
THαβ Immunological Pathway as Protective Immune Response against Prion Diseases: An Insight for Prion Infection Therapy


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Sporadic Human Prion Disease
2.1. Sporadic Creutzfeldt–Jakob Disease (sCJD)
2.2. Sporadic Fatal Insomnia
2.3. Variably Protease-Sensitive Prionopathy
3. Genetic Human Prion Diseases
3.1. Familial Creutzfeldt–Jakob Disease
3.2. Gerstmann–Straussler–Scheinker Syndrome
3.3. Familial Fatal Insomnia
3.4. Other PRNP Mutations
4. Acquired Human Prion Diseases
4.1. Kuru
4.2. Iatrogenic Creutzfeldt–Jakob Disease (iaCJD)
4.3. Variant Creutzfeldt–Jakob Disease (vCJD)
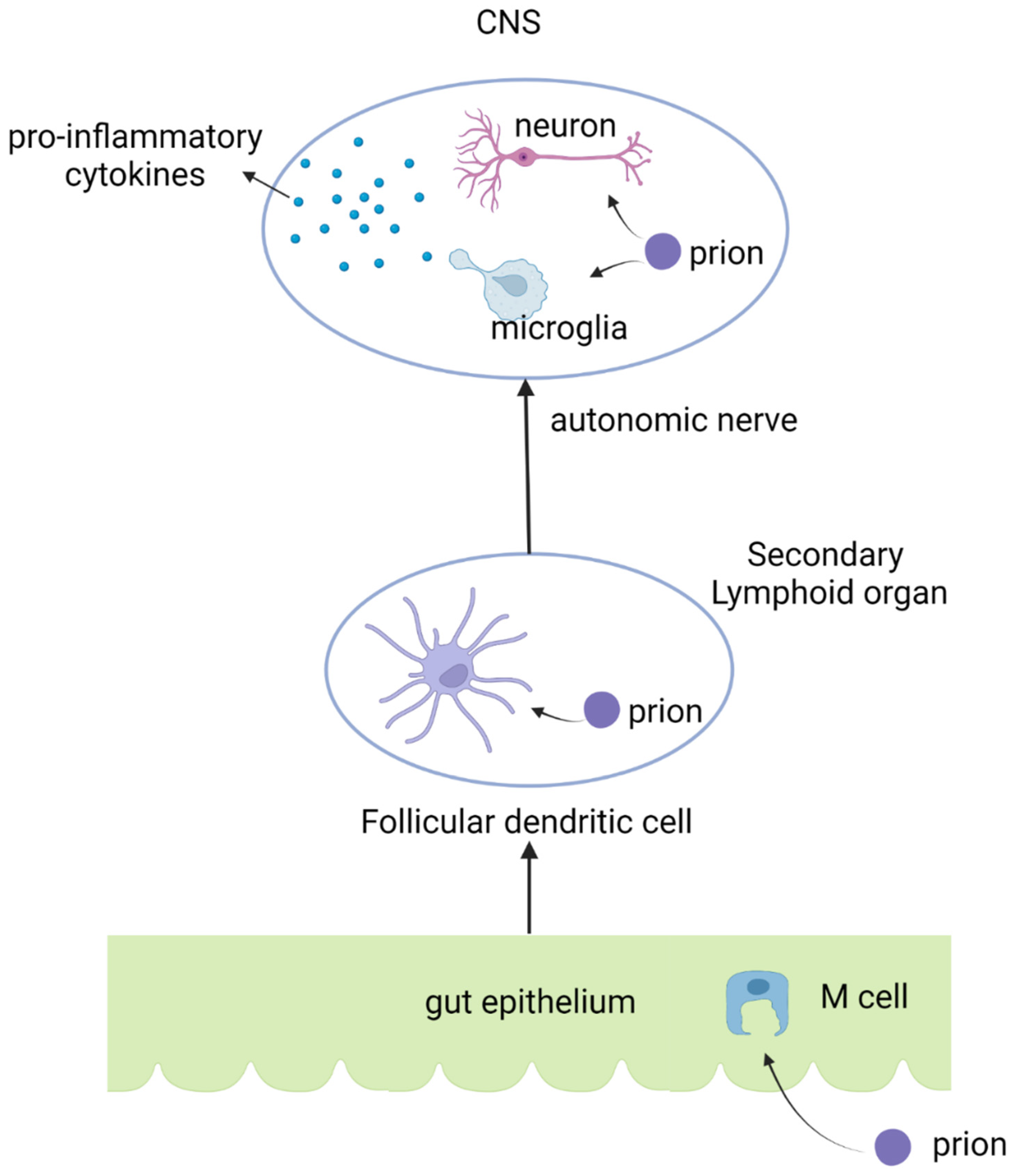
5. Host Immune Reaction against Prion Diseases
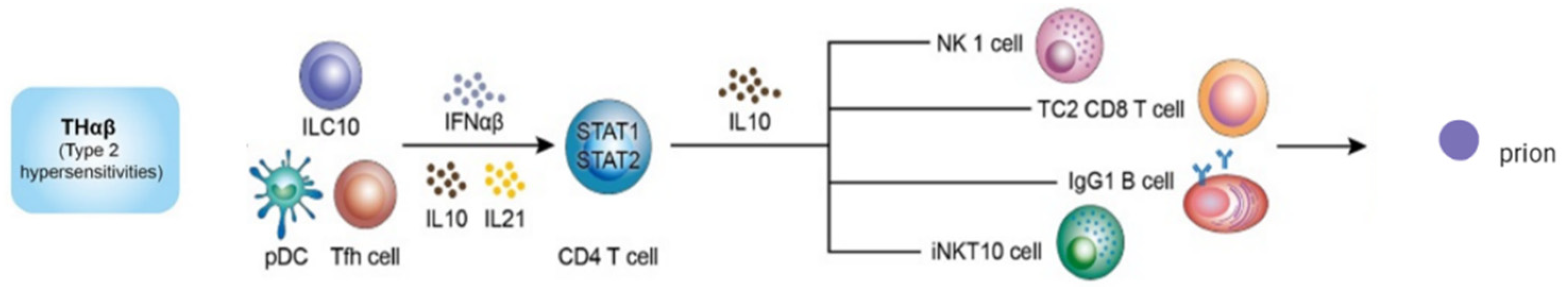
6. Protective Immunity against Prion Diseases
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. Molecular Biology of Prion Diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B.; Scott, M.; Foster, D.; Pan, K.-M.; Groth, D.; Mirenda, C.; Torchia, M.; Yang, S.-L.; Serban, D.; Carlson, G.A.; et al. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 1990, 63, 673–686. [Google Scholar] [CrossRef]
- Belay, E.D. Transmissible Spongiform Encephalopathies in Humans. Annu. Rev. Microbiol. 1999, 53, 283–314. [Google Scholar] [CrossRef] [Green Version]
- Zerr, I.; Parchi, P. Sporadic Creutzfeldt-Jakob disease. Handb. Clin. Neurol. 2018, 153, 155–174. [Google Scholar] [CrossRef]
- Parchi, P.; Strammiello, R.; Notari, S.; Giese, A.; Langeveld, J.P.M.; Ladogana, A.; Zerr, I.; Roncaroli, F.; Cras, P.; Ghetti, B.; et al. Incidence and spectrum of sporadic Creutzfeldt–Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: An updated classification. Acta. Neuropathol. 2009, 118, 659–671. [Google Scholar] [CrossRef] [Green Version]
- Rabinovici, G.D.; Wang, P.N.; Levin, J.; Cook, L.; Pravdin, M.; Davis, J.; DeArmond, S.J.; Barbaro, N.M.; Martindale, J.; Miller, B.L.; et al. First symptom in sporadic Creutzfeldt-Jakob disease. Neurology 2006, 66, 286–287. [Google Scholar] [CrossRef]
- Brown, P.; Castaigne, P.; Gajdusek, D.C. Creutzfeldt-Jakob disease: Clinical analysis of a consecutive series of 230 neuropathologically verified cases. Ann. Neurol. 1986, 20, 597–602. [Google Scholar] [CrossRef]
- Paterson, R.W.; Torres-Chae, C.C.; Kuo, A.L.; Ando, T.; Nguyen, E.A.; Wong, K.; DeArmond, S.J.; Haman, A.; Garcia, P.; Johnson, D.Y.; et al. Differential diagnosis of Jakob-Creutzfeldt disease. Arch. Neurol. 2012, 69, 1578–1582. [Google Scholar] [CrossRef]
- Damavandi, P.T.; Dove, M.; Pickersgill, R.W. A review of drug therapy for sporadic fatal insomnia. Prion 2017, 11, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Montagna, P.; Gambetti, P.; Cortelli, P.; Lugaresi, E. Familial and sporadic fatal insomnia. Lancet Neurol. 2003, 2, 167–176. [Google Scholar] [CrossRef]
