
The Persistence of HIV Diversity, Transcription, and Nef Protein in Kaposi’s Sarcoma Tumors during Antiretroviral Therapy
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Source of Specimens Derived from KS Subjects
2.2. RNA/DNA Extraction
2.3. HIV Sequencing
2.4. Sequence Alignment
2.5. Phylogenetic Analysis
2.6. Immunohistochemistry (IHC)
3. Results
3.1. Clinical Characteristics of the Study Cohort and Sample Integrity
3.2. HIV DNA and RNA Sequences Were Obtained from KS Tumors at Baseline and Post-Treatment
3.3. Post-Treatment Viral Diversity Remained High despite a Significant Reduction in HIV DNA
3.4. Completely Spliced RNA Transcripts Were Amplified More Frequently Post-Treatment
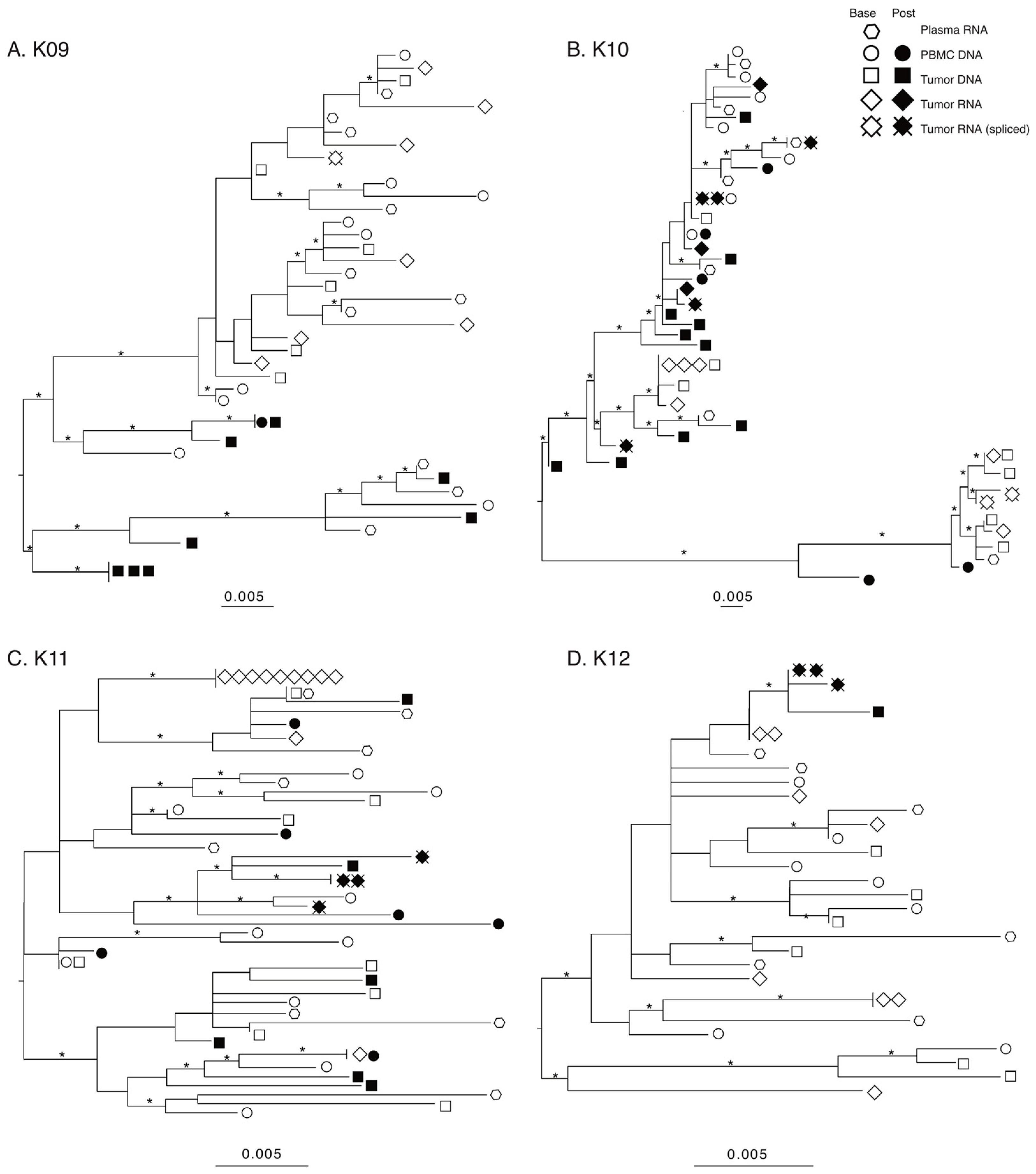
3.5. Lack of Compartmentalization and Maintenance of HIV Genetic Diversity in KS Tumors after 180–280 Days of ART
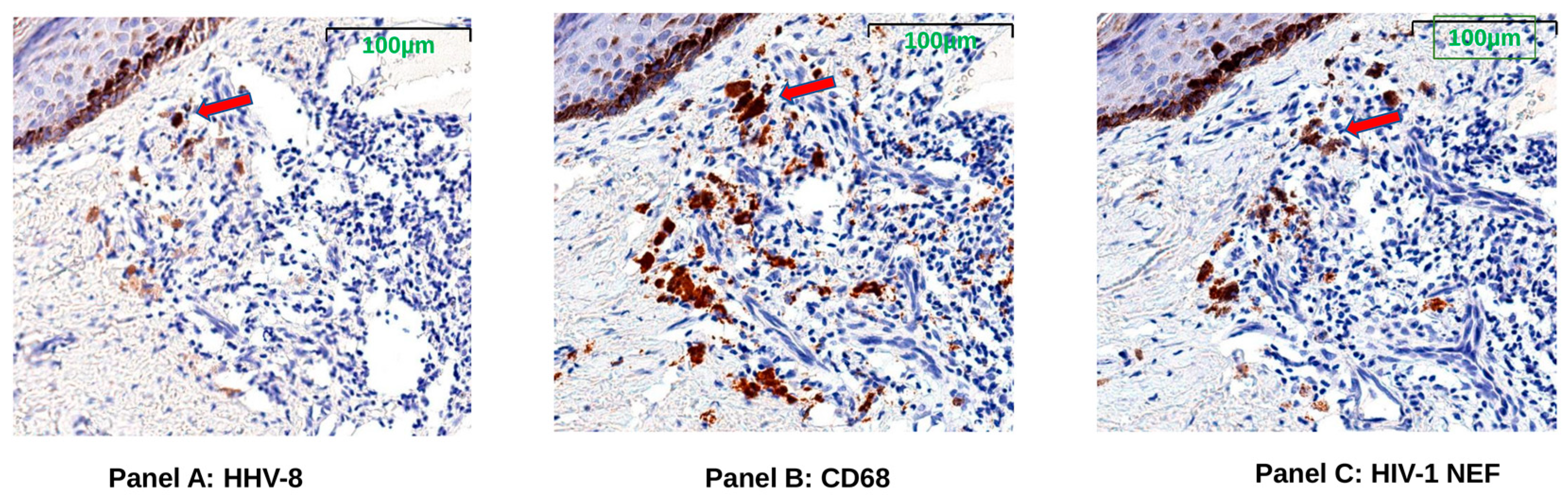
3.6. HIV Nef Protein Persists in CD68+ Macrophages after ART
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.W.; Dittmer, D.P. Diagnosis and Treatment of Kaposi Sarcoma. Am. J. Clin. Dermatol. 2017, 18, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Ngalamika, O.; Munsaka, S.; Lidenge, S.J.; West, J.T.; Wood, C. Antiretroviral Therapy for HIV-Associated Cutaneous Kaposi’s Sarcoma: Clinical, HIV-Related, and Sociodemographic Predictors of Outcome. AIDS Res. Hum. Retrovir. 2021, 37, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Dalla Pria, A.; Pinato, D.J.; Bracchi, M.; Bower, M. Recent advances in HIV-associated Kaposi sarcoma. F1000Research 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Dorsey, G.; Hladik, W.; Rosenthal, P.J.; Brander, C.; Neilands, T.B.; Mbisa, G.; Whitby, D.; Kiepiela, P.; Mosam, A.; et al. Kaposi sarcoma-associated herpesvirus (KSHV) seroprevalence in population-based samples of African children: Evidence for at least 2 patterns of KSHV transmission. J. Infect. Dis. 2009, 200, 430–438. [Google Scholar] [CrossRef]
- Krown, S.E.; Moser, C.B.; MacPhail, P.; Matining, R.M.; Godfrey, C.; Caruso, S.R.; Hosseinipour, M.C.; Samaneka, W.; Nyirenda, M.; Busakhala, N.W.; et al. Treatment of advanced AIDS-associated Kaposi sarcoma in resource-limited settings: A three-arm, open-label, randomised, non-inferiority trial. Lancet 2020, 395, 1195–1207. [Google Scholar] [CrossRef]
- Freeman, E.E.; Busakhala, N.; Regan, S.; Asirwa, F.C.; Wenger, M.; Seth, D.; Moon, K.C.; Semeere, A.; Maurer, T.; Wools-Kaloustian, K.; et al. Real-world use of chemotherapy for Kaposi’s sarcoma in a large community-based HIV primary care system in Kenya. BMC Cancer 2020, 20, 71. [Google Scholar] [CrossRef]
- Nyirenda, M.; Ngongondo, M.; Kang, M.; Umbleja, T.; Krown, S.E.; Godfrey, C.; Samaneka, W.; Mngqibisa, R.; Hoagland, B.; Mwelase, N.; et al. Early Progression and Immune Reconstitution Inflammatory Syndrome during Treatment of Mild-To-Moderate Kaposi Sarcoma in Sub-Saharan Africa and South America: Incidence, Long-Term Outcomes, and Effects of Early Chemotherapy. J. Acquir. Immune Defic. Syndr. 2020, 84, 422–429. [Google Scholar] [CrossRef]
- Ensoli, B.; Sgadari, C.; Barillari, G.; Sirianni, M.C.; Sturzl, M.; Monini, P. Biology of Kaposi’s sarcoma. Eur. J. Cancer 2001, 37, 1251–1269. [Google Scholar] [CrossRef]
- Lamers, S.L.; Rose, R.; Nolan, D.J.; Fogel, G.B.; Barbier, A.E.; Salemi, M.; McGrath, M.S. HIV-1 Evolutionary Patterns Associated with Metastatic Kaposi’s Sarcoma during AIDS. Sarcoma 2016, 2016, 4510483. [Google Scholar] [CrossRef]
