
Phylodynamic Dispersal of SARS-CoV-2 Lineages Circulating across Polish–German Border Provinces


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Sequence Data and Down-Sampling
2.2. Inference of a Time-Scaled Phylogenetic Trees
2.3. Preliminary Discrete Phylogeographic Inference
2.4. Discrete Phylogeographic Analyses at the Polish–German Border Regions
3. Results
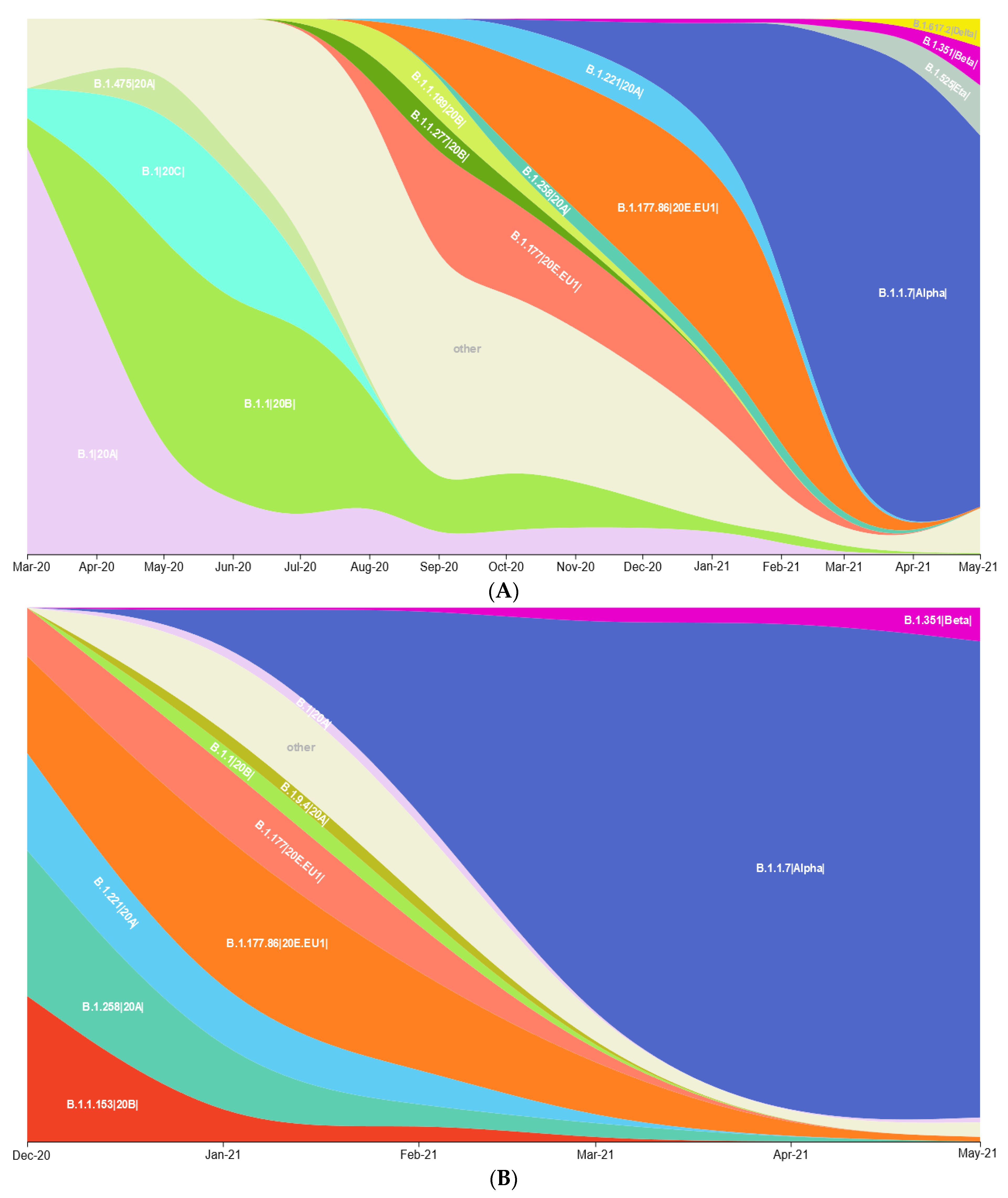
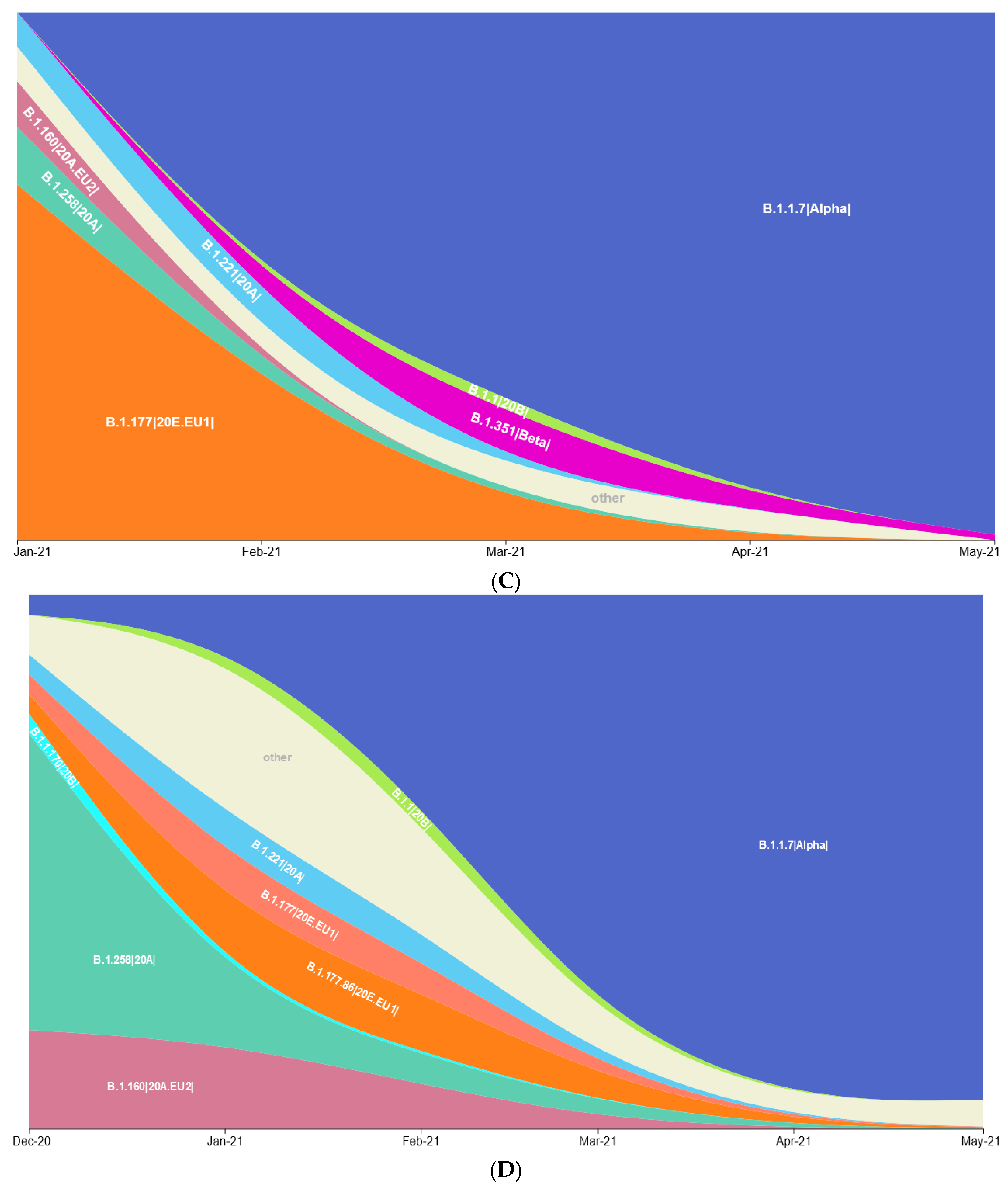
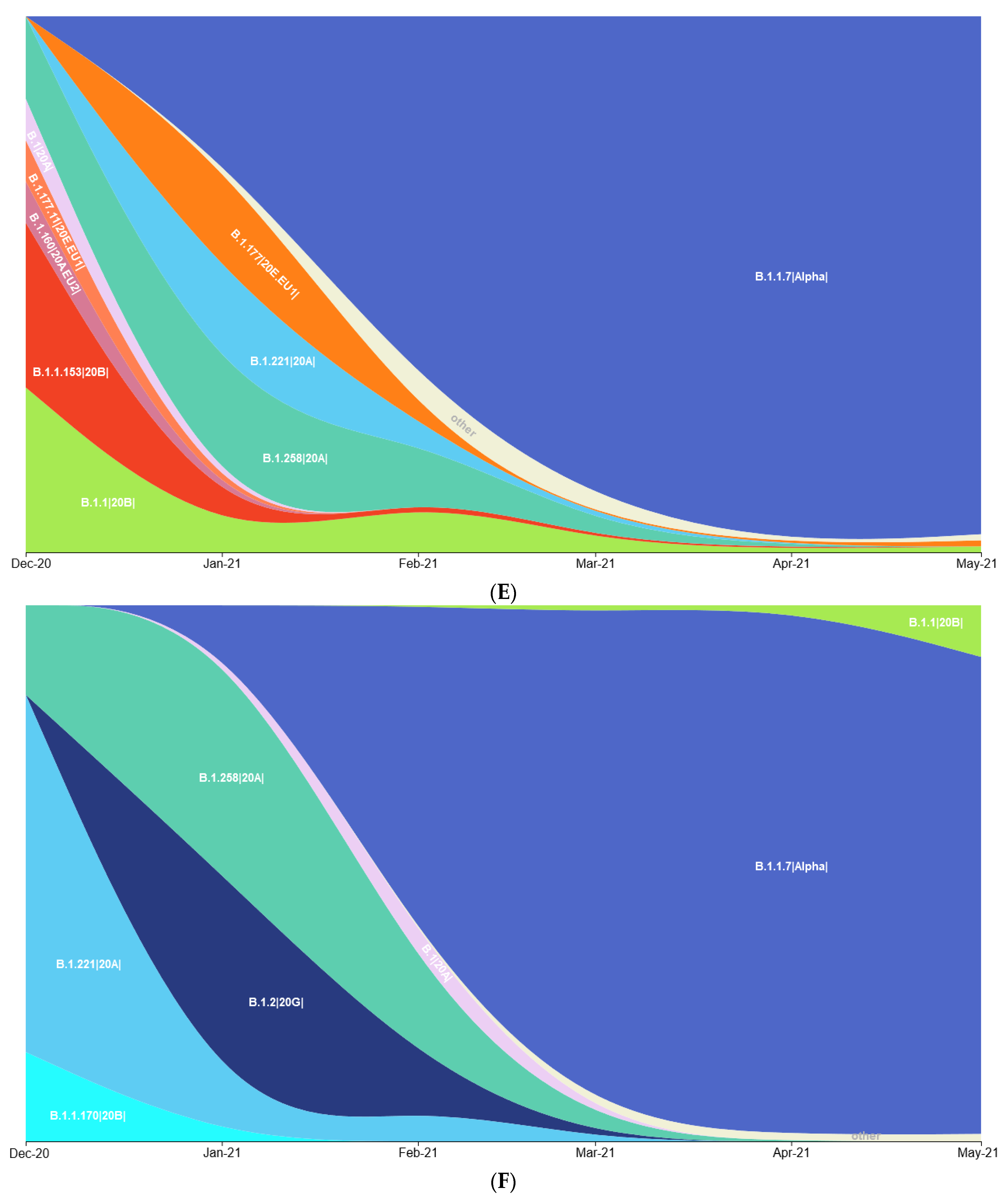
