1. Introduction
For many years, the modified virus Ankara (MVA) has been widely used as a viral vector for the development of numerous vaccines that are presently at different stages of clinical and preclinical trials. Vaccine candidates against tuberculosis, malaria, flu, HIV and MERS expressing transgenic antigens have been developed [
1].
Some features of the virus have contributed to that popularity of the vector. MVA was obtained in Germany as a result of multiple passages of vaccinia virus (VACV) Ankara in chicken embryo fibroblasts, which led to the loss of some part of the genome and the ability to replicate in mammalian cells, including human cells (except for the cell line BHK-21) [
2].
Such an attenuated phenotype is beneficial from a safety perspective, which was proven on more than 120,000 people during vaccination against smallpox [
3]. At the same time, MVA as a viral vector provides a high level of expression of the delivered antigen, and as a result, it exerts a potent immunogenic effect, thus provoking humoral and cellular responses [
4]. Along with the high capacity of a vector for recombinant DNA and the simplicity of handling vectors in laboratory conditions, these features make MVA a promising viral vector for the development of effective vaccines.
Recombinant strains of MVA are obtained by homologous recombination between the genome of the virus and the vector plasmid that carries the target gene. Recombination occurs in host cells during the replication of the virus. However, the rate of homologous recombination is very low, and the obtained virus progeny primarily consists of a parental virus (99.9%) [
5]; therefore, it is necessary to enrich and isolate the recombinant virus after recombination.
A traditional method of recombinant virus purification is the isolation of individual virus plaques. This method contains several stages and is labor- and time-consuming, particularly because of the difficulty of visualizing and analyzing plaques. Thus, other methods were developed that simplified this process. For example, some of them were based on co-expression of genes allowing resistance to some substance, such as mycophenolic acid (xanthine transferase gene-based selection (
gpt) from
E. coli ) [
6] or puromycin [
7]. Another method of selection involved the application of different markers that stain the plaques. The application of β-galactosidase and chromogenic substrate (X-gal) stains plaques blue [
8,
9], and the application of fluorescent proteins enables the selection of plaques by fluorescence [
10].
However, even when selective markers are used, the obtained plaques contain a significant share of the initial virus, and the isolation of pure recombinants can be time-consuming. Attempts have been made to accelerate the procedure of plaque selection; in particular, additional approaches to the acceleration of recombinant plaque selection using a semiquantitative PCR test were proposed [
11]. The proposed approach allowed us to select plaques with the maximal content of recombinant virus, thus accelerating the procedure of selection of the recombinant MVA viruses. However, the laborious character of the process associated with the regular selection of plaques limits the application of this approach.
The attempts to simplify the procedure of recombinant virus purification led to the development of selection methods based on altering of MVA replication process. For this, some of the Poxvirus genes that rescue MVA growth in certain host cells are inserted into the MVA genome along with the target gene.
For example, the insertion into the MVA of a Poxvirus K1L gene, which is lost during attenuation, allows a recombinant virus to replicate in rabbit kidney cell line RK-13. During passaging of a viral mixture of a wild-type and a recombinant MVA only the latter will replicate in this cell line, which provides the selection of rMVA [
12].
In addition, approaches based on the application of MVA with weakened growth obtained due to the deletion of certain genes have been developed. One such gene is F13L. Its deletion slows the rate of virus reproduction and results in a small plaques phenotype [
5]. A deletion of another gene, D4R, leads to a complete loss of replication ability in cells permissive for MVA [
13]. The restoration of a deleted gene in the MVA genome during recombination leads to the reacquisition of the growth rate of a wild strain of the virus. During further passages of the mixture of the initial and recombinant viruses, the growth of the parental strain with a deletion will be suppressed.
The aim of the study was to test and compare the above-mentioned selection methods.
