

Bovine Papillomavirus Type 1 or 2 Virion-Infected Primary Fibroblasts Constitute a Near-Natural Equine Sarcoid Model
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
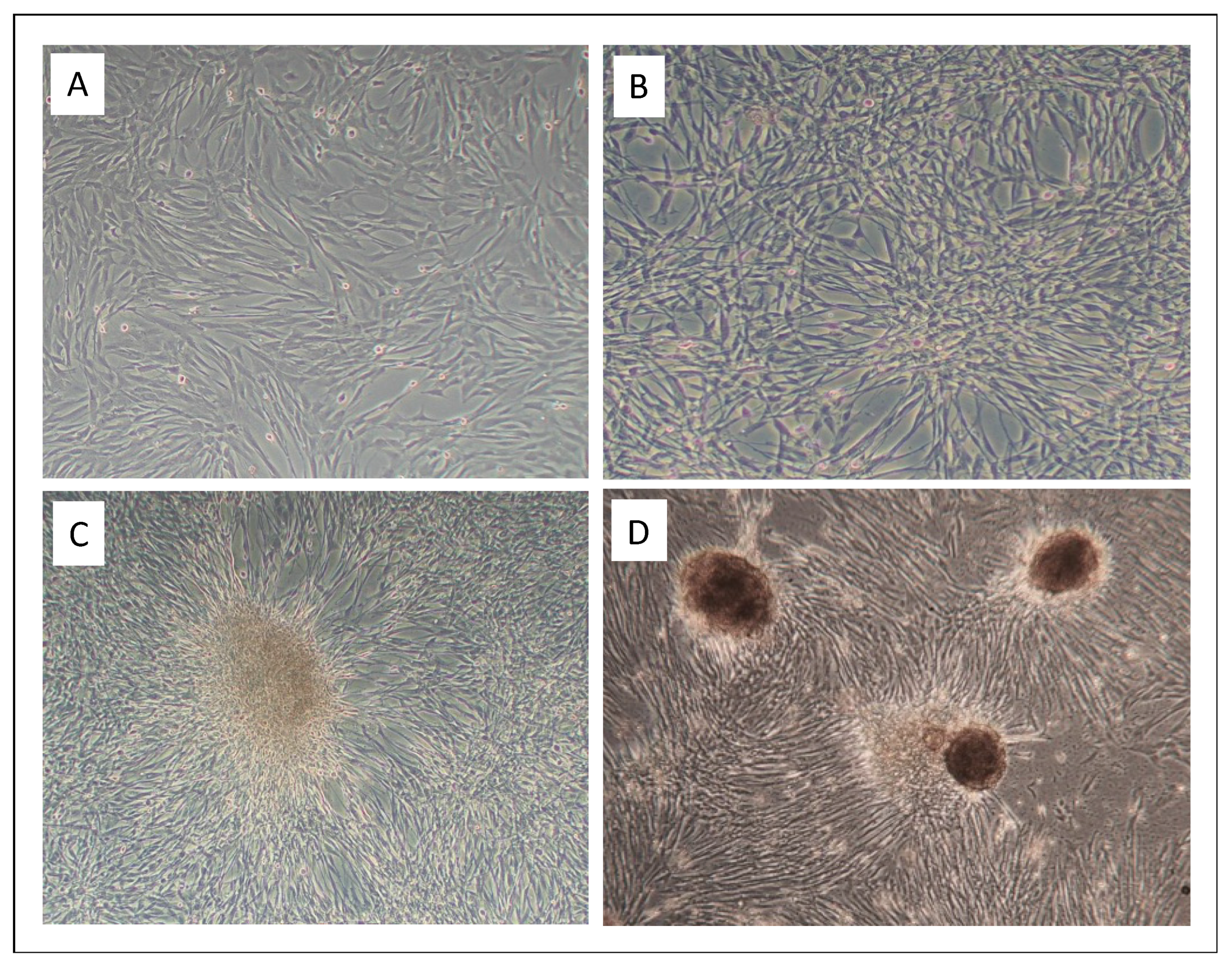
3.1. Infection of Equine Primary Fibroblasts with BPV1 and -2 Virions Induced Hyperproliferation, Loss of Contact Inhibition and Most Probably Immortalization
3.2. Low Virion Numbers Sufficed to Produce Infection
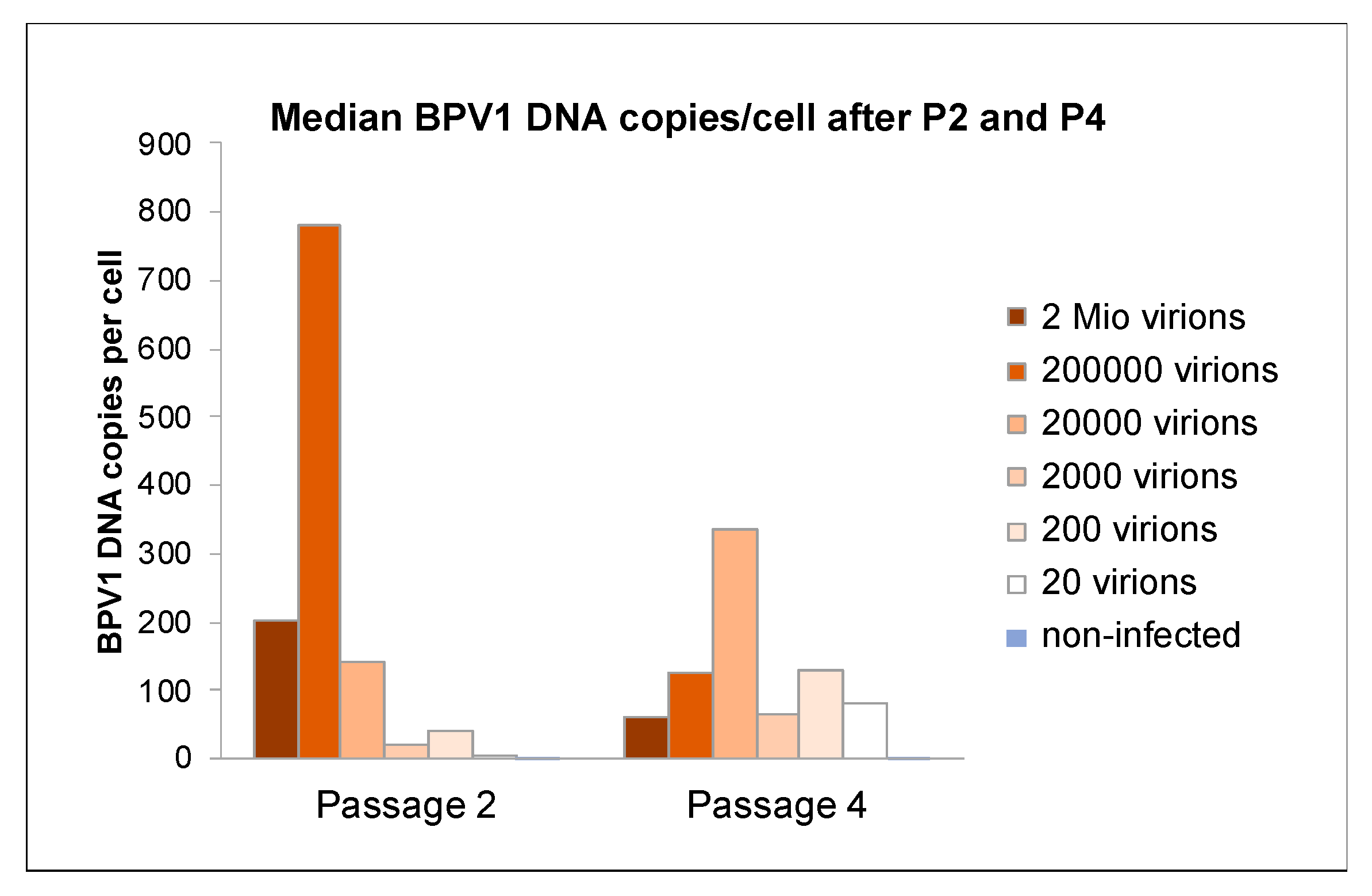
3.3. Viral DNA Loads Reached a Physiological Level after Four Passages
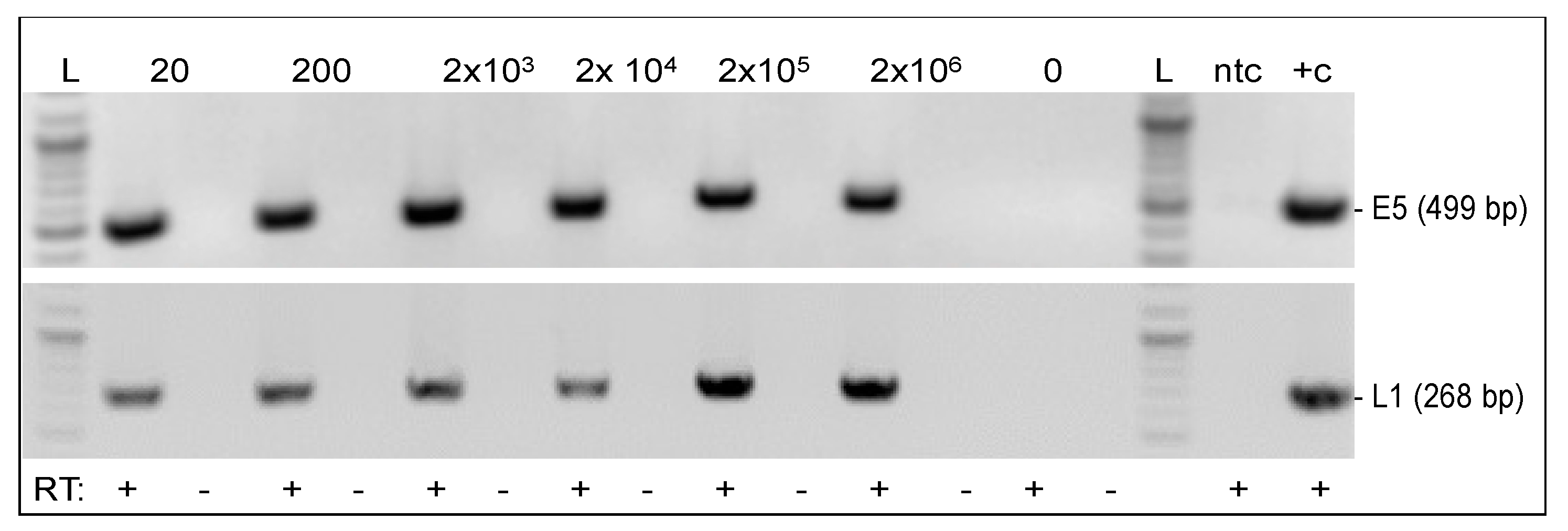
3.4. Viral Genes E5 and L1 Were Transcribed in Infected Fibroblasts
3.5. Infected Fibroblasts Were Not Permissive for Productive BPV1 and BPV2 Infection
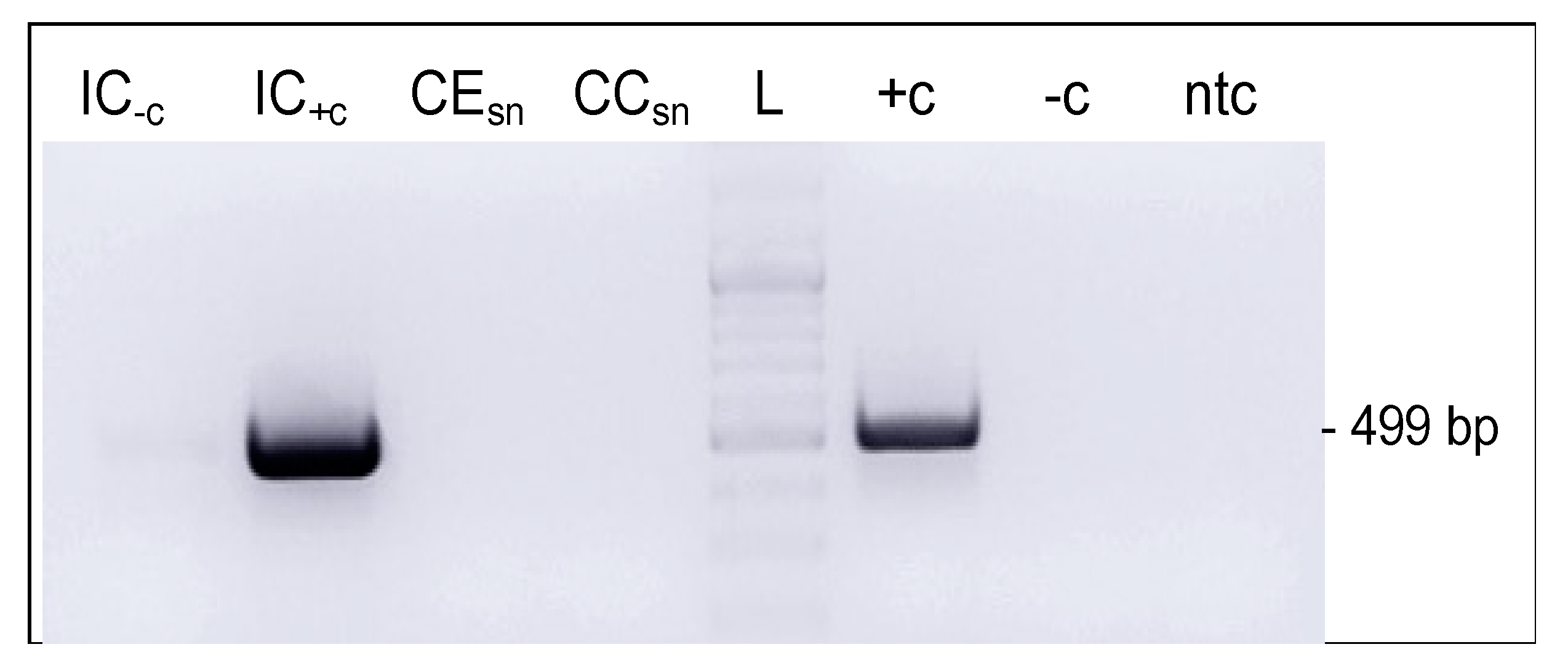
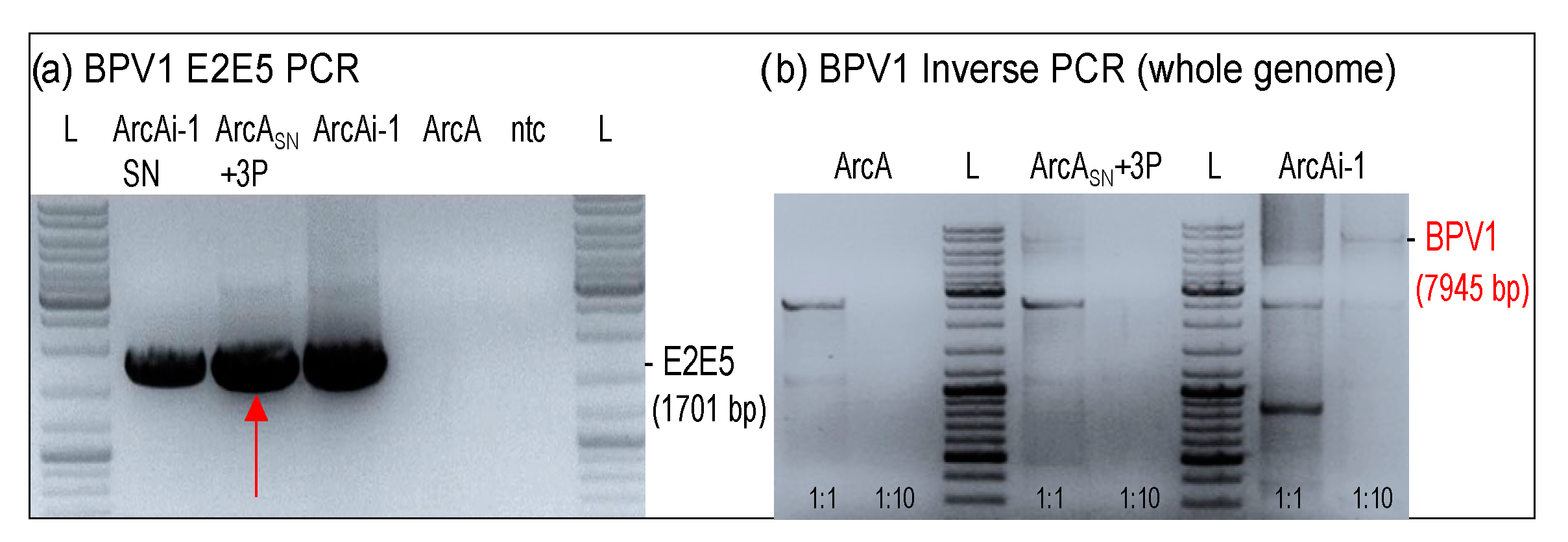
3.6. BPV1 DNA Could Be Transferred from Infected to Non-Infected Cells via Cell-Free Culture Medium
3.7. Extracellular Vesicles Secreted by Infected Cells Harbored Viral DNA
4. Discussion
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campo, M.S. Introduction. In Papillomavirus Research: From Natural History to Vaccines and Beyond, 1st ed.; Campo, M.S., Ed.; Caister Academic Press: Norfolk, UK, 2006; pp. 1–2. [Google Scholar]
- de Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, G.; Ellsmore, V.A.; O’Brien, P.M.; Reid, S.W.; Love, S.; Campo, M.S.; Nasir, L. Association of bovine papillomavirus with the equine sarcoid. J. Gen. Virol. 2003, 84, 1055–1062. [Google Scholar] [CrossRef]
- Lunardi, M.; de Alcantara, B.K.; Otonel, R.A.; Rodrigues, W.B.; Alfieri, A.F.; Alfieri, A.A. Bovine papillomavirus type 13 DNA in equine sarcoids. J. Clin. Microbiol. 2013, 51, 2167–2171. [Google Scholar] [CrossRef] [Green Version]