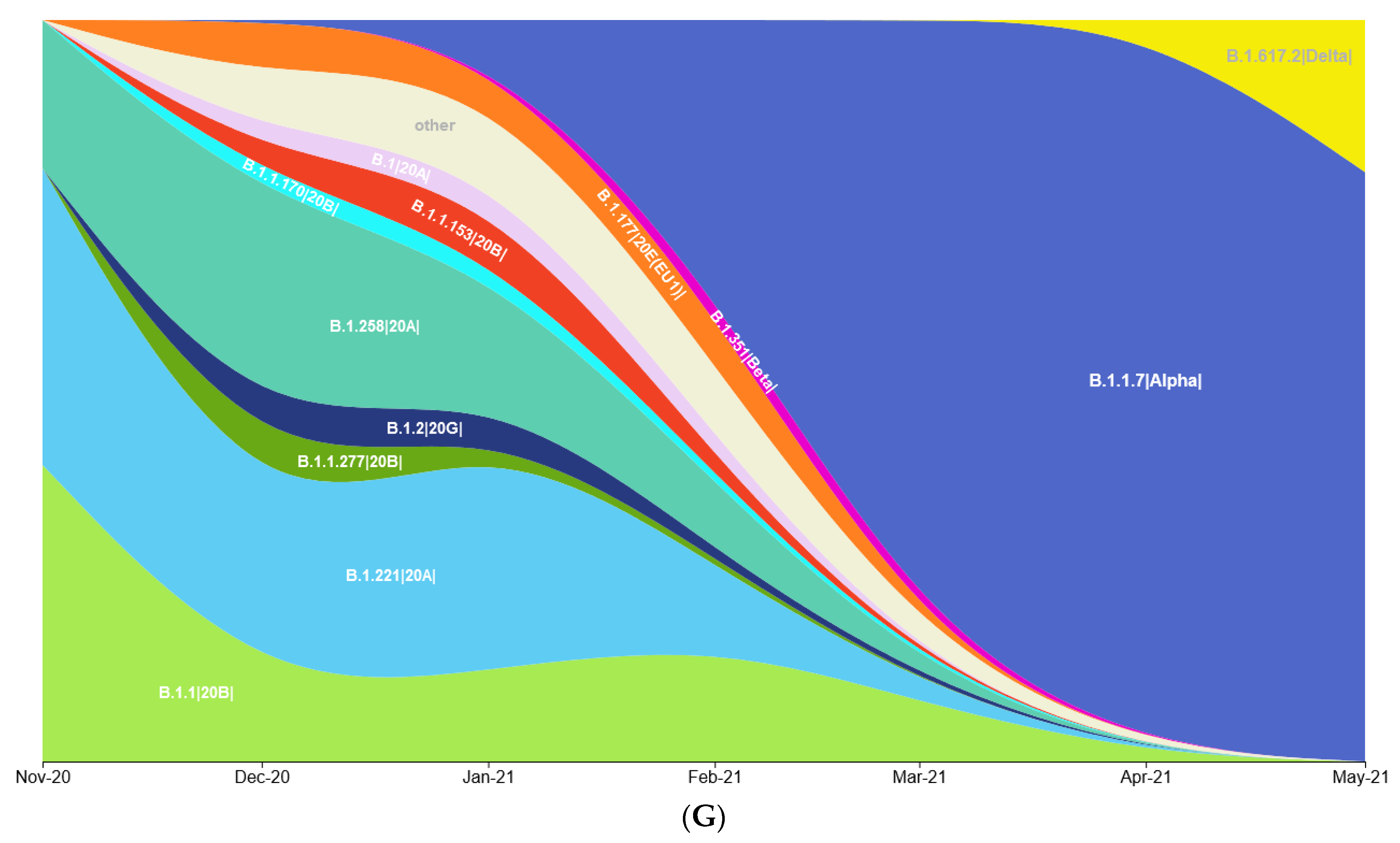
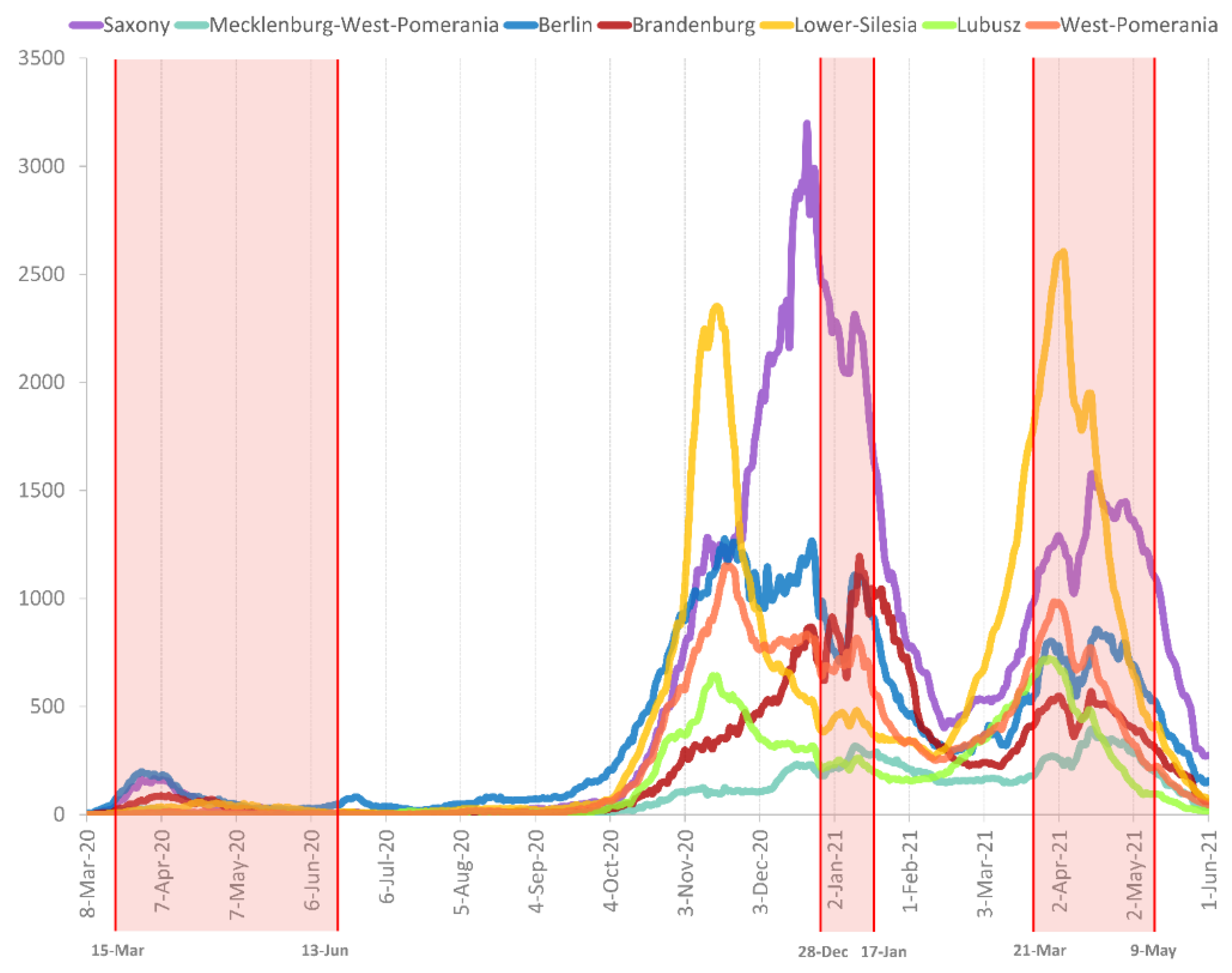
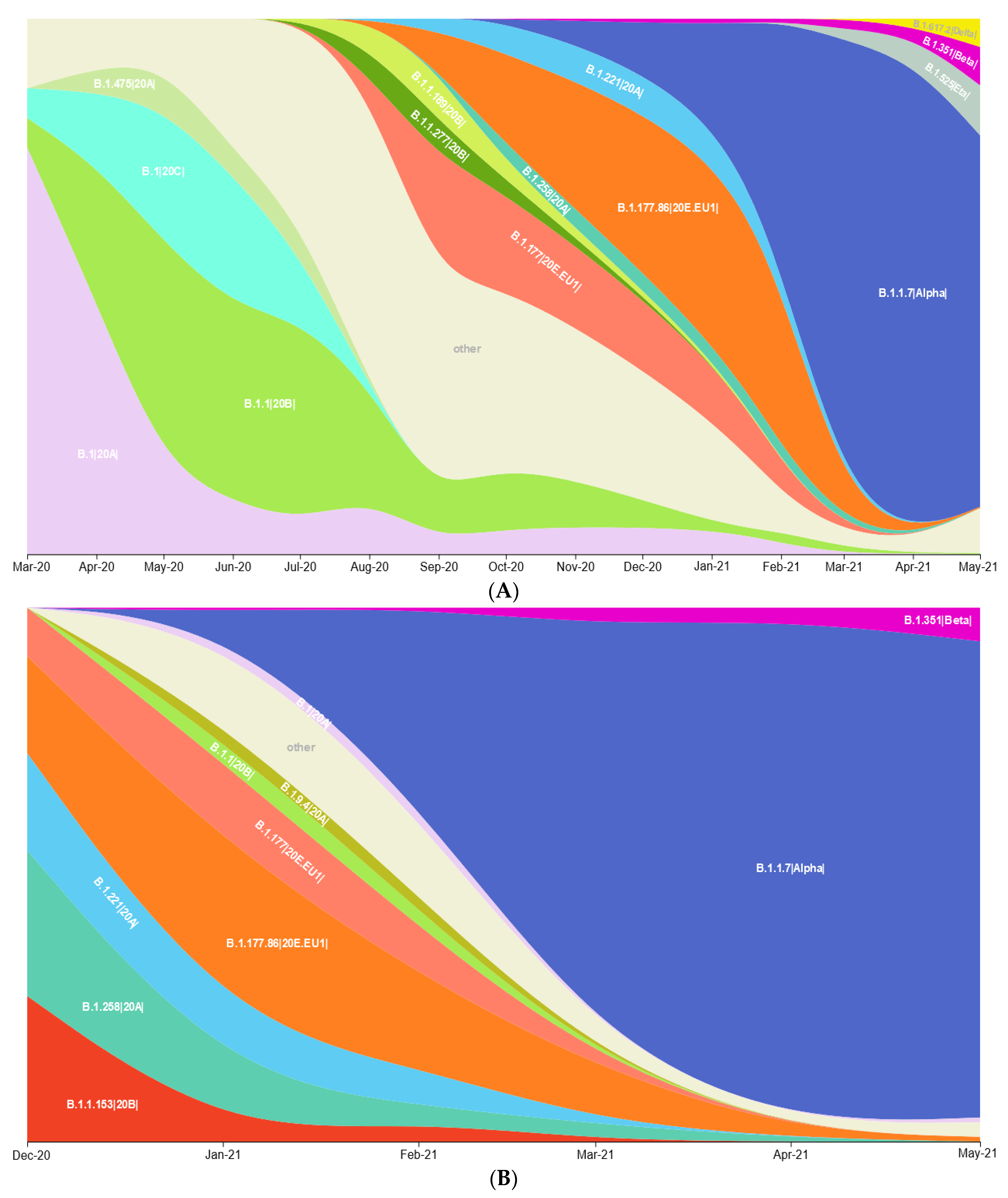