2. Materials and Methods
2.1. Cells and Viruses
Baby hamster kidney cells BHK-21 (ATCC ® CCL-10) and BHK-21-D4R and human embryonic kidney HEK293FT (Invitrogen, Carlsbad, CA, USA) cells were grown in DMEM (Gibco, Waltham, MA, USA) supplemented with 4.5 g/L D-glucose, 10% fetal bovine serum (FBS) (Gibco, Waltham, MA, USA) and 0.01 M HEPES (Gibco, Waltham, MA, USA). Rabbit kidney cells RK-13 (ATCC® CCL-37) were grown in MEM (Gibco, Waltham, MA, USA) supplemented with 10% FBS (Gibco, Waltham, MA, USA), 0.01 M HEPES (Gibco, Waltham, MA, USA) and 0.1 mM nonessential amino acids (Gibco, Waltham, MA, USA). The cells were maintained in a 5% CO2 humidified atmosphere at 37 °C.
MVA and VACV strain WR were obtained from ATCC (VR-1508 and VR-2035, respectively).
2.2. Plasmids
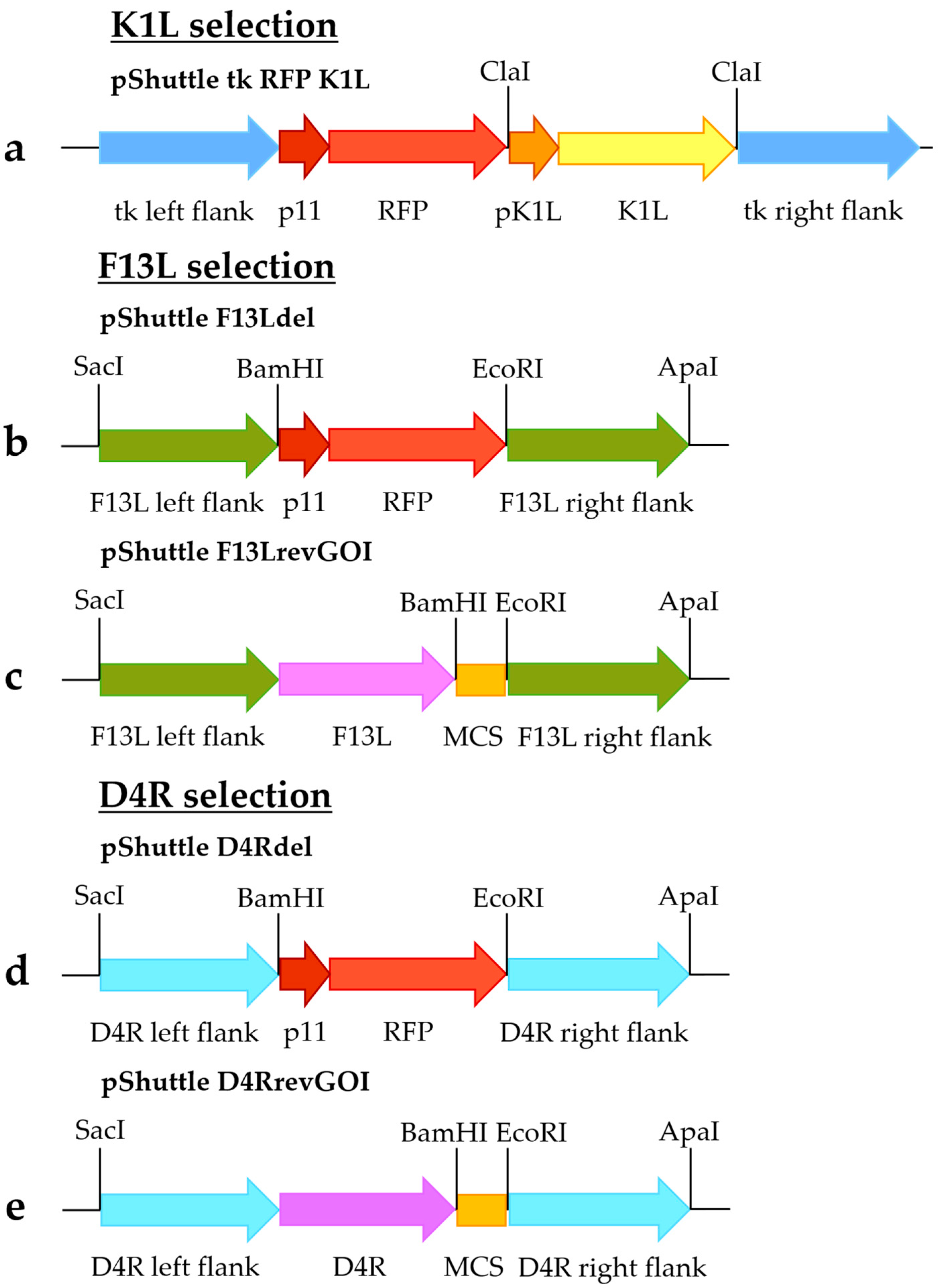
The shuttle vector pShuttle tk RFP K1L (
Figure 1a) was constructed as previously described [
12]. The sequence containing K1L controlled by its promoter was amplified using primers 1 and 2 specified in
Table 1 and VACV strain Western Reserve as a template. The PCR product was digested by ClaI and inserted into the corresponding site in the pShuttle tk RFP plasmid, which contained the red fluorescent protein (RFP) gene flanked by the MVA thymidine kinase gene flanking sequences. The insert orientation was checked by sequencing. Generation of the pShuttle tk RFP vector was previously described [
11].
To obtain pShuttle F13Ldel (
Figure 1b) and pShuttle D4Rdel (
Figure 1d) shuttle vectors, flanking sequences of the F13L and D4R genes were amplified. Fragments containing left recombination arms of F13L and D4R (using primers 3 and 4 for F13L [