- Lunardi, M.; Alfieri, A.A.; Otonel, R.A.; de Alcantara, B.K.; Rodrigues, W.B.; de Miranda, A.B.; Alfieri, A.F. Genetic characterization of a novel bovine papillomavirus member of the Deltapapillomavirus genus. Vet. Microbiol. 2013, 162, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Campo, M.S. Bovine papillomavirus: Old system, new lessons? In Papillomavirus Research: From Natural History to Vaccines and Beyond, 1st ed.; Campo, M.S., Ed.; Caister Academic Press: Norfolk, UK, 2006; pp. 373–387. [Google Scholar]
- Goodrich, L.; Gerber, H.; Marti, E.; Antczak, D.F. Equine sarcoids. Vet. Clin. North Am. Equine Pract. 1998, 14, 607–623. [Google Scholar] [CrossRef] [PubMed]
- Studer, S.; Gerber, V.; Straub, R.; Brehm, W.; Gaillard, C.; Luth, A.; Burger, D. Prevalence of hereditary diseases in three-year-old Swiss Warmblood horses. Schweiz. Arch. Tierheilkd. 2007, 149, 161–171. [Google Scholar] [CrossRef]
- Knottenbelt, D.C. A suggested clinical classification for the equine sarcoid. Clin. Tech. Equine Pract. 2005, 4, 278–295. [Google Scholar] [CrossRef]
- Bogaert, L.; Van Poucke, M.; De Baere, C.; Dewulf, J.; Peelman, L.; Ducatelle, R.; Gasthuys, F.; Martens, A. Bovine papillomavirus load and mRNA expression, cell proliferation and p53 expression in four clinical types of equine sarcoid. J. Gen. Virol. 2007, 88, 2155–2161. [Google Scholar] [CrossRef]
- Haralambus, R.; Burgstaller, J.; Klukowska-Rotzler, J.; Steinborn, R.; Buchinger, S.; Gerber, V.; Brandt, S. Intralesional bovine papillomavirus DNA loads reflect severity of equine sarcoid disease. Equine Vet. J. 2010, 42, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Amtmann, E.; Muller, H.; Sauer, G. Equine connective tissue tumors contain unintegrated bovine papilloma virus DNA. J. Virol. 1980, 35, 962–964. [Google Scholar] [CrossRef]