3.1. Prevalence of SARS-CoV-2 Variants between Polish–German Border Regions
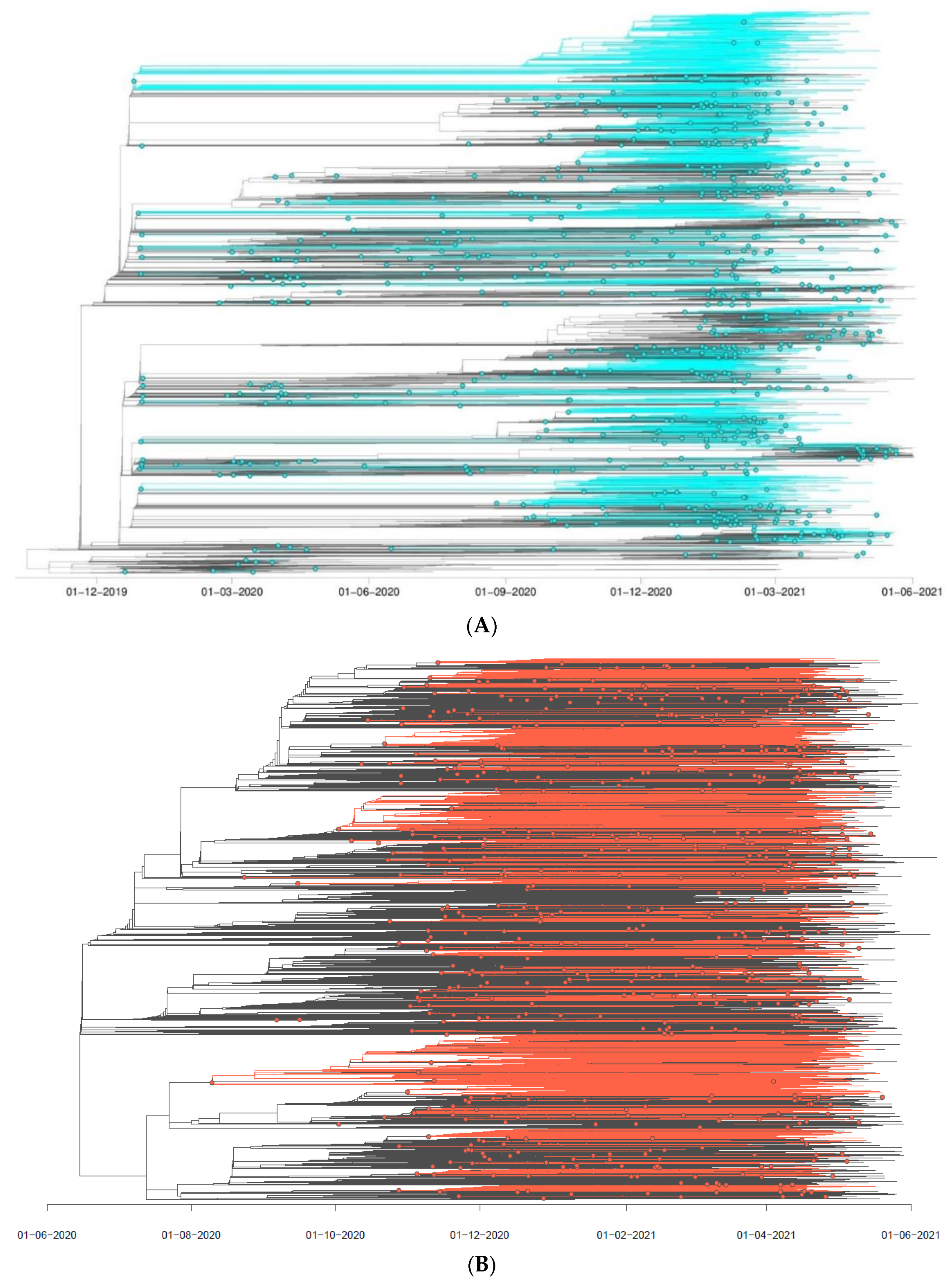