5] and primers 13 and 14 for D4R [
13],
Table 1) and right recombination arms (primers 5 and 6 for F13L and primers 15 and 16 for D4R,
Table 1) were amplified using the MVA genome as a template. In addition, a fragment containing RFP controlled by the tk promoter was amplified using primers 7 and 8 and plasmid pShuttle tk RFP as a template. The fragments were digested with restriction enzymes specified in
Table 1 and
Figure 1 and cloned between SacI/ApaI restriction sites of the pGEM-T Easy vector (Promega, Madison, WI, USA) (
Figure 1b,d).
Shuttle vectors pShuttle F13LrevGOI (
Figure 1c) and pShuttle D4RrevGOI (
Figure 1e) were constructed by replacing the left flanking sequences in the pShuttle F13Ldel and pShuttle D4Rdel shuttle vectors with fragments containing the left F13L/D4R flanking sequences along with complete F13L/D4R genes, respectively. These fragments were amplified using the MVA genome as a template, with primers 9 and 10 for the F13L fragment and primers 19 and 20 for the D4R fragment (specified in
Table 1) and digested by SacI/BamHI sites. In addition, a fragment containing a multiple cloning site was obtained by restriction of the plasmid pShuttle tk RFP by BamHI/EcoRI sites. The fragments were cloned into SacI/EcoRI-digested shuttle vectors pShuttle F13Ldel (
Figure 1b) or pShuttle D4Rdel (
Figure 1d).
To generate the lentiviral vector pLV D4R, a fragment containing D4R controlled by its promoter was amplified using primers 11 and 12 and the MVA genome as a template and cloned into the XbaI/BamHI-digested vector pG with EGFP as a marker gene (previously described in [
14]).