- Cracco, L.; Appleby, B.S.; Gambetti, P. Fatal familial insomnia and sporadic fatal insomnia. Handb. Clin. Neurol. 2018, 153, 271−299. [Google Scholar] [CrossRef]
- Hamlin, C.; Puoti, G.; Berri, S.; Sting, E.; Harris, C.; Cohen, M.; Spear, C.; Bizzi, A.; Debanne, S.M.; Rowland, D.Y. A comparison of tau and 14-3-3 protein in the diagnosis of Creutzfeldt-Jakob disease. Neurology 2012, 79, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Muayqil, T.; Gronseth, G.; Camicioli, R. Evidence-based guideline: Diagnostic accuracy of CSF 14-3-3 protein in sporadic Creutzfeldt-Jakob disease: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2012, 79, 1499–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, M.; Ebert, E.; Stoeck, K.; Karch, A.; Collins, S.; Calero, M.; Sklaviadis, T.; Laplanche, J.-L.; Golanska, E.; Baldeiras, I.; et al. Validation of 14-3-3 Protein as a Marker in Sporadic Creutzfeldt-Jakob Disease Diagnostic. Mol. Neurobiol. 2015, 53, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Puopolo, M.; Ladogana, A.; Pocchiari, M.; Budka, H.; Van Duijn, C.; Collins, S.J.; Boyd, A.; Giulivi, A.; Coulthart, M.; et al. Genetic prion disease: The EUROCJD experience. Qual. Life Res. 2005, 118, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Trabattoni, G.; Hainfellner, J.A.; Ironside, J.; Knight, R.S.G.; Budka, H. Mutations of the Prion Protein Gene. J. Neurol. 2002, 249, 1567–1582. [Google Scholar] [CrossRef]
- Tesar, A.; Matej, R.; Kukal, J.; Msc, S.J.; Rektorova, I.; Vyhnalek, M.; Keller, J.; Eliasova, I.; Parobkova, E.; Msc, M.S.; et al. Clinical Variability in P102L Gerstmann–Sträussler–Scheinker Syndrome. Ann. Neurol. 2019, 86, 643–652. [Google Scholar] [CrossRef]
- Liberski, P.P. Gerstmann-Sträussler-Scheinker Disease. Adv. Exp. Med. Biol. 2012, 724, 128–137. [Google Scholar] [CrossRef]
- Lugaresi, E.; Medori, R.; Montagna, P.; Baruzzi, A.; Cortelli, P.; Lugaresi, A.; Tinuper, P.; Zucconi, M.; Gambetti, P. Fatal Familial Insomnia and Dysautonomia with Selective Degeneration of Thalamic Nuclei. N. Engl. J. Med. 1986, 315, 997–1003. [Google Scholar] [CrossRef]
- Krasnianski, A.; Bartl, M.; Juan, P.J.S.; Heinemann, U.; Meissner, B.; Varges, D.; Schulze-Sturm, U.; Kretzschmar, H.A.; Schulz-Schaeffer, W.J.; Zerr, I. Fatal familial insomnia: Clinical features and early identification. Ann. Neurol. 2008, 63, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Sforza, E.; Montagna, P.; Tinuper, P.; Cortelli, P.; Avoni, P.; Ferrillo, F.; Petersen, R.; Gambetti, P.; Lugaresi, E. Sleep-wake cycle abnormalities in fatal familial insomnia. Evidence of the role of the thalamus in sleep regulation. Electroencephalogr. Clin. Neurophysiol. 1995, 94, 398–405. [Google Scholar] [CrossRef]