- Lidenge, S.J.; Tso, F.Y.; Ngalamika, O.; Kolape, J.; Ngowi, J.R.; Mwaiselage, J.; Wood, C.; West, J.T. Lack of CD8+ T-cell co-localization with Kaposi’s sarcoma-associated herpesvirus infected cells in Kaposi’s sarcoma tumors. Oncotarget 2020, 11, 1556–1572. [Google Scholar] [CrossRef] [PubMed]
- Joest, B.; Kempf, W.; Berisha, A.; Peyk, P.; Tronnier, M.; Mitteldorf, C. Stage-related PD-L1 expression in Kaposi sarcoma tumor microenvironment. J. Cutan. Pathol. 2020, 47, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Lin, T.Y.; Chang, L.; Yeh, W.W.; Huang, S.C.; Chen, T.Y.; Hsieh, Y.T.; Chang, P.C. HIV-1 Tat Interacts with a Kaposi’s Sarcoma-Associated Herpesvirus Reactivation-Upregulated Antiangiogenic Long Noncoding RNA, LINC00313, and Antagonizes Its Function. J. Virol. 2020, 94, e01280-19. [Google Scholar] [CrossRef] [PubMed]
- Ensoli, B.; Gendelman, R.; Markham, P.; Fiorelli, V.; Colombini, S.; Raffeld, M.; Cafaro, A.; Chang, H.-K.; Brady, J.N.; Gallo, R.C. Synergy between basic fibroblast growth factor and HIV-1 Tat protein in induction of Kaposi’s sarcoma. Nature 1994, 371, 674–680. [Google Scholar] [CrossRef]
- Chen, L.; Feng, Z.; Yue, H.; Bazdar, D.; Mbonye, U.; Zender, C.; Harding, C.V.; Bruggeman, L.; Karn, J.; Sieg, S.F.; et al. Exosomes derived from HIV-1-infected cells promote growth and progression of cancer via HIV TAR RNA. Nat. Commun. 2018, 9, 4585. [Google Scholar] [CrossRef]
- Valentín-Guillama, G.; López, S.; Kucheryavykh, Y.V.; Chorna, N.E.; Pérez, J.; Ortiz-Rivera, J.; Inyushin, M.; Makarov, V.; Valentín-Acevedo, A.; Quinones-Hinojosa, A.; et al. HIV-1 Envelope Protein gp120 Promotes Proliferation and the Activation of Glycolysis in Glioma Cell. Cancers 2018, 10, 301. [Google Scholar] [CrossRef]
- Caccuri, F.; Giordano, F.; Barone, I.; Mazzuca, P.; Giagulli, C.; Andò, S.; Caruso, A.; Marsico, S. HIV-1 matrix protein p17 and its variants promote human triple negative breast cancer cell aggressiveness. Infect. Agents Cancer 2017, 12, 49. [Google Scholar] [CrossRef][Green Version]
- Zhu, X.; Guo, Y.; Yao, S.; Yan, Q.; Xue, M.; Hao, T.; Zhou, F.; Zhu, J.; Qin, D.; Lu, C. Synergy between Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6 and HIV-1 Nef protein in promotion of angiogenesis and oncogenesis: Role of the AKT signaling pathway. Oncogene 2014, 33, 1986–1996. [Google Scholar] [CrossRef]
- Kazer, S.W.; Walker, B.D.; Shalek, A.K. Evolution and Diversity of Immune Responses during Acute HIV Infection. Immunity 2020, 53, 908–924. [Google Scholar] [CrossRef]
- Dittmer, D.P.; Damania, B. Kaposi sarcoma-associated herpesvirus: Immunobiology, oncogenesis, and therapy. J. Clin. Investig. 2016, 126, 3165–3175. [Google Scholar] [CrossRef]
- Duette, G.; Hiener, B.; Morgan, H.; Mazur, F.G.; Mathivanan, V.; Horsburgh, B.A.; Fisher, K.; Tong, O.; Lee, E.; Ahn, H.; et al. The HIV-1 proviral landscape reveals Nef contributes to HIV-1 persistence in effector memory CD4+ T-cells. J. Clin. Investig. 2022, 132, 7. [Google Scholar] [CrossRef] [PubMed]
- Mwimanzi, F.; Toyoda, M.; Mahiti, M.; Mann, J.K.; Martin, J.N.; Bangsberg, D.; Brockman, M.A.; Goulder, P.; Kirchhoff, F.; Brumme, Z.L.; et al. Resistance of Major Histocompatibility Complex Class B (MHC-B) to Nef-Mediated Downregulation Relative to that of MHC-A Is Conserved among Primate Lentiviruses and Influences Antiviral T Cell Responses in HIV-1-Infected Individuals. J. Virol. 2018, 92, e01409-17. [Google Scholar] [CrossRef] [PubMed]