2.3. Generation of Recombinant MVA
Recombinant MVA viruses (rMVA) were generated by homologous recombination [
15]. A confluent monolayer of BHK-21 cells grown in a 6-well plate was infected with MVA (0.05 PFU/cell). Ninety minutes after infection, the medium was replaced with 2 mL of fresh DMEM supplemented with 2% FBS, and the cells were transfected with 3 µg of plasmid DNA using FuGENE HD Transfection Reagent (Promega, Madison, WI, USA). After 48 h of incubation, the cells were detached and lysed with three cycles of freeze/thawing. For subsequent infection, the virus stocks were sonicated.
2.4. RK-13 Cells Infection and Plaques Isolation
A confluent monolayer of RK-13 cells grown in a 6-well plate was infected in a total volume of 2 mL per well with serial dilutions of viral lysates obtained from homologous recombination or separate plaques and incubated for 72 h. Separate plaques were picked with a pipette directly from the cell monolayer, transferred into 500 µL of MEM and lysed with three cycles of freeze/thawing. For subsequent infection, the virus stocks were sonicated.
2.5. BHK-21 Cells Infection (Virus Serial Passaging)
A confluent monolayer of BHK-21 cells grown in a 6-well plate was infected with 100 µL of the virus stock after recombination (total volume was 2 mL of medium per well). The cells were incubated for 48 h, detached and lysed with three cycles of freeze/thawing. The obtained stock was used for the next passage (50 µL per well of a 6-well plate) [
13,
16].
2.6. Purification of MVA by Plaque Isolation
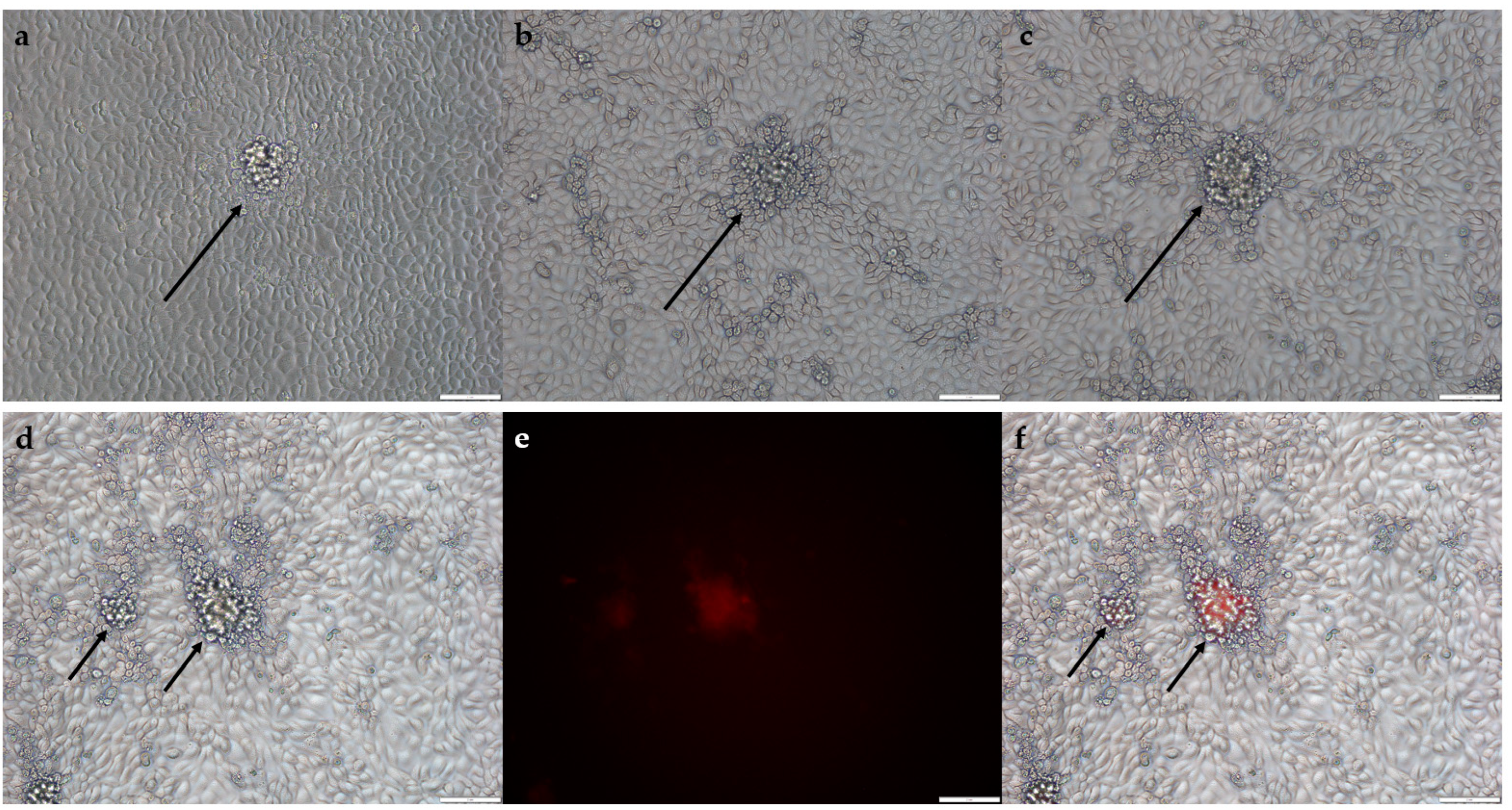
A confluent monolayer of BHK-21 or BHK-21-D4R cells grown in a 6-well plate was infected with serial dilutions of viral lysates obtained from homologous recombination or separate plaques. Ninety minutes after infection, the medium was replaced with 2 mL of fresh DMEM supplemented with 2% FBS and 1% low-melting point agarose. Forty-eight hours after infection, the fluorescent plaques were picked through agarose overlay.
2.7. Cloning by the Limiting Dilution Method