- Benarroch, E.E.; Stotz-Potter, E.H. Dysautonomia in Fatal Familial Insomnia as an Indicator of the Potential Role of the Thalamus in Autonomie Control. Brain Pathol. 2006, 8, 527–530. [Google Scholar] [CrossRef]
- Cortelli, P.; Perani, D.; Montagna, P.; Gallassi, R.; Tinuper, P.; Federica, P.; Avoni, P.; Ferrillo, F.; Anchisi, D.; Moresco, R.M.; et al. Pre-symptomatic diagnosis in fatal familial insomnia: Serial neurophysiological and 18FDG-PET studies. Brain 2006, 129, 668–675. [Google Scholar] [CrossRef]
- Schmitz, M.; Dittmar, K.; Llorens, F.; Gelpi, E.; Ferrer, I.; Schulz-Schaeffer, W.J.; Zerr, I. Hereditary Human Prion Diseases: An Update. Mol. Neurobiol. 2016, 54, 4138–4149. [Google Scholar] [CrossRef]
- Krasnianski, A.; Sanchez Juan, P.; Ponto, C.; Bartl, M.; Heinemann, U.; Varges, D.; Schulz-Schaeffer, W.J.; Kretzschmar, H.A.; Zerr, I. A proposal of new diagnostic pathway for fatal familial insomnia. J. Neurol. Neurosurg. Psychiatry 2014, 85, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.; Mastrianni, J.A. The Prion Diseases. J. Geriatr. Psychiatry Neurol. 2010, 23, 277–298. [Google Scholar] [CrossRef]
- Liberski, P.P.; Sikorska, B.; Lindenbaum, S.; Goldfarb, L.G.; McLean, C.; Hainfellner, J.A.; Brown, P. Kuru: Genes, cannibals and neuropathology. J. Neuropathol. Exp. Neurol. 2012, 71, 92–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seed, C.R.; Hewitt, P.E.; Dodd, R.Y.; Houston, F.; Cervenakova, L. Creutzfeldt-Jakob disease and blood transfusion safety. Vox. Sang. 2018, 113, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Rudge, P.; Jaunmuktane, Z.; Adlard, P.; Bjurstrom, N.; Caine, D.; Lowe, J.; Norsworthy, P.; Hummerich, H.; Druyeh, R.; Wadsworth, J.; et al. Iatrogenic CJD due to pituitary-derived growth hormone with genetically determined incubation times of up to 40 years. Brain 2015, 138, 3386–3399. [Google Scholar] [CrossRef]
- Holman, R.C.; Belay, E.; Christensen, K.Y.; Maddox, R.A.; Minino, A.M.; Folkema, A.M.; Haberling, D.; Hammett, T.A.; Kochanek, K.D.; Sejvar, J.J.; et al. Human Prion Diseases in the United States. PLoS ONE 2010, 5, e8521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandel, J.-P.; Preece, M.; Brown, P.; Croes, E.; Laplanche, J.-L.; Agid, Y.; Will, R.; Alpérovitch, A. Distribution of codon 129 genotype in human growth hormone-treated CJD patients in France and the UK. Lancet 2003, 362, 128–130. [Google Scholar] [CrossRef]