- Dirk, B.S.; Pawlak, E.N.; Johnson, A.L.; Van Nynatten, L.R.; Jacob, R.A.; Heit, B.; Dikeakos, J.D. HIV-1 Nef sequesters MHC-I intracellularly by targeting early stages of endocytosis and recycling. Sci. Rep. 2016, 6, 37021. [Google Scholar] [CrossRef]
- Williams, M.; Roeth, J.F.; Kasper, M.R.; Fleis, R.I.; Przybycin, C.G.; Collins, K.L. Direct binding of human immunodeficiency virus type 1 Nef to the major histocompatibility complex class I (MHC-I) cytoplasmic tail disrupts MHC-I trafficking. J. Virol. 2002, 76, 12173–12184. [Google Scholar] [CrossRef]
- Collins, K.L.; Chen, B.K.; Kalams, S.A.; Walker, B.D.; Baltimore, D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 1998, 391, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.B.; Gandhi, R.T.; Davis, D.M.; Mandelboim, O.; Chen, B.K.; Strominger, J.L.; Baltimore, D. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 1999, 10, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, S.; Erdtmann, L.; Benichou, S.; Berlioz-Torrent, C.; Liu, L.; Benarous, R.; Heard, J.M.; Schwartz, O. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 1998, 8, 483–495. [Google Scholar] [CrossRef]
- Chaillon, A.; Gianella, S.; Dellicour, S.; Rawlings, S.A.; Schlub, T.E.; de Oliveira, M.F.; Ignacio, C.; Porrachia, M.; Vrancken, B.; Smith, D.M. HIV persists throughout deep tissues with repopulation from multiple anatomical sources. J. Clin. Investig. 2020, 130, 1699–1712. [Google Scholar] [CrossRef]
- Rose, R.; Lamers, S.L.; Nolan, D.J.; Maidji, E.; Faria, N.R.; Pybus, O.G.; Dollar, J.J.; Maruniak, S.A.; McAvoy, A.C.; Salemi, M.; et al. HIV Maintains an Evolving and Dispersed Population in Multiple Tissues during Suppressive Combined Antiretroviral Therapy in Individuals with Cancer. J. Virol. 2016, 90, 8984–8993. [Google Scholar] [CrossRef]
- Simonetti, F.R.; Sobolewski, M.D.; Fyne, E.; Shao, W.; Spindler, J.; Hattori, J.; Anderson, E.M.; Watters, S.A.; Hill, S.; Wu, X. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 1883–1888. [Google Scholar] [CrossRef]
- Salemi, M.; Lamers, S.L.; Huysentruyt, L.C.; Galligan, D.; Gray, R.R.; Morris, A.; McGrath, M.S. Distinct patterns of HIV-1 evolution within metastatic tissues in patients with non-Hodgkins lymphoma. PLoS ONE 2009, 4, e8153. [Google Scholar] [CrossRef] [PubMed]
- Lamers, S.L.; Salemi, M.; Galligan, D.C.; De Oliveira, T.; Fogel, G.B.; Granier, S.C.; Zhao, L.; Brown, J.N.; Morris, A.; Masliah, E.; et al. Extensive HIV-1 intra-host recombination is common in tissues with abnormal histopathology. PLoS ONE 2009, 4, e5065. [Google Scholar] [CrossRef] [PubMed]
- Byakwaga, H.; Hunt, P.W.; Laker-Oketta, M.; Glidden, D.; Huang, Y.; Bwana, B.M.; Mocello, A.R.; Bennett, J.; Walusansa, V.; Dollard, S.C.; et al. The Kynurenine Pathway of Tryptophan Catabolism and AIDS-Associated Kaposi Sarcoma in Africa. J. Acquir. Immune Defic. Syndr. 2015, 70, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Amerson, E.; Woodruff, C.M.; Forrestel, A.; Wenger, M.; McCalmont, T.; LeBoit, P.; Maurer, T.; Laker-Oketta, M.; Muyindike, W.; Bwana, M.; et al. Accuracy of Clinical Suspicion and Pathologic Diagnosis of Kaposi Sarcoma in East Africa. J. Acquir. Immune Defic. Syndr. 2016, 71, 295–301. [Google Scholar] [CrossRef]