A confluent monolayer of BHK-21 or BHK-21-D4R or RK-13 cells grown in a 48-well plate was infected with serial dilutions of viral lysates and incubated for 72 h. Then, cells in wells that contained single plaques of the respective phenotype were detached and used as a crude virus stock.
2.8. Determination of Infectious Titers
A virus titer was determined using BHK-21/BHK-21-D4R cell lines and the TCID50 method previously described [
15].
2.9. Generation of a BHK-21-Based Cell Line Expressing the D4R Gene
The D4R gene was introduced into BHK-21 cells via lentiviral vector transduction.
Lentiviral vector particles were obtained by a standard method of transient transfection (previously described in [
17]) of HEK293FT cells with packaging plasmids Lenti-X HT Packaging Mix (Clontech, Mountain View, CA, USA) and vector plasmid pLV D4R. After 72 h, the supernatant containing vector particles was collected and filtered through 0.45 μm filters.
For target cell line generation, BHK-21 cells (105 per well in a 24-well plate) were infected with 200 µL of viral supernatant with the addition of 5 µg/mL polybrene (Sigma Aldrich, Burlington, MA, USA). On the next day, the medium was replaced with a fresh medium. Furthermore, the cells were seeded in a 96-well plate at a density of one cell per well, and EGFP-positive cells were selected. The selected EGFP+ clones were grown into separate cell lines.
2.10. Isolation of Virus DNA and PCR Performance
Viral DNA was isolated with the isopropanol precipitation method using the commercial “AmpliTest RIBO-prep” kit (Centre for Strategic Planning of FMBA of Russia, Moscow, Russia) according to the manufacturer’s recommendations.
For each selection system, we selected primers that anneal adjacent to the shuttle vector insertion sites in the MVA genome (tk locus, F13L locus and D4R locus). During amplification, fragments corresponding to rMVA and a wild-type virus are obtained that differ by length. All primers and amplification programs are listed in
Table 2. The products of amplification were analyzed with gel electrophoresis. By the presence of products, the presence of the parent and target viruses in the viral mixture was determined.
