
Analysis of the Origin and Dissemination of HIV-1 Subtype C in Bulgaria

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Specimen Preparation
2.2. Sequence Analysis and Data Set
2.3. Likelihood Mapping and Temporal Signal Analysis
2.4. Bayesian Phylogenetics and Molecular Clock Calibration
2.5. Metapopulation Analysis
2.6. Ethics Statement
3. Results
3.1. Study Population Demographics
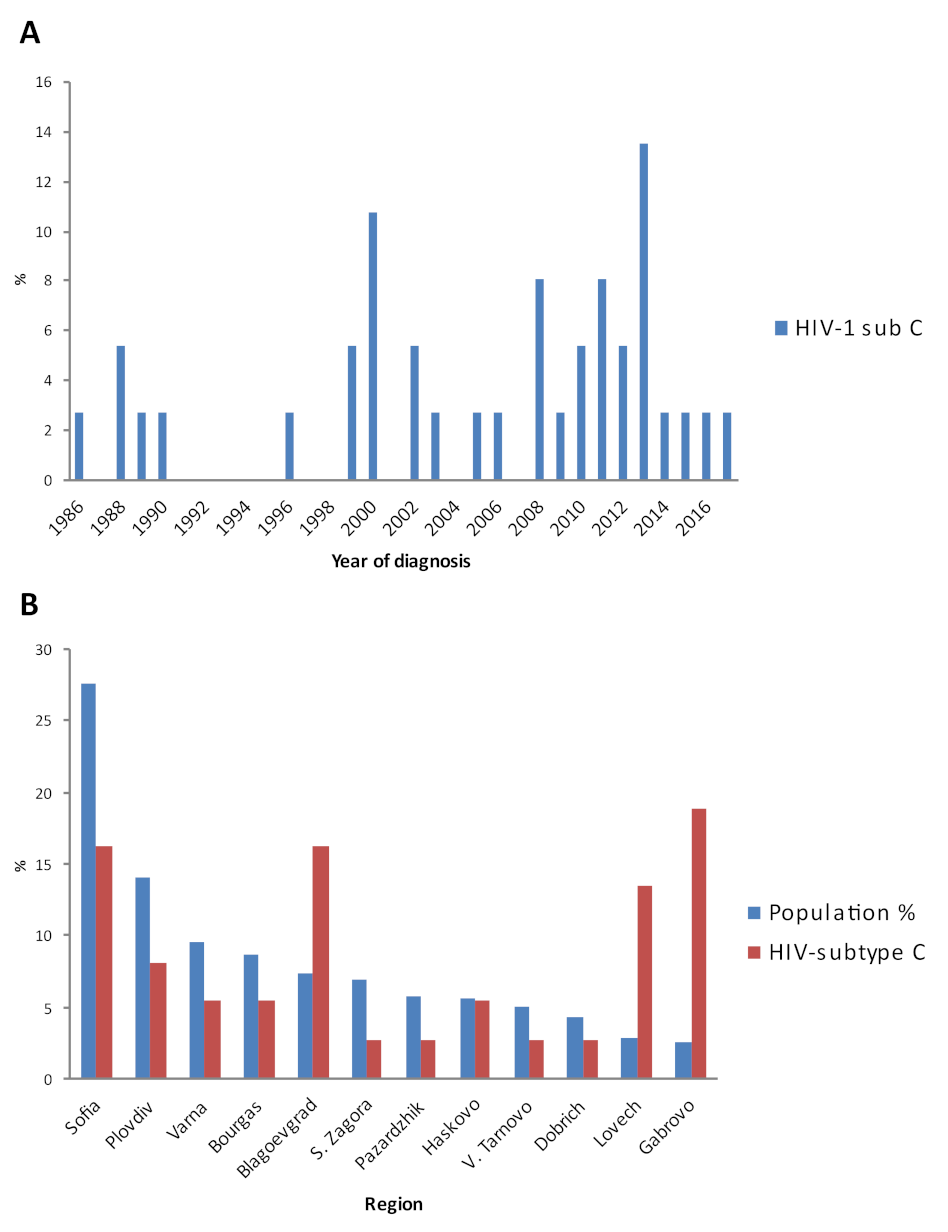
3.2. Introduction and Distribution of HIV-1 Subtype C in the Country
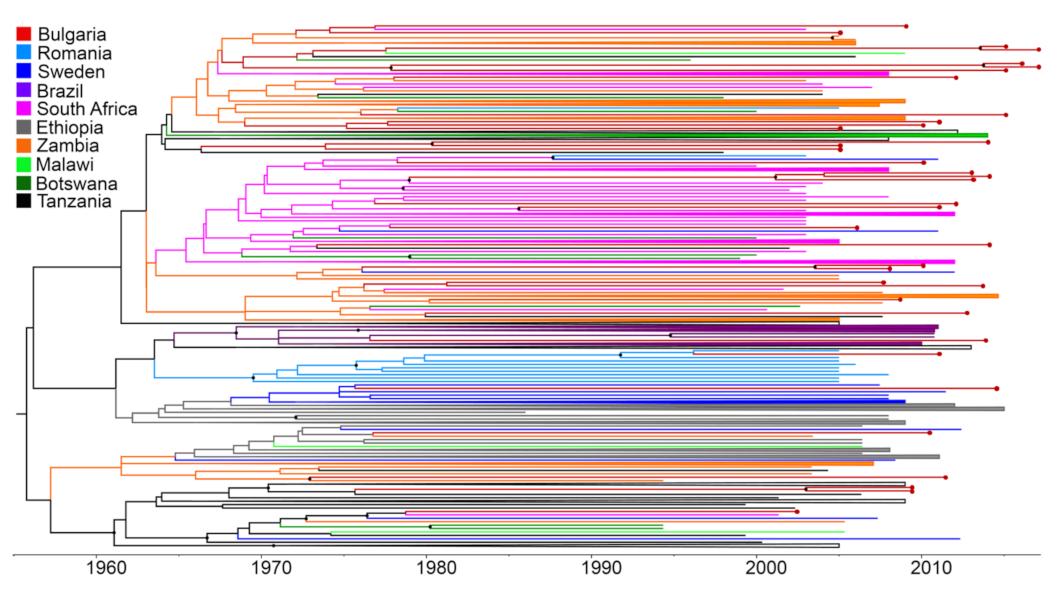
3.3. Multiple Introductions of HIV-1 Subtype C in Bulgaria, Countries of Origin, and Transmission within the Country
3.4. Time-Scale of HIV-1 Subtype C Introductions in Bulgaria
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heuverswyn, F.V.; Li, Y.; Bailes, E.; Neel, C.; Lafay, B.; Keele, B.F.; Shaw, K.S.; Takehisa, J.; Kraus, M.H.; Loul, S.; et al. Genetic diversity and phylogeographic clustering of SIVcpzPtt in wild chimpanzees in Cameroon. Virology 2007, 368, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef] [Green Version]
- Hemelaar, J.; Gouws, E.; Ghys, P.D.; Osmanov, S. Global trends in molecular epidemiology of HIV-1 during 2000–2007. AIDS 2011, 25, 679. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.A.; Hogan, J.W.; Huang, A.; DeLong, A.; Salemi, M.; Mayer, K.H.; Kantor, R. Phylogenetic investigation of a statewide HIV-1 epidemic reveals ongoing and active transmission networks among men who have sex with men. J. Acquir. Immune Defic. Syndr. 2015, 70, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Rife, B.D.; Salemi, M. On the early dynamics and spread of HIV-1. Trends Microbiol. 2015, 23, 3–4. [Google Scholar] [CrossRef]
- Murillo, W.; Veras, N.M.C.; Prosperi, M.C.F.; de Rivera, I.L.; Paz-Bailey, G.; Morales-Miranda, S.; Juarez, S.I.; Yang, C.; Devos, J.; Marín, J.P.; et al. A single early introduction of HIV-1 subtype B into Central America accounts for most current cases. J. Virol. 2013, 87, 7463–7470. [Google Scholar] [CrossRef] [Green Version]
- Tatem, J.; Hemelaar, J.; Gray, R.R.; Salemi, M. Spatial accessibility and the spread of HIV-1 subtypes and recombinants. AIDS 2013, 26, 2351–2360. [Google Scholar] [CrossRef] [PubMed]