- Brown, P.; Brandel, J.-P.; Sato, T.; Nakamura, Y.; MacKenzie, J.; Will, R.G.; Ladogana, A.; Pocchiari, M.; Leschek, E.W.; Schonberger, L.B. Iatrogenic Creutzfeldt-Jakob Disease, Final Assessment. Emerg. Infect. Dis. 2012, 18, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Heath, C.A.; Cooper, S.A.; Murray, K.; Lowman, A.; Henry, C.; MacLeod, M.; E Stewart, G.; Zeidler, M.; McKenzie, J.M.; Knight, R.S.G.; et al. Diagnosing variant Creutzfeldt-Jakob disease: A retrospective analysis of the first 150 cases in the UK. J. Neurol. Neurosurg. Psychiatry 2010, 82, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Binelli, S.; Agazzi, P.; Giaccone, G.; Will, R.G.; Bugiani, O.; Franceschetti, S.; Tagliavini, F. Periodic electroencephalogram complexes in a patient with variant Creutzfeldt–Jakob disease. Ann. Neurol. 2006, 59, 423–427. [Google Scholar] [CrossRef]
- Collie, D.A.; Summers, D.M.; Sellar, R.J.; Ironside, J.W.; Cooper, S.; Zeidler, M.; Knight, R.; Will, R.G. Diagnosing Variant Creutzfeldt-Jakob Disease with the Pulvinar Sign: MR Imaging Findings in 86 Neuropathologically Confirmed Cases. Am. J. Neuroradiol. 2003, 24, 1560–1569. [Google Scholar]
- Hill, A.; Butterworth, R.; Joiner, S.; Jackson, G.; Rossor, M.; Thomas, D.; Frosh, A.; Tolley, N.; Bell, J.; Spencer, M.; et al. Investigation of variant Creutzfeldt-Jakob disease and other human prion diseases with tonsil biopsy samples. Lancet 1999, 353, 183–189. [Google Scholar] [CrossRef]
- Aguzzi, A.; Nuvolone, M.; Zhu, C. The immunobiology of prion diseases. Nat. Rev. Immunol. 2013, 13, 888–902. [Google Scholar] [CrossRef]
- Aguzzi, A.; Heikenwalder, M. Pathogenesis of prion diseases: Current status and future outlook. Nat. Rev. Genet. 2006, 4, 765–775. [Google Scholar] [CrossRef]
- Jeon, J.-W.; Park, B.-C.; Jung, J.-G.; Jang, Y.-S.; Shin, E.-C.; Park, Y.W. The Soluble Form of the Cellular Prion Protein Enhances Phagocytic Activity and Cytokine Production by Human Monocytes Via Activation of ERK and NF-κB. Immune Netw. 2013, 13, 148–156. [Google Scholar] [CrossRef]
- Sharief, M.K.; Green, A.; Dick, J.P.R.; Gawler, J.; Thompson, E.J. Heightened intrathecal release of proinflammatory cytokines in Creutzfeldt-Jakob disease. Neurology 1999, 52, 1289. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.L.; Eddleston, M.; Kemper, P.; Oldstone, M.B.; Hobbs, M.V. Activation of cerebral cytokine gene expression and its correlation with onset of reactive astrocyte and acute-phase response gene expression in scrapie. J. Virol. 1994, 68, 2383–2387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinz, M.; Huber, G.; Macpherson, A.J.; Heppner, F.; Glatzel, M.; Eugster, H.-P.; Wagner, N.; Aguzzi, A. Oral Prion Infection Requires Normal Numbers of Peyer’s Patches but Not of Enteric Lymphocytes. Am. J. Pathol. 2003, 162, 1103–1111. [Google Scholar] [CrossRef]