- Nasir, L.; Brandt, S. Papillomavirus associated diseases of the horse. Vet. Microbiol. 2013, 167, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Bogaert, L.; Martens, A.; Van Poucke, M.; Ducatelle, R.; De Cock, H.; Dewulf, J.; De Baere, C.; Peelman, L.; Gasthuys, F. High prevalence of bovine papillomaviral DNA in the normal skin of equine sarcoid-affected and healthy horses. Vet. Microbiol. 2008, 129, 58–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, S.; Haralambus, R.; Shafti-Keramat, S.; Steinborn, R.; Stanek, C.; Kirnbauer, R. A subset of equine sarcoids harbours BPV-1 DNA in a complex with L1 major capsid protein. Virology 2008, 375, 433–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, E.A.; Theon, A.P.; Madewell, B.R.; Griffey, S.M.; Hitchcock, M.E. Bovine papillomavirus DNA in neoplastic and nonneoplastic tissues obtained from horses with and without sarcoids in the western United States. Am. J. Vet. Res. 2001, 62, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Martens, A.; De Moor, A.; Demeulemeester, J.; Ducatelle, R. Histopathological characteristics of five clinical types of equine sarcoid. Res. Vet. Sci. 2000, 69, 295–300. [Google Scholar] [CrossRef]
- Trenfield, K.; Spradbrow, P.B.; Vanselow, B. Sequences of papillomavirus DNA in equine sarcoids. Equine Vet. J. 1985, 17, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Knottenbelt, D.C. Sarcoid. In Pascoe’s Principles and Practice of Equine Dermatology; Knottenbelt, D.C., Ed.; Saunders Elsevier: London, UK, 2009; pp. 387–407. [Google Scholar]