- Ciccozzi, M.; Madeddu, G.; Lo Presti, A.; Cella, E.; Giovanetti, M.; Budroni, C.; Babudieri, S.; Mura, M.S.; Zehender, G.; Salemi, M. HIV type 1 origin and transmission dynamics among different risk groups in sardinia: Molecular epidemiology within the close boundaries of an Italian island. AIDS Res. Hum. Retrovir. 2013, 29, 404–410. [Google Scholar] [CrossRef]
- Veras, N.M.C.; Gray, R.R.; de Macedo Brigido, L.F.; Rodrigues, R.; Salemi, M. High-resolution phylogenetics and phylogeography of HIV-1 subtype C epidemic in South America. J. Gen. Virol. 2011, 92, 1698–1709. [Google Scholar] [CrossRef]
- Gray, R.R.; Tatem, A.J.; Lamers, S.L.; Hou, W.; Laeyendecker, O.; Serwadda, D.; Serwankambo, N.; Gray, R.H.; Wawer, M.; Quinn, T.C.; et al. Spatial phylodynamics of HIV-1 epidemic emergence in east Africa. AIDS 2009, 23, F9–F17. [Google Scholar] [CrossRef] [PubMed]
- Hemelaar, J. The origin and diversity of the HIV-1 pandemic. Trends Mol. Med. 2012, 18, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Salemi, M.; de Oliveira, T.; Ciccozzi, M.; Rezza, G.; Goodenow, M.M. High-resolution molecular epidemiology and evolutionary history of HIV-1 subtypes in Albania. PLoS ONE 2008, 3, e1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemelaar, J.; Elangovan, R.; Yun, J.; Fleminger, A.; Kirtley, S.; Williams, B.; Gouws-Williams, E.; Ghys, P.D. Global and regional molecular epidemiology of HIV-1, 1990–2015: A systematic review, global survey, and trend analysis. Lancet Infect. Dis. 2019, 19, 143–155. [Google Scholar] [CrossRef]
- Magiorkinisa, G.; Angelisb, K.; Mamais, I.; Katzourakis, A.; Hatzakisb, A.; Albert, J.; Lawyer, G.; Hamoudae, O.; Struckf, D.; Vercauterenet, J.; et al. The global spread of HIV-1 subtype B epidemic. Infect. Genet. Evol. 2016, 46, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, E.; Engelbrecht, S.; De Oliveira, T. History and origin of the HIV-1 subtype C epidemic in South Africa and the greater southern African region. Sci. Rep. 2015, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Alaeus, A. Significance of HIV-1 Genetic Subtypes. Scand. J. Infect. Dis. 2000, 32, 455–463. [Google Scholar] [CrossRef]
- Kijak, G.H.; McCutchan, F.E. HIV diversity, molecular epidemiology, and the role of recombination. Curr. Infect. Dis. Rep. 2005, 7, 480–488. [Google Scholar] [CrossRef]
- Angelis, K.; Albert, J.; Mamais, I.; Magiorkinis, G.; Hatzakis, A.; Hamouda, O.; Struck, D.; Vercauteren, J.; Wensing, A.M.; Alexiev, I.; et al. Global dispersal pattern of HIV-1 CRF01_AE: A genetic trace of human mobility related to heterosexual activities centralized in South-East Asia. J. Infect. Dis. 2015, 11, 1735–1744. [Google Scholar] [CrossRef]