- Donaldson, D.S.; Kobayashi, A.; Ohno, H.; Yagita, H.; Williams, I.R.; A Mabbott, N. M cell-depletion blocks oral prion disease pathogenesis. Mucosal Immunol. 2012, 5, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Liu, A.; Zhou, X.; Kouadir, M.; Zhao, W.; Zhang, S.; Yin, X.; Yang, L.; Zhao, D. Prion Peptide PrP106-126 Induces Inducible Nitric Oxide Synthase and Proinflammatory Cytokine Gene Expression Through the Activation of NF-κB in Macrophage Cells. DNA Cell Biol. 2012, 31, 833–838. [Google Scholar] [CrossRef]
- Beringue, V.; Demoy, M.; Gouritin, B.; Weingarten, C.; Deslys, J.-P.; Andreux, J.-P.; Couvreur, P.; Dormont, D. Role of spleen macrophages in the clearance of scrapie agent early in pathogenesis. J. Pathol. 2000, 190, 495–502. [Google Scholar] [CrossRef]
- Beringue, V.; Couvreur, P.; Dormont, D. Involvement of Macrophages in the Pathogenesis of Transmissible Spongiform Encephalopathies. Dev. Immunol. 2002, 9, 19–27. [Google Scholar] [CrossRef]
- Carp, R.I.; Callahan, S.M. In vitro Interaction of Scrapie Agent and Mouse Peritoneal Macrophages. Intervirology 1981, 16, 8–13. [Google Scholar] [CrossRef]
- Mabbott, N.A.; Bradford, B. The Good, the Bad, and the Ugly of Dendritic Cells during Prion Disease. J. Immunol. Res. 2015, 2015, 168574. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.L.; Stewart, K.; Ritchie, D.L.; Mabbott, N.A.; Williams, A.; Fraser, H.; Morrison, W.I.; Bruce, M.E. Scrapie replication in lymphoid tissues depends on prion protein-expressing follicular dendritic cells. Nat. Med. 1999, 5, 1308–1312. [Google Scholar] [CrossRef]
- Kitamoto, T.; Muramoto, T.; Mohri, S.; Doh-Ura, K.; Tateishi, J. Abnormal isoform of prion protein accumulates in follicular dendritic cells in mice with Creutzfeldt-Jakob disease. J. Virol. 1991, 65, 6292–6295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krautler, N.J.; Kana, V.; Kranich, J.; Tian, Y.; Perera, D.; Lemm, D.; Schwarz, P.; Armulik, A.; Browning, J.; Tallquist, M.; et al. Follicular Dendritic Cells Emerge from Ubiquitous Perivascular Precursors. Cell 2012, 150, 194–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabbott, N.A.; Mackay, F.; Minns, F.; Bruce, M.E. Temporary inactivation of follicular dendritic cells delays neuroinvasion of scrapie. Nat. Med. 2000, 6, 719–720. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, L.; Brown, K.L.; Bradford, B.M.; Hopkins, J.; Bailey, M.; Rajewsky, K.; Manson, J.C.; Mabbott, N.A. Follicular Dendritic Cell-Specific Prion Protein (PrPc) Expression Alone Is Sufficient to Sustain Prion Infection in the Spleen. PLoS Pathog. 2011, 7, e1002402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, J.; Brown, K.L.; Farquhar, C.F.; E Bruce, M.; A Mabbott, N. Scrapie transmission following exposure through the skin is dependent on follicular dendritic cells in lymphoid tissues. J. Dermatol. Sci. 2004, 35, 101–111. [Google Scholar] [CrossRef]