4. Discussion
To obtain an MVA-based vaccine, it is necessary to integrate the sequence encoding antigens into the viral genome. It is carried out by homologous recombination between the initial virus and shuttle vector with further isolation of the target recombinant MVA by plaque purification. This process is rather laborious and inefficient. Previously, we tried to optimize a conventional method of rMVA selection by adding a real-time PCR test of single plaques [
11], which allowed for the acceleration of recombinant virus selection. However, the process remained laborious and did not allow us to obtain numerous recombinants simultaneously.
Several methods of recombinant virus purification based on the enrichment of rMVA during replication have been developed to simplify the process. However, until now these methods have not been compared directly. Thus, the aim of the present study was to compare the efficiency and usability of some of them.
The first tested method of rMVA isolation was based on the host range gene, K1L, as the selective growth marker in rabbit kidney RK-13 cells. We showed that such a method of rMVA selection did not perform well. The publications mentioned various periods required for isolation of pure recombinant virus (from 2 to 5 selective passages in RK-13) [
18,
19]. In our turn, we did not manage to obtain pure recombinant after four rounds of plaque selection, which is a more effective selection than just blind passaging. Even after the application of additional limiting dilution cloning, each of the tested clones had residual traces of wild-type MVA. The problem of rMVA selection using RK-13 was also described in the work by [
19], who suggested that the recombinant virus (K1L
+) allows the rescue of the wild-type virus (K1L
−) in the same cell.
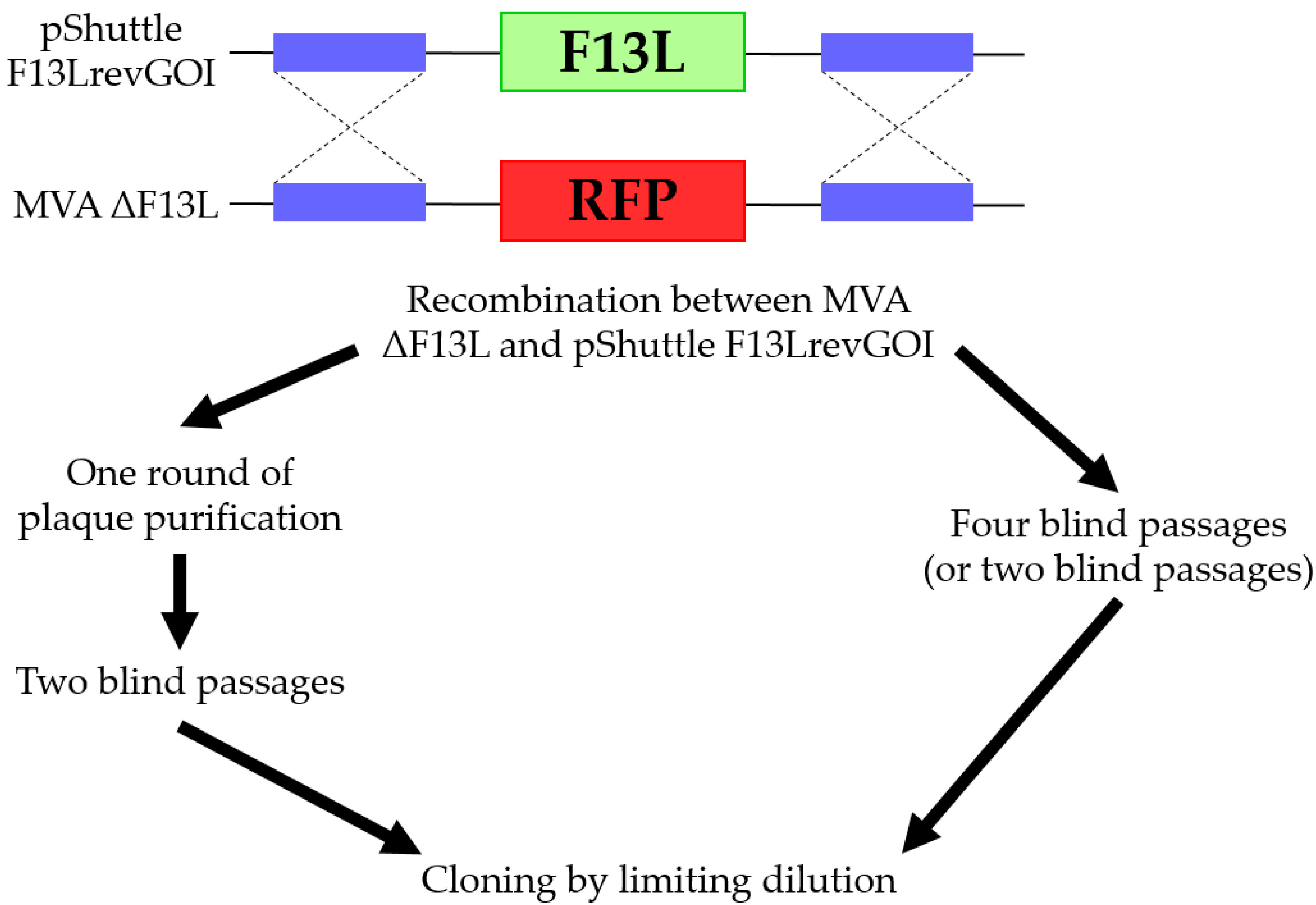
Further, we tested two similar systems of selection based on the deletion of F13L or D4R from the MVA genome and further reinsertion of these genes, which are necessary for successful replication. The main difference between MVA ΔF13L and MVA ΔD4R was in the rate of replication. MVA ΔF13L replication is dramatically reduced in the BHK-21 cell culture compared to wild-type MVA, while MVA without essential D4R gene could not replicate in these cells at all. Since MVA ΔD4R was not capable of replicating in cells, for its propagating it was required to additionally generate a cell line expressing the D4R to complement the absent gene function.