- Alexiev, I.; Beshkov, D.; Georgieva, V.; Varleva, T. Diversity of HIV-1 in Bulgaria Phylogenetic analisis. Probl. Infect. Parasit. Dis. 2009, 37, 9–12. [Google Scholar]
- Stanojevic, M.; Alexiev, I.; Beshkov, D.; Gökengin, D.; Mezei, M.; Minarovits, J.; Otelea, D.; Paraschiv, S.; Poljak, M.; Zidovec-Lepej, S.; et al. HIV1 Molecular Epidemiology in the Balkans—A Melting Pot for High Genetic Diversity. AIDS Rev. 2012, 14, 28–36. [Google Scholar]
- Alexiev, I.; Beshkov, D.; Shankar, A.; Hanson, D.; Paraskevis, D.; Georgieva, V.; Karamacheva, L.; Taskov, H.; Varleva, T.; Elenkov, I.; et al. Detailed Molecular Epidemiologic Characterization of HIV-1 Infection in Bulgaria. Reveals Broad Diversity and Evolving Phylodynamics. PLoS ONE 2013, 8, e59666. [Google Scholar]
- Salemi, M.; Goodenow, M.M.; Montieri, S.; de Oliveira, T.; Santoro, M.M.; Beshkov, D.; Alexiev, I.; Elenkov, I.; Elenkov, I.; Ya-kimova, T.; et al. The HIV type 1 epidemic in Bulgaria involves multiple subtypes and is sustained by continuous viral inflow from West and East European countries. AIDS Res Hum Retrovir. 2008, 24, 771–779. [Google Scholar] [CrossRef] [Green Version]
- Alexiev, I.; Shankar, A.; Wensing, A.M.J.; Beshkov, D.; Elenkov, I.; Stoycheva, M.; Nikolova, D.; Nikolova, M.; Switzer, W.M. Low HIV-1 transmitted drug resistance in Bulgaria against a background of high clade diversity. J. Antimicrob. Chemother. 2015, 70, 1874–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexiev, I.; Shankar, A.; Dimitrova, R.; Gancheva, A.; Kostadinova, A.; Teoharov, P.; Golkocheva, E.; Nikolova, M.; Muhtarova, M.; Elenkov, E.; et al. Origin and spread of HIV-1 in persons who inject drugs in Bulgaria. Infect. Genet. Evol. 2016, 46, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Alexiev, I.; Lo Presti, A.; Dimitrova, R.; Foley, B.; Gancheva, A.; Kostadinova, A.; Nikolova, L.; Angeletti, S.; Cella, E.; Elenkov, I.; et al. Origin and Spread of HIV-1 Subtype B Among Heterosexual Individuals in Bulgaria. AIDS Res. Hum. Retrovir. 2018, 34, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Struck, D.; Lawyer, G.; Ternes, A.-M.; Schmit, J.-C.; Bercoff, D.P. Comet: Adaptive context-based modeling for ultrafast HIV-1 subtype identification. Nucleic Acids Res. 2014, 42, e144. [Google Scholar] [CrossRef] [PubMed]