- Montrasio, F.; Frigg, R.; Glatzel, M.; Klein, M.A.; Mackay, F.; Aguzzi, A.; Weissmann, C. Impaired Prion Replication in Spleens of Mice Lacking Functional Follicular Dendritic Cells. Science 2000, 288, 1257–1259. [Google Scholar] [CrossRef]
- Prinz, M.; Montrasio, F.; Klein, M.A.; Schwarz, P.; Priller, J.; Odermatt, B.; Pfeffer, K.; Aguzzi, A. Lymph nodal prion replication and neuroinvasion in mice devoid of follicular dendritic cells. Proc. Natl. Acad. Sci. USA 2002, 99, 919–924. [Google Scholar] [CrossRef] [Green Version]
- Prinz, M.; Heikenwalder, M.; Junt, T.; Schwarz, P.E.H.; Glatzel, M.; Heppner, F.; Fu, Y.-X.; Lipp, M.; Aguzzi, A. Positioning of follicular dendritic cells within the spleen controls prion neuroinvasion. Nature 2003, 425, 957–962. [Google Scholar] [CrossRef]
- Mabbott, N.; Young, J.; McConnell, I.; Bruce, M.E. Follicular Dendritic Cell Dedifferentiation by Treatment with an Inhibitor of the Lymphotoxin Pathway Dramatically Reduces Scrapie Susceptibility. J. Virol. 2003, 77, 6845–6854. [Google Scholar] [CrossRef] [Green Version]
- Heikenwalder, M.; Kurrer, M.O.; Margalith, I.; Kranich, J.; Zeller, N.; Haybaeck, J.; Polymenidou, M.; Matter, M.; Bremer, J.; Jackson, W.S.; et al. Lymphotoxin-Dependent Prion Replication in Inflammatory Stromal Cells of Granulomas. Immunity 2008, 29, 998–1008. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, T.; Frei, N.; Sponarova, J.; Schwarz, P.; Heikenwälder, M.; Aguzzi, A. Lymphotoxin, but Not TNF, Is Required for Prion Invasion of Lymph Nodes. PLoS Pathog. 2012, 8, e1002867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glatzel, M.; Heppner, F.L.; Albers, K.M.; Aguzzi, A. Sympathetic Innervation of Lymphoreticular Organs Is Rate Limiting for Prion Neuroinvasion. Neuron 2001, 31, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Haik, S.; A Faucheux, B.; Sazdovitch, V.; Privat, N.; Kemeny, J.-L.; Perret-Liaudet, A.; Hauw, J.-J. The sympathetic nervous system is involved in variant Creutzfeldt-Jakob disease. Nat. Med. 2003, 9, 1121–1122. [Google Scholar] [CrossRef] [PubMed]
- McBride, P.A.; Beekes, M. Pathological PrP is abundant in sympathetic and sensory ganglia of hamsters fed with scrapie. Neurosci. Lett. 1999, 265, 135–138. [Google Scholar] [CrossRef]
- Sigurdson, C.J.; Spraker, T.R.; Miller, M.W.; Oesch, B.; Hoover, E.A. PrPCWD in the myenteric plexus, vagosympathetic trunk and endocrine glands of deer with chronic wasting disease. J. Gen. Virol. 2001, 82, 2327–2334. [Google Scholar] [CrossRef]