3.2. SARS-CoV-2 Introduction Events in the Polish–German Interregional Area
3.3. SARS-CoV-2 Circulation in the Polish–German Interregional Area
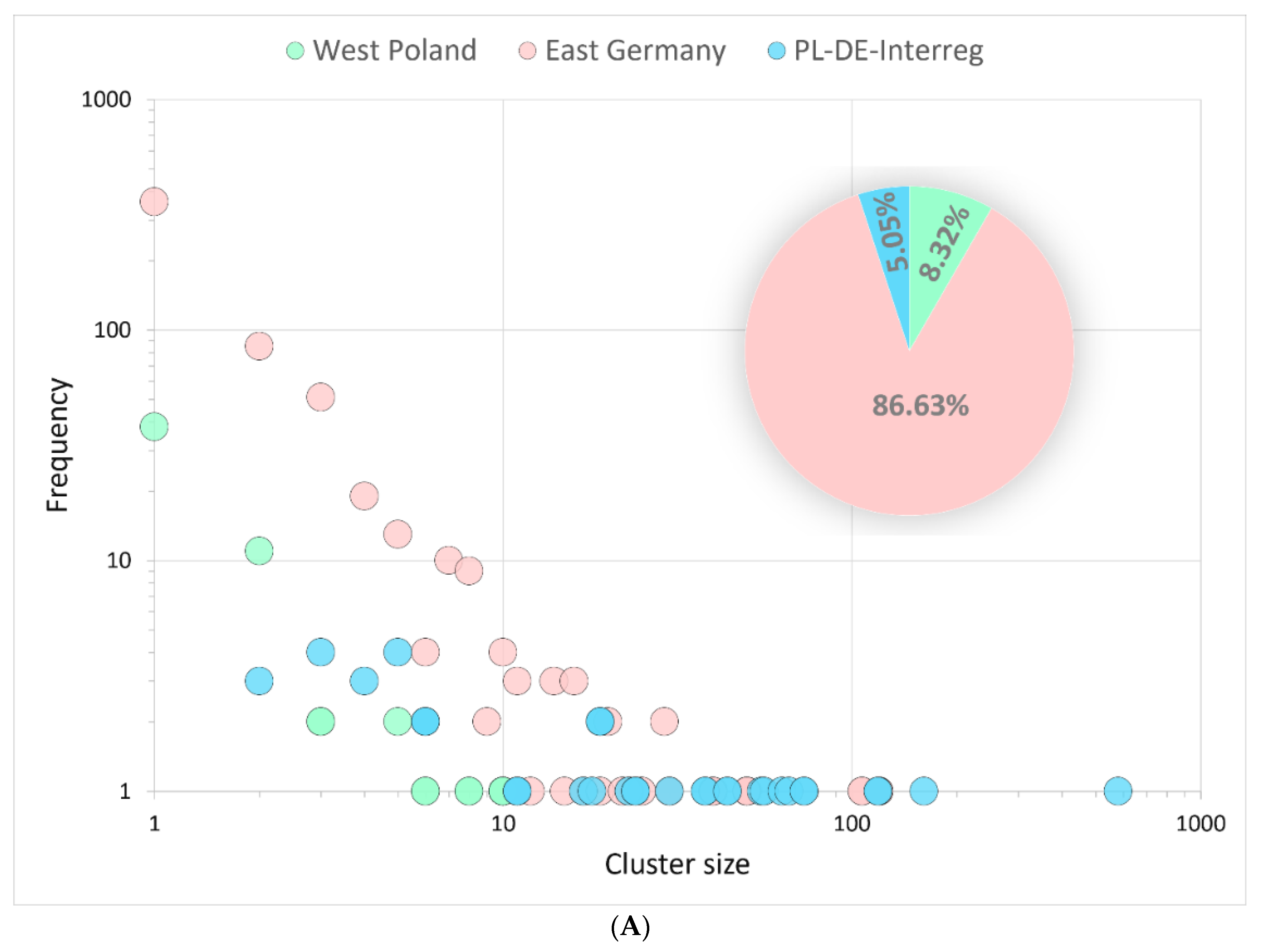
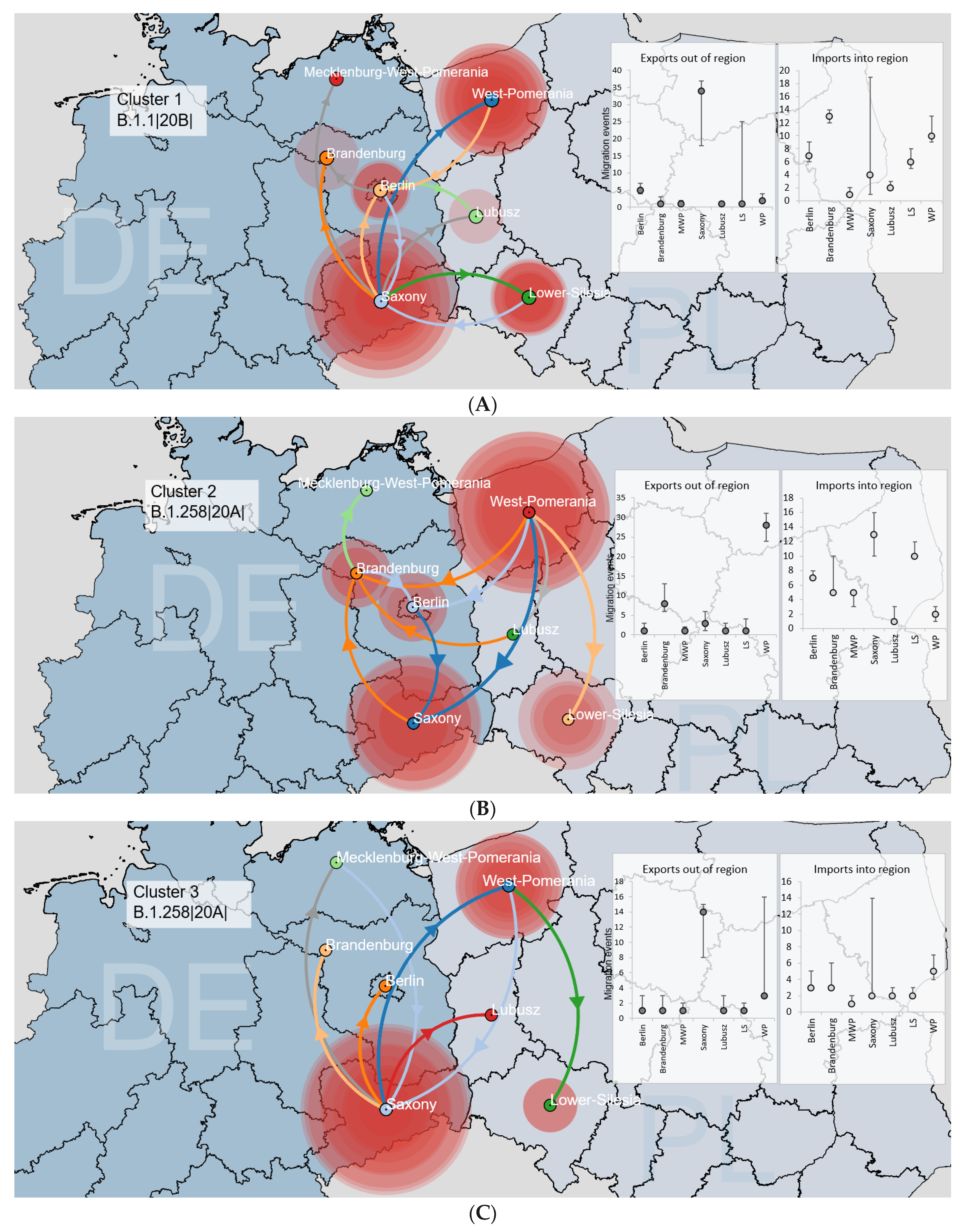