- Peña, A.C.P.; Faria, N.R.; Imbrechts, S.; Libin, P.; Abecasis, A.B.; Deforche, K.; Gomez, A.; Camacho, R.J.; de Oliveira, T.; Vandamme, A.-M. Automated subtyping of HIV-1 genetic sequences for clinical and surveillance purposes: Performance evaluation of the new REGA version 3 and seven other tools. Infect. Genet. Evol. 2013, 19, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Schultz, A.-K.; Zhang, M.; Bulla, I.; Leitner, T.; Korber, B.; Morgenstern, B.; Stanke, M. jpHMM: Improving the reliability of re-combination prediction in HIV-1. Nucleic Acids Res. 2009, 37, W647–W651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maxi-mum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.T.; Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [Green Version]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large data sets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, H.A.; Strimmer, K.; Vingron, M.; Haeseler, A. TREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 2002, 18, 502–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Lam, T.; Carvalho, L.; Pybus, O. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baele, G.; Lemey, P.; Vansteelandt, S. Make the most of your samples: Bayes factor estimators for high-dimensional models of sequence evolution. BMC Bioinform. 2013, 14, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Kass, R.E.; Raftery, A.E. Bayes factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- Salemi, M.; Strimmer, K.; Hall, W.W.; Duffy, M.; Delaporte, E.; Mboup, S.; Peeters, M.; Vandamme, A.M. Dating the common ancestor of SIVcpz and HIV-1 group M and the origin of HIV-1 subtypes using a new method to uncover clock-like molecular evolution. FASEB J. 2001, 15, 276–278. [Google Scholar] [CrossRef] [Green Version]
- Faria, N.R.; Rambaut, A.; Suchard, M.A.; Baele, G.; Bedford, T.; Ward, M.J.; Tatem, A.J.; Sousa, J.D.; Arinaminpathy, N.; Pépin, J.; et al. The early spread and epidemic ignition of HIV-1 in human populations. Science 2014, 346, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Marcus, U.; Nöstlinger, C.; Rosińska, M.; Sherriff, N.; Gios, L.; Dias, S.F.; Gama, A.F.; Toskin, I.; Alexiev, I.; Naseva, E.; et al. Be-havioural and demographic correlates of undiagnosed HIV infection in a MSM sample recruited in 13 European cities. BMC Infect. Dis. 