- Obst, J.; Simon, E.; Mancuso, R.; Gomez-Nicola, D. The Role of Microglia in Prion Diseases: A Paradigm of Functional Diversity. Front. Aging Neurosci. 2017, 9, 207. [Google Scholar] [CrossRef] [Green Version]
- Baker, C.A.; Lu, Z.Y.; Zaitsev, I.; Manuelidis, L. Microglial Activation Varies in Different Models of Creutzfeldt-Jakob Disease. J. Virol. 1999, 73, 5089–5097. [Google Scholar] [CrossRef] [Green Version]
- Baker, C.A.; Martin, D.; Manuelidis, L. Microglia from Creutzfeldt-Jakob Disease-Infected Brains Are Infectious and Show Specific mRNA Activation Profiles. J. Virol. 2002, 76, 10905–10913. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.M.; Field, R.H.; Perry, V.H.; Murray, C.L.; Cunningham, C. Microglia in the degenerating brain are capable of phagocytosis of beads and of apoptotic cells, but do not efficiently remove PrPSc, even upon LPS stimulation. Glia 2010, 58, 2017–2030. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, A.; Hirato, J.; Nakazato, Y. Immunohistochemical study of microglia in the Creutzfeldt-Jakob diseased brain. Acta Neuropathol. 1993, 86, 337–344. [Google Scholar] [CrossRef]
- Williams, A.E.; Lawson, L.J.; Perry, V.H.; Fraser, H. Characterization of the microglial response in murine scrapie. Neuropathol. Appl. Neurobiol. 1994, 20, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Chida, J.; Hara, H.; Uchiyama, K.; Takahashi, E.; Miyata, H.; Kosako, H.; Tomioka, Y.; Ito, T.; Horiuchi, H.; Matsuda, H.; et al. Prion protein signaling induces M2 macrophage polarization and protects from lethal influenza infection in mice. PLoS Pathog. 2020, 16, e1008823. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, D.; Homma, T.; Nakagaki, T.; Fuse, T.; Sano, K.; Satoh, K.; Mori, T.; Atarashi, R.; Nishida, N. Type I interferon protects neurons from prions inin vivomodels. Brain 2019, 142, 1035–1050. [Google Scholar] [CrossRef] [Green Version]
- Malachin, G.; Reiten, M.R.; Salvesen, Ø; Aanes, H.; Kamstra, J.H.; Skovgaard, K.; Heegaard, P.M.H.; Ersdal, C.; Espenes, A.; Tranulis, M.A.; et al. Loss of prion protein induces a primed state of type I interferon-responsive genes. PLoS ONE 2017, 12, e0179881. [Google Scholar] [CrossRef] [Green Version]
- Thackray, A.M.; McKenzie, A.N.; Klein, M.A.; Lauder, A.; Bujdoso, R. Accelerated Prion Disease in the Absence of Interleukin-10. J. Virol. 2004, 78, 13697–13707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.-C. A Framework of All Discovered Immunological Pathways and Their Roles for Four Specific Types of Pathogens and Hypersensitivities. Front. Immunol. 2020, 11, 1992. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.-C. The Central THαβ Immunity Associated Cytokine: IL-10 Has a Strong Anti-Tumor Ability Toward Established Cancer Models In Vivo and Toward Cancer Cells In Vitro. Front. Oncol. 2021, 11, 1024. [Google Scholar] [CrossRef]