We successfully obtained pure recombinant viruses without admixtures of the parental viruses using both systems. Although D4R-based selection was more effective. Nine out of ten (90%) viral clones, isolated by the limiting dilution method, did not contain the admixtures of the initial MVA ΔD4R after two blind passages. On the other hand, for F13L selection approximately 12% and 50% of such samples were obtained after two and four blind passages correspondingly.
Both systems were simple and convenient in practice. First, there is no need for the application of antibiotics, selective agents, agar medium, etc. Second, a recombinant virus can be obtained after two to four blind passages without laborious plaque isolation and analysis of plaques at each stage. Third, since passaging is not time-consuming, several recombinant viruses can be obtained simultaneously. Fourth, the advantage of this method compared to the conventional method is that it does not require additional marker genes to select a recombinant sequence and thus does not need further removal of these sequences.
The disadvantage of both systems is that the insertion of the target gene is possible only in the initial locus, where the genes F13L and D4R are located. Furthermore, this type of selection can be used for the integration of only one insert because the reinsertion of F13L/D4R along with the target gene restores the native growth properties of the virus.
When it is necessary to obtain rMVA with the additional insert in a different locus, the methods of rMVA selection should be combined. For example, F13L/D4R-based selection can be used for the first integration. For the second integration, it is possible to use an accelerated method of selection of single plaques using real-time PCR [
11]. The application of this approach allowed us to obtain rMVA that had insertions in the F13L locus and 136/137 intergene area. In this case, we omitted the limiting dilution cloning during the first recombinant purification (F13L selection), which did not influence the further process of selection of double recombinants (unpublished data). As a result, double recombinants were obtained in 16 days.
Additionally, the approach based on the application of MVA with the deletion of both genes (MVA ΔF13L ΔD4R) is promising because it can reduce the time for isolation of double recombinants. However, the prospects of this approach have yet to be elucidated.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}