding Virus Membrane Fusion Complex and Chemokine Genes Shows High Protection from HSV-2. Viruses. 2022; 14(11):2317. https://doi.org/10.3390/v14112317
Chicago/Turabian StyleGompels, Ursula A., Fernando J. Bravo, Sean Briggs, Shima Ameri, Rhonda D. Cardin, and David I. Bernstein. 2022. "Immunisation Using Novel DNA Vaccine Encoding Virus Membrane Fusion Complex and Chemokine Genes Shows High Protection from HSV-2" Viruses 14, no. 11: 2317. https://doi.org/10.3390/v14112317
APA StyleGompels, U. A., Bravo, F. J., Briggs, S., Ameri, S., Cardin, R. D., & Bernstein, D. I. (2022). Immunisation Using Novel DNA Vaccine Encoding Virus Membrane Fusion Complex and Chemokine Genes Shows High Protection from HSV-2. Viruses, 14(11), 2317. https://doi.org/10.3390/v14112317