- Laker-Oketta, M.O.; Wenger, M.; Semeere, A.; Castelnuovo, B.; Kambugu, A.; Lukande, R.; Asirwa, F.C.; Busakhala, N.; Buziba, N.; Diero, L.; et al. Task Shifting and Skin Punch for the Histologic Diagnosis of Kaposi’s Sarcoma in Sub-Saharan Africa: A Public Health Solution to a Public Health Problem. Oncology 2015, 89, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Banin, A.N.; Tuen, M.; Bimela, J.S.; Tongo, M.; Zappile, P.; Khodadadi-Jamayran, A.; Nanfack, A.J.; Meli, J.; Wang, X.; Mbanya, D.; et al. Development of a Versatile, Near Full Genome Amplification and Sequencing Approach for a Broad Variety of HIV-1 Group M Variants. Viruses 2019, 11, 317. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Struck, D.; Lawyer, G.; Ternes, A.M.; Schmit, J.C.; Bercoff, D.P. COMET: Adaptive context-based modeling for ultrafast HIV-1 subtype identification. Nucleic Acids Res. 2014, 42, e144. [Google Scholar] [CrossRef]
- Slatkin, M.; Maddison, W.P. A cladistic measure of gene flow inferred from the phylogenies of alleles. Genetics 1989, 123, 603–613. [Google Scholar] [CrossRef]
- Pond, S.L.; Frost, S.D.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Brillen, A.L.; Walotka, L.; Hillebrand, F.; Müller, L.; Widera, M.; Theiss, S.; Schaal, H. Analysis of Competing HIV-1 Splice Donor Sites Uncovers a Tight Cluster of Splicing Regulatory Elements within Exon 2/2b. J. Virol. 2017, 91, e00389-17. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.; Zhou, S.; Pollom, E.; Swanstrom, R. Characterizing HIV-1 Splicing by Using Next-Generation Sequencing. J. Virol. 2017, 91, e02515–e02516. [Google Scholar] [CrossRef]
- Joussef-Piña, S.; Nankya, I.; Nalukwago, S.; Baseke, J.; Rwambuya, S.; Winner, D.; Kyeyune, F.; Chervenak, K.; Thiel, B.; Asaad, R.; et al. Reduced and highly diverse peripheral HIV-1 reservoir in virally suppressed patients infected with non-B HIV-1 strains in Uganda. Retrovirology 2022, 19, 1. [Google Scholar] [CrossRef]
- Laskey, S.B.; Pohlmeyer, C.W.; Bruner, K.M.; Siliciano, R.F. Evaluating Clonal Expansion of HIV-Infected Cells: Optimization of PCR Strategies to Predict Clonality. PLoS Pathog. 2016, 12, e1005689. [Google Scholar] [CrossRef]
- Lau, C.Y.; Adan, M.A.; Maldarelli, F. Why the HIV Reservoir Never Runs Dry: Clonal Expansion and the Characteristics of HIV-Infected Cells Challenge Strategies to Cure and Control HIV Infection. Viruses 2021, 13, 2512. [Google Scholar] [CrossRef]
- Yeh, Y.J.; Yang, K.; Razmi, A.; Ho, Y.C. The Clonal Expansion Dynamics of the HIV-1 Reservoir: Mechanisms of Integration Site-Dependent Proliferation and HIV-1 Persistence. Viruses 2021, 13, 1858. [Google Scholar] [CrossRef]
- Mohammadi, P.; Desfarges, S.; Bartha, I.; Joos, B.; Zangger, N.; Muñoz, M.; Günthard, H.; Beerenwinkel, N.; Telenti, A.; Ciuffi, A. 24 hours in the life of HIV-1 in a T cell line. PLoS Pathog. 2013, 9, e1003161. [Google Scholar] [CrossRef]
- Klotman, M.E.; Kim, S.; Buchbinder, A.; DeRossi, A.; Baltimore, D.; Wong-Staal, F. Kinetics of expression of multiply spliced RNA in early human immunodeficiency virus type 1 infection of lymphocytes and monocytes. Proc. Natl. Acad. Sci. USA 1991, 88, 5011–5015. [Google Scholar] [CrossRef]
- Kim, S.Y.; Byrn, R.; Groopman, J.; Baltimore, D. Temporal aspects of DNA and RNA synthesis during human immunodeficiency virus infection: Evidence for differential gene expression. J. Virol. 1989, 63, 3708–3713. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, A.O.; Berkhout, B. The Splice of Life: Does RNA Processing Have a Role in HIV-1 Persistence? Viruses 2021, 13, 1751. [Google Scholar] [CrossRef]
- Emery, A.; Swanstrom, R. HIV-1: To Splice or Not to Splice, That Is the Question. Viruses 2021, 13, 181. [Google Scholar] [CrossRef]