- Knottenbelt, D.C. The Equine Sarcoid: Why Are There so Many Treatment Options? Vet. Clin. N. Am. Equine Pract. 2019, 35, 243–262. [Google Scholar] [CrossRef] [PubMed]
- Tarwid, J.N.; Fretz, P.B.; Clark, E.G. Equine sarcoids: A study with emphasis on pathological diagnosis. Compend. Contin. Educ. Pract. Vet. 1985, 7, 293–300. [Google Scholar]
- Chow, L.T.; Broker, T.R. Mechanisms and regulation of papillomavirus DNA replication. In Papillomavirus Research: From Natural History to Vaccines and Beyond, 1st ed.; Campo, M.S., Ed.; Caister Academic Press: Norfolk, UK, 2006; pp. 53–71. [Google Scholar]
- Doorbar, J. Model systems of human papillomavirus-associated disease. J. Pathol. 2016, 238, 166–179. [Google Scholar] [CrossRef] [Green Version]
- Christensen, N.D.; Budgeon, L.R.; Cladel, N.M.; Hu, J. Recent advances in preclinical model systems for papillomaviruses. Virus Res. 2017, 231, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Biryukov, J.; Meyers, C. Papillomavirus Infectious Pathways: A Comparison of Systems. Viruses 2015, 7, 4303–4325. [Google Scholar] [CrossRef]
- Marchetti, B.; Gault, E.A.; Cortese, M.S.; Yuan, Z.; Ellis, S.A.; Nasir, L.; Campo, M.S. Bovine papillomavirus type 1 oncoprotein E5 inhibits equine MHC class I and interacts with equine MHC I heavy chain. J. Gen. Virol. 2009, 90, 2865–2870. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Gault, E.A.; Campo, M.S.; Nasir, L. Different contribution of bovine papillomavirus type 1 oncoproteins to the transformation of equine fibroblasts. J. Gen. Virol. 