2018, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gios, L.; Mirandola, M.; Toskin, I.; Marcus, U.; Dudareva-Vizule, S.; Sherriff, N.; Breveglieri, M.; Furegato, M.; Folch, C.; Ferrer, L.; et al. Bio-behavioural HIV and STI surveillance among men who have sex with men in Europe: The Sialon II protocols. BMC Public Health 2016, 16, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varleva, T.; Zamfirova, M.; Georgieva, V.; Nikolova, M.; Alexiev, I.; Taskov, H. Bulgaria HIV surveillance system in the context of continuum of care 90-90-90 strategy. Probl. Infect. Parasit. Dis. 2018, 46, 9–13. [Google Scholar]
- Alexiev, I.; Alexandrova, M.; Golkocheva-Markova, E.; Teoharov, P.; Gancheva, A.; Kostadinova, A.; Dimitrova, R.; Elenkov, I.; Chervenjakova, T.; Stoycheva, M.; et al. High rate of hepatitis B and C co-infections among people living with HIV-1 in Bul-garia: 2010–2014. AIDS Res. Hum. Retrovir. 2017, 33, 228–229. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients Characteristics | Patients with HIV-1 Subtype C | |
|---|---|---|
| (n) | (%) | |
| Total | 37 | 100 |
| Men | 19 | 51.4 |
| Women | 18 | 48.6 |
| Age (years) | ||
| ≤20 | 1 | 2.7 |
| 20–29 | 15 | 40.5 |
| 30–39 | 8 | 21.6 |
| 40–49 | 8 | 21.6 |
| ≥50 | 5 | 13.5 |
| Mean age (years) | 34.3 | |
| Likely country of infection | ||
| Bulgaria | 28 | 75.7 |
| Other country | 9 (2 Germany, 1 Somalia, 1 South Africa, 1 Congo, 4 Uncnown) | 24.3 |
| HET | 34 | 91.9 |
| MSM | 1 | 2.7 |
| Blood transfusion | 1 | 2.7 |
| Mother to child | 1 | 2.7 |
| Diagnosis period | ||
| 1986–1995 | 5 | 13.5 |
| 1996–2005 | 11 | 29.7 |
| 2006–2017 | 21 | 56.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alexiev, I.; Mavian, C.; Paisie, T.; Ciccozzi, M.; Dimitrova, R.; Gancheva, A.; Kostadinova, A.; Seguin-Devaux, C.; Salemi, M. Analysis of the Origin and Dissemination of HIV-1 Subtype C in Bulgaria. Viruses 2022, 14, 263. https://doi.org/10.3390/v14020263
Alexiev I, Mavian C, Paisie T, Ciccozzi M, Dimitrova R, Gancheva A, Kostadinova A, Seguin-Devaux C, Salemi M. Analysis of the Origin and Dissemination of HIV-1 Subtype C in Bulgaria. Viruses. 2022; 14(2):263. https://doi.org/10.3390/v14020263
Chicago/Turabian StyleAlexiev, Ivailo, Carla Mavian, Taylor Paisie, Massimo Ciccozzi, Reneta Dimitrova, Anna Gancheva, Asya Kostadinova, Carole Seguin-Devaux, and Marco Salemi. 2022. "Analysis of the Origin and Dissemination of HIV-1 Subtype C in Bulgaria" Viruses 14, no. 2: 263. https://doi.org/10.3390/v14020263