- Ocwieja, K.; Sherrill-Mix, S.; Mukherjee, R.; Custers-Allen, R.; David, P.; Brown, M.; Wang, S.; Link, D.R.; Olson, J.; Travers, K.; et al. Dynamic regulation of HIV-1 mRNA populations analyzed by single-molecule enrichment and long-read sequencing. Nucleic Acids Res. 2012, 40, 10345–10355. [Google Scholar] [CrossRef] [PubMed]
- Scherpenisse, M.; Kootstra, N.A.; Bakker, M.; Berkhout, B.; Pasternak, A.O. Cell-Associated HIV-1 Unspliced-to-Multiply-Spliced RNA Ratio at 12 Weeks of ART Predicts Immune Reconstitution on Therapy. mBio 2021, 12, e00099-21. [Google Scholar] [CrossRef] [PubMed]
- Uprety, P.; Chadwick, E.G.; Rainwater-Lovett, K.; Ziemniak, C.; Luzuriaga, K.; Capparelli, E.V.; Yenokyan, G.; Persaud, D. Cell-Associated HIV-1 DNA and RNA Decay Dynamics during Early Combination Antiretroviral Therapy in HIV-1-Infected Infants. Clin. Infect. Dis. 2015, 61, 1862–1870. [Google Scholar] [CrossRef]
- Zanchetta, M.; Walker, S.; Burighel, N.; Bellanova, D.; Rampon, O.; Giaquinto, C.; Rossi, A.D. Long-term decay of the HIV-1 reservoir in HIV-1-infected children treated with highly active antiretroviral therapy. J. Infect. Dis. 2006, 193, 1718–1727. [Google Scholar] [CrossRef]
- For the Swiss HIV Cohort Study (members listed in Acknowledgements); Fischer, M.; Wong, J.K.; Russenberger, D.; Joos, B.; Opravil, M.; Hirschel, B.; Trkola, A.; Kuster, H.; Weber, R.; et al. Residual cell-associated unspliced HIV-1 RNA in peripheral blood of patients on potent antiretroviral therapy represents intracellular transcripts. Antivir. Ther. 2002, 7, 91–103. [Google Scholar]
- Vesanen, M.; Markowitz, M.; Cao, Y.; Ho, D.D.; Saksela, K. Human immunodeficiency virus type-1 mRNA splicing pattern in infected persons is determined by the proportion of newly infected cells. Virology 1997, 236, 104–109. [Google Scholar] [CrossRef]
- Moron-Lopez, S.; Xie, G.; Kim, P.; Siegel, D.A.; Lee, S.; Wong, J.K.; Price, J.C.; Elnachef, N.; Greenblatt, R.M.; Tien, P.C.; et al. Tissue-specific differences in HIV DNA levels and mechanisms that govern HIV transcription in blood, gut, genital tract and liver in ART-treated women. J. Int. AIDS Soc. 2021, 24, e25738. [Google Scholar] [CrossRef]
- Telwatte, S.; Lee, S.; Somsouk, M.; Hatano, H.; Baker, C.; Kaiser, P.; Kim, P.; Chen, T.-H.; Milush, J.; Hunt, P.W.; et al. Gut and blood differ in constitutive blocks to HIV transcription, suggesting tissue-specific differences in the mechanisms that govern HIV latency. PLoS Pathog. 2018, 14, e1007357. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Joos, B.; Wong, J.K.; Ott, P.; Opravil, M.; Hirschel, B.; Weber, R.; Günthard, H.F.; Swiss HIV Cohort Study. Attenuated and nonproductive viral transcription in the lymphatic tissue of HIV-1-infected patients receiving potent antiretroviral therapy. J. Infect. Dis. 2004, 189, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Telwatte, S.; Kim, P.; Chen, T.-H.; Milush, J.M.; Somsouk, M.; Deeks, S.G.; Hunt, P.W.; Wong, J.K.; Yukl, S.A. Mechanistic differences underlying HIV latency in the gut and blood contribute to differential responses to latency-reversing agents. AIDS 2020, 34, 2013–2024. [Google Scholar] [CrossRef]
- Pushkarsky, T.; Ward, A.; Ivanov, A.; Lin, X.; Sviridov, D.; Nekhai, S.; Bukrinsky, M.I. Abundance of Nef and p-Tau217 in Brains of Individuals Diagnosed with HIV-Associated Neurocognitive Disorders Correlate with Disease Severance. Mol. Neurobiol. 2022, 59, 1088–1097. [Google Scholar] [CrossRef]