2011, 92, 773–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Z.Q.; Bennett, L.; Campo, M.S.; Nasir, L. Bovine papillomavirus type 1 E2 and E7 proteins down-regulate Toll Like Receptor 4 (TLR4) expression in equine fibroblasts. Virus Res. 2010, 149, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.Q.; Gault, E.A.; Gobeil, P.; Nixon, C.; Campo, M.S.; Nasir, L. Establishment and characterization of equine fibroblast cell lines transformed in vivo and in vitro by BPV-1: Model systems for equine sarcoids. Virology 2008, 373, 352–361. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.Q.; Nicolson, L.; Marchetti, B.; Gault, E.A.; Campo, M.S.; Nasir, L. Transcriptional changes induced by bovine papillomavirus type 1 in equine fibroblasts. J. Virol. 2008, 82, 6481–6491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corteggio, A.; Di Geronimo, O.; Roperto, S.; Roperto, F.; Borzacchiello, G. Bovine papillomavirus E7 oncoprotein binds to p600 in naturally occurring equine sarcoids. J. Gen. Virol. 2011, 92, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Campo, M.S.; Spandidos, D.A. Molecularly cloned bovine papillomavirus DNA transforms mouse fibroblasts in vitro. J. Gen. Virol. 1983, 64, 549–557. [Google Scholar] [CrossRef]
- Jarrett, W.F.; O’Neil, B.W.; Gaukroger, J.M.; Laird, H.M.; Smith, K.T.; Campo, M.S. Studies on vaccination against papillomaviruses: A comparison of purified virus, tumour extract and transformed cells in prophylactic vaccination. Vet. Rec. 1990, 126, 449–452. [Google Scholar] [PubMed]
- Jindra, C.; Hainisch, E.K.; Rummele, A.; Wolschek, M.; Muster, T.; Brandt, S. Influenza virus vector iNS1 expressing bovine papillomavirus 1 (BPV1) antigens efficiently induces tumour regression in equine sarcoid patients. PLoS ONE 2021, 16, e0260155. [Google Scholar] [CrossRef] [PubMed]