- Ishibashi, D.; Atarashi, R.; Fuse, T.; Nakagaki, T.; Yamaguchi, N.; Satoh, K.; Honda, K.; Nishida, N. Protective Role of Interferon Regulatory Factor 3-Mediated Signaling against Prion Infection. J. Virol. 2012, 86, 4947–4955. [Google Scholar] [CrossRef] [Green Version]
- Spinner, D.S.; Kascsak, R.B.; LaFauci, G.; Meeker, H.C.; Ye, X.; Flory, M.J.; Kim, J.I.; Schuller-Levis, G.B.; Levis, W.R.; Wisniewski, T.; et al. CpG oligodeoxynucleotide-enhanced humoral immune response and production of antibodies to prion protein PrPScin mice immunized with 139A scrapie-associated fibrils. J. Leukoc. Biol. 2007, 81, 1374–1385. [Google Scholar] [CrossRef]
- Oumata, N.; Nguyen, P.H.; Beringue, V.; Soubigou, F.; Pang, Y.; Desban, N.; Massacrier, C.; Morel, Y.; Paturel, C.; Contesse, M.-A.; et al. The Toll-Like Receptor Agonist Imiquimod Is Active against Prions. PLoS ONE 2013, 8, e72112. [Google Scholar] [CrossRef]
- Prinz, M.; Heikenwalder, M.; Schwarz, P.; Takeda, K.; Akira, S.; Aguzzi, A. Prion pathogenesis in the absence of Toll-like receptor signalling. EMBO Rep. 2003, 4, 195–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riemer, C.; Schultz, J.; Burwinkel, M.; Schwarz, A.; Mok, S.W.F.; Gültner, S.; Bamme, T.; Norley, S.; van Landeghem, F.; Lu, B.; et al. Accelerated Prion Replication in, but Prolonged Survival Times of, Prion-Infected CXCR3 −/− Mice. J. Virol. 2008, 82, 12464–12471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aucouturier, P.; Carp, R.I.; Carnaud, C.; Wisniewski, T. Prion Diseases and the Immune System. Clin. Immunol. 2000, 96, 79–85. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsou, A.; Chen, P.-J.; Tsai, K.-W.; Hu, W.-C.; Lu, K.-C. THαβ Immunological Pathway as Protective Immune Response against Prion Diseases: An Insight for Prion Infection Therapy. Viruses 2022, 14, 408. https://doi.org/10.3390/v14020408
Tsou A, Chen P-J, Tsai K-W, Hu W-C, Lu K-C. THαβ Immunological Pathway as Protective Immune Response against Prion Diseases: An Insight for Prion Infection Therapy. Viruses. 2022; 14(2):408. https://doi.org/10.3390/v14020408
Chicago/Turabian StyleTsou, Adam, Po-Jui Chen, Kuo-Wang Tsai, Wan-Chung Hu, and Kuo-Cheng Lu. 2022. "THαβ Immunological Pathway as Protective Immune Response against Prion Diseases: An Insight for Prion Infection Therapy" Viruses 14, no. 2: 408. https://doi.org/10.3390/v14020408
APA StyleTsou, A., Chen, P.-J., Tsai, K.-W., Hu, W.-C., & Lu, K.-C. (2022). THαβ Immunological Pathway as Protective Immune Response against Prion Diseases: An Insight for Prion Infection Therapy. Viruses, 14(2), 408. https://doi.org/10.3390/v14020408