- Huysentruyt, L.C.; McGrath, M.S. The role of macrophages in the development and progression of AIDS-related non-Hodgkin lymphoma. J. Leukoc. Biol. 2010, 87, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Mack, K.D.; Jin, X.; Yu, S.; Wei, R.; Kapp, L.; Green, C.; Herndier, B.; Abbey, N.W.; Elbaggari, A.; Liu, Y.; et al. HIV insertions within and proximal to host cell genes are a common finding in tissues containing high levels of HIV DNA and macrophage-associated p24 antigen expression. J. Acquir. Immune Defic. Syndr. 2003, 33, 308–320. [Google Scholar] [CrossRef]
- Ferdin, J.; Goričar, K.; Dolžan, V.; Plemenitaš, A.; Martin, J.N.; Peterlin, B.M.; Deeks, S.G.; Lenassi, M. Viral protein Nef is detected in plasma of half of HIV-infected adults with undetectable plasma HIV RNA. PLoS ONE 2018, 13, e0191613. [Google Scholar] [CrossRef]
- Anyanwu, S.I.; Doherty, A.; Powell, M.D.; Obialo, C.; Huang, M.B.; Quarshie, A.; Mitchell, C.; Bashir, K.; Newman, G.W. Detection of HIV-1 and Human Proteins in Urinary Extracellular Vesicles from HIV+ Patients. Adv. Virol. 2018, 2018, 7863412. [Google Scholar] [CrossRef]
- Khan, M.B.; Lang, M.J.; Huang, M.B.; Raymond, A.; Bond, V.C.; Shiramizu, B.; Powell, M.D. Nef exosomes isolated from the plasma of individuals with HIV-associated dementia (HAD) can induce Abeta(1-42) secretion in SH-SY5Y neural cells. J. Neurovirol. 2016, 22, 179–190. [Google Scholar] [CrossRef]
- Raymond, A.; Campbell-Sims, T.; Khan, M.; Lang, M.; Huang, M.; Bond, V.; Powell, M. HIV Type 1 Nef is released from infected cells in CD45+ microvesicles and is present in the plasma of HIV-infected individuals. AIDS Res. Hum. Retrovir. 2011, 27, 167–178. [Google Scholar] [CrossRef]
- Chelvanambi, S.; Gupta, S.K.; Chen, X.; Ellis, B.W.; Maier, B.F.; Colbert, T.M.; Kuriakose, J.; Zorlutuna, P.; Jolicoeur, P.; Obukhov, A.G.; et al. HIV-Nef Protein Transfer to Endothelial Cells Requires Rac1 Activation and Leads to Endothelial Dysfunction Implications for Statin Treatment in HIV Patients. Circ. Res. 2019, 125, 805–820. [Google Scholar] [CrossRef]
- Wang, T.; Green, L.A.; Gupta, S.K.; Amet, T.; Byrd, D.J.; Yu, Q.; Twigg, H.L.; Clauss, M. Intracellular Nef detected in peripheral blood mononuclear cells from HIV patients. AIDS Res. Hum. Retrovir. 2015, 31, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Chelvanambi, S.; Bogatcheva, N.; Bednorz, M.; Agarwal, S.; Maier, B.; Alves, N.J.; Li, W.; Syed, F.; Saber, M.M.; Dahl, N.; et al. HIV-Nef Protein Persists in the Lungs of Aviremic Patients with HIV and Induces Endothelial Cell Death. Am. J. Respir. Cell Mol. Biol. 2019, 60, 357–366. [Google Scholar] [CrossRef]
- Stevenson, E.M.; Ward, A.R.; Truong, R.; Thomas, A.S.; Huang, S.-H.; Dilling, T.R.; Terry, S.; Bui, J.K.; Mota, T.M.; Danesh, A.; et al. HIV-specific T cell responses reflect substantive in vivo interactions with antigen despite long-term therapy. J. Clin. Investig. 2021, 6, e142640. [Google Scholar] [CrossRef] [PubMed]
- Greenway, A.L.; McPhee, D.A.; Allen, K.; Johnstone, R.; Holloway, G.; Mills, J.; Azad, A.; Sankovich, S.; Lambert, P. Human immunodeficiency virus type 1 Nef binds to tumor suppressor p53 and protects cells against p53-mediated apoptosis. J. Virol. 2002, 76, 2692–2702. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Witte, V.; Laffert, B.; Blume, K.; Stromer, E.; Trapp, S.; D’Aloja, P.; Schurmann, A.; Baur, A.S. HIV-1 Nef associated PAK and PI3-kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat. Med. 2001, 7, 1217–1224. [Google Scholar] [CrossRef]