- Shafti-Keramat, S.; Handisurya, A.; Kriehuber, E.; Meneguzzi, G.; Slupetzky, K.; Kirnbauer, R. Different heparan sulfate proteoglycans serve as cellular receptors for human papillomaviruses. J. Virol. 2003, 77, 13125–13135. [Google Scholar] [CrossRef]
- Dvoretzky, I.; Shober, R.; Chattopadhyay, S.K.; Lowy, D.R. A quantitative in vitro focus assay for bovine papilloma virus. Virology 1980, 103, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Roden, R.B.; Greenstone, H.L.; Kirnbauer, R.; Booy, F.P.; Jessie, J.; Lowy, D.R.; Schiller, J.T. In vitro generation and type-specific neutralization of a human papillomavirus type 16 virion pseudotype. J. Virol. 1996, 70, 5875–5883. [Google Scholar] [CrossRef] [Green Version]
- Brandt, S.; Haralambus, R.; Schoster, A.; Kirnbauer, R.; Stanek, C. Peripheral blood mononuclear cells represent a reservoir of bovine papillomavirus DNA in sarcoid-affected equines. J. Gen. Virol. 2008, 89, 1390–1395. [Google Scholar] [CrossRef]
- Brodesser, D.; Walter, I.; Tschulenk, W.; Brandt, S.; Brem, G.; Trautinger, F.; Burgstaller, J.P. Analysis of DNA Content from Human Melanoma Cell Line Derived Extracellular Vesicles. In Proceedings of the 9th Gene Quantification Event, qPCR, dPCR & NGS, Freising, Germany, 18–22 March 2019. [Google Scholar]
- Howley, P.M.; Lowy, D.R. Papillomaviruses and their replication. In Fields Virology, 4th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; Volume 2, pp. 2197–2230. [Google Scholar]
- Nasir, L.; Reid, S.W.J. Bovine papillomaviruses and equine sarcoids. In Papillomavirus Research: From Natural History to Vaccines and Beyond, 1st ed.; Campo, M.S., Ed.; Caister Academic Press: Norfolk, UK, 2006; Volume 1, pp. 389–397. [Google Scholar]