- Geleziunas, R.; Xu, W.; Takeda, K.; Ichijo, H.; Greene, W.C. HIV-1 Nef inhibits ASK1-dependent death signalling providing a potential mechanism for protecting the infected host cell. Nature 2001, 410, 834–838. [Google Scholar] [CrossRef]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotic, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitas, A.; Peterlin, B.M. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 2010, 11, 110–122. [Google Scholar] [CrossRef]
- Rasola, A.; Gramaglia, D.; Boccaccio, C.; Comoglio, P.M. Apoptosis enhancement by the HIV-1 Nef protein. J. Immunol. 2001, 166, 81–88. [Google Scholar] [CrossRef]
- Painter, M.M.; Zimmerman, G.E.; Merlino, M.S.; Robertson, A.W.; Terry, V.H.; Ren, X.; McLeod, M.R.; Gomez-Rodriguez, L.; Garcia, K.A.; Leonard, J.A.; et al. Concanamycin A counteracts HIV-1 Nef to enhance immune clearance of infected primary cells by cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 2020, 117, 23835–23846. [Google Scholar] [CrossRef]
- Shi, H.; Tice, C.M.; Emert-Sedlak, L.; Chen, L.; Li, W.F.; Carlsen, M.; Wrobel, J.E.; Reitz, A.B.; Smithgall, T.E. Tight-Binding Hydroxypyrazole HIV-1 Nef Inhibitors Suppress Viral Replication in Donor Mononuclear Cells and Reverse Nef-Mediated MHC-I Downregulation. ACS Infect. Dis. 2020, 6, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, X.; Zhang, W.; Wu, L.; Jing, S.; Liu, W.; Xia, B.; Zou, F.; Lu, L.; Ma, X.; et al. Lovastatin Inhibits HIV-1-Induced MHC-I Downregulation by Targeting Nef-AP-1 Complex Formation: A New Strategy to Boost Immune Eradication of HIV-1 Infected Cells. Front. Immunol. 2019, 10, 2151. [Google Scholar] [CrossRef]
- Mujib, S.; Saiyed, A.; Fadel, S.; Bozorgzad, A.; Aidarus, N.; Yue, F.Y.; Benko, E.; Kovacs, C.; Emert-Sedlak, L.A.; Smithgall, T.E.; et al. Pharmacologic HIV-1 Nef blockade promotes CD8 T cell-mediated elimination of latently HIV-1-infected cells in vitro. JCI Insight. 2017, 2, e93684. [Google Scholar] [CrossRef]
- Luo, X.; Fan, Y.; Park, I.W.; He, J.J. Exosomes are unlikely involved in intercellular Nef transfer. PLoS ONE 2015, 10, e0124436. [Google Scholar] [CrossRef] [PubMed]
- UcUccini, S.; Sirianni, M.C.; Vincenzi, L.; Topino, S.; Stoppacciaro, A.; La Parola, I.L.; Capuano, M.; Masini, C.; Cerimele, D.; Cella, M.; et al. Kaposi’s sarcoma cells express the macrophage-associated antigen mannose receptor and develop in peripheral blood cultures of Kaposi’s sarcoma patients. Am. J. Pathol. 1997, 150, 929–938. [Google Scholar]
- Zhou, F.; Xue, M.; Qin, D.; Zhu, X.; Wang, C.; Zhu, J.; Hao, T.; Cheng, L.; Chen, X.; Bai, Z.; et al. HIV-1 Tat promotes Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6-induced angiogenesis and tumorigenesis by regulating PI3K/PTEN/AKT/GSK-3β signaling pathway. PLoS ONE 2013, 8, e53145. [Google Scholar] [CrossRef] [PubMed]
- Sgadari, C.; Barillari, G.; Palladino, C.; Bellino, S.; Taddeo, B.; Toschi, E.; Ensoli, B. Fibroblast Growth Factor-2 and the HIV-1 Tat Protein Synergize in Promoting Bcl-2 Expression and Preventing Endothelial Cell Apoptosis: Implications for the Pathogenesis of AIDS-Associated Kaposi’s Sarcoma. Int. J. Vasc. Med. 2011, 2011, 452729. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Subject ID | Gender | Age (Years) | Days of Pre-Randomization | HIV Load (Copies/mL) | CD4 Cells Count (Cells/mm3) |
|---|---|---|---|---|---|
| K09 | Male | 44 | 11 | 556,826 | 124 |
| K10 | Male | 38 | 6 | 39,996 | 113 |
| K11 | Female | 23 | 12 | 479,742 | 142 |
| K12 | Female | 37 | 14 | 272,745 | 624 |
| Baseline Tumors | |||||||
| ID | ACSR ID | Biopsy (mg) | DNA Conc (ng/µL) | Total DNA (ng) | RNA Conc (ng/µL) | Total RNA (ng) | RIN * |
| K09 | KPA5178 | 21.8 | 93.8 | 37,520 | 57.6 | 5760 | 5.7 |
| K10 | KPA5552 | 34.5 | 250 | 100,000 | 224 | 22,400 | 7.0 |
| K11 | KPA5763 | 21.6 | 80.4 | 32,160 | 71.2 | 7120 | 5.1 |
| K12 | KPA6014 | 24.7 | 177 | 70,800 | 90.4 | 9040 | 7.2 |
| averages | 26.3 | 158.3 | 63,306.7 | 101.7 | 10,173.3 | 6.6 | |
| Post-Treatment Tumors | |||||||
| ID | ACSR ID | Biopsy (mg) | DNA Conc (ng/µL) | Total DNA (ng) | RNA Conc (ng/µL) | Total RNA (ng) | RIN * |
| K09 | KPA5178 | 30.1 | 184 | 73,600 | 182.0 | 18,200 | 9.3 |
| K10 | KPA5552 | 27.4 | 169 | 67,600 | 212.2 | 21,224 | 8.7 |
| K11 | KPA5763 | 21.6 | 122 | 48,800 | 140.6 | 14,056 | 7.2 |
| K12 | KPA6014 | 18.9 | 61.6 | 24,640 | 91.8 | 9184 | 7.6 |
| averages | 23.0 | 119.7 | 47,866.7 | 132.1 | 13,206.7 | 7.4 | |
| Subject ID | ACSR ID | COMET Subtype | Timepoint | Plasma | PBMC | Tumor | Tumor |
|---|---|---|---|---|---|---|---|
| RNA | DNA | DNA | RNA * | ||||
| K09 | KPA5178 | A1 | baseline | 10 | 9 | 6 | 7 (1) |
| post-treatment | 0 | 1 | 8 | 0 | |||
| K10 | KPA5552 | A1 | baseline | 7 | 7 | 7 | 6 (2) |
| post-treatment | 0 | 5 | 10 | 3 (5) | |||
| K11 | KPA5763 | D | baseline | 8 | 10 | 8 | 11 |
| post-treatment | 0 | 6 | 6 | 0 (4) | |||
| K12 | KPA6014 | A1 | baseline | 6 | 7 | 8 | 6 |
| post-treatment | 0 | 0 | 1 | 0 (3) |
| Baseline Tumor | ||||
| BIE | ACSR | SGS * | µg DNA Used for PCR | SGS/µg DNA |
| K09 | KPA5178 | 6 | 0.49 | 12.30 |
| K10 | KPA5552 | 7 | 1.30 | 5.38 |
| K11 | KPA5763 | 8 | 1.58 | 5.08 |
| K12 | KPA6014 | 6 | 3.47 | 1.73 |
| averages | 7.0 | 1.66 | 5.61 | |
| Post-Treatment Tumor | ||||
| BIE | ACSR | SGS * | µg DNA Used for PCR | SGS/µg DNA |
| K09 | KPA5178 | 8 | 8.10 | 0.99 |
| K10 | KPA5552 | 10 | 7.10 | 1.41 |
| K11 | KPA5763 | 6 | 10.25 | 0.59 |
| K12 | KPA6014 | 1 | 5.17 | 0.19 |
| averages | 4.3 | 7.64 | 0.57 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nolan, D.J.; Rose, R.; Zhang, R.; Leong, A.; Fogel, G.B.; Scholte, L.L.S.; Bethony, J.M.; Bracci, P.; Lamers, S.L.; McGrath, M.S. The Persistence of HIV Diversity, Transcription, and Nef Protein in Kaposi’s Sarcoma Tumors during Antiretroviral Therapy. Viruses 2022, 14, 2774. https://doi.org/10.3390/v14122774
Nolan DJ, Rose R, Zhang R, Leong A, Fogel GB, Scholte LLS, Bethony JM, Bracci P, Lamers SL, McGrath MS. The Persistence of HIV Diversity, Transcription, and Nef Protein in Kaposi’s Sarcoma Tumors during Antiretroviral Therapy. Viruses. 2022; 14(12):2774. https://doi.org/10.3390/v14122774
Chicago/Turabian StyleNolan, David J., Rebecca Rose, Rongzhen Zhang, Alan Leong, Gary B. Fogel, Larissa L. S. Scholte, Jeffrey M. Bethony, Paige Bracci, Susanna L. Lamers, and Michael S. McGrath. 2022. "The Persistence of HIV Diversity, Transcription, and Nef Protein in Kaposi’s Sarcoma Tumors during Antiretroviral Therapy" Viruses 14, no. 12: 2774. https://doi.org/10.3390/v14122774
APA StyleNolan, D. J., Rose, R., Zhang, R., Leong, A., Fogel, G. B., Scholte, L. L. S., Bethony, J. M., Bracci, P., Lamers, S. L., & McGrath, M. S. (2022). The Persistence of HIV Diversity, Transcription, and Nef Protein in Kaposi’s Sarcoma Tumors during Antiretroviral Therapy. Viruses, 14(12), 2774. https://doi.org/10.3390/v14122774