- Aksoy, P.; Gottschalk, E.Y.; Meneses, P.I. HPV entry into cells. Mutat. Res. Rev. Mutat. Res. 2017, 772, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiller, J.T.; Day, P.M.; Kines, R.C. Current understanding of the mechanism of HPV infection. Gynecol. Oncol. 2010, 118, S12–S17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fons, N.R.; Kines, R.C.; Thompson, C.D.; Day, P.M.; Lowy, D.R.; Schiller, J.T. Chondroitin Sulfate Proteoglycans Are De Facto Cellular Receptors for Human Papillomavirus 16 under High Serum Conditions. J. Virol. 2022, 96, e0185721. [Google Scholar] [CrossRef] [PubMed]
- Bogaert, L.; Martens, A.; Kast, W.M.; Van Marck, E.; De Cock, H. Bovine papillomavirus DNA can be detected in keratinocytes of equine sarcoid tumors. Vet. Microbiol. 2010, 146, 269–275. [Google Scholar] [CrossRef]
- Brandt, S.; Tober, R.; Corteggio, A.; Burger, S.; Sabitzer, S.; Walter, I.; Kainzbauer, C.; Steinborn, R.; Nasir, L.; Borzacchiello, G. BPV-1 infection is not confined to the dermis but also involves the epidermis of equine sarcoids. Vet. Microbiol. 2011, 150, 35–40. [Google Scholar] [CrossRef]
- Wilson, A.D.; Armstrong, E.L.; Gofton, R.G.; Mason, J.; De Toit, N.; Day, M.J. Characterisation of early and late bovine papillomavirus protein expression in equine sarcoids. Vet. Microbiol. 2013, 162, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Nasir, L.; Campo, M.S. Bovine papillomaviruses: Their role in the aetiology of cutaneous tumours of bovids and equids. Vet. Dermatol. 2008, 19, 243–254. [Google Scholar] [CrossRef]
- Venuti, A.; Paolini, F.; Nasir, L.; Corteggio, A.; Roperto, S.; Campo, M.S.; Borzacchiello, G. Papillomavirus E5: The smallest oncoprotein with many functions. Mol. Cancer 2011, 10, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishnaiah, V.; Thumann, C.; Fofana, I.; Habersetzer, F.; Pan, Q.; de Ruiter, P.E.; Willemsen, R.; Demmers, J.A.; Stalin Raj, V.; Jenster, G.; et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13109–13113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acevedo-Sanchez, V.; Rodriguez-Hernandez, R.M.; Aguilar-Ruiz, S.R.; Torres-Aguilar, H.; Romero-Tlalolini, M.L.A. Extracellular Vesicles in Cervical Cancer and HPV Infection. Membranes 2021, 11, 453. [Google Scholar] [CrossRef] [PubMed]
- Guenat, D.; Hermetet, F.; Pretet, J.L.; Mougin, C. Exosomes and Other Extracellular Vesicles in HPV Transmission and Carcinogenesis. Viruses 2017, 9, 211. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hainisch, E.K.; Jindra, C.; Reicher, P.; Miglinci, L.; Brodesser, D.M.; Brandt, S. Bovine Papillomavirus Type 1 or 2 Virion-Infected Primary Fibroblasts Constitute a Near-Natural Equine Sarcoid Model. Viruses 2022, 14, 2658. https://doi.org/10.3390/v14122658
Hainisch EK, Jindra C, Reicher P, Miglinci L, Brodesser DM, Brandt S. Bovine Papillomavirus Type 1 or 2 Virion-Infected Primary Fibroblasts Constitute a Near-Natural Equine Sarcoid Model. Viruses. 2022; 14(12):2658. https://doi.org/10.3390/v14122658
Chicago/Turabian StyleHainisch, Edmund K., Christoph Jindra, Paul Reicher, Lea Miglinci, Daniela M. Brodesser, and Sabine Brandt. 2022. "Bovine Papillomavirus Type 1 or 2 Virion-Infected Primary Fibroblasts Constitute a Near-Natural Equine Sarcoid Model" Viruses 14, no. 12: 2658. https://doi.org/10.3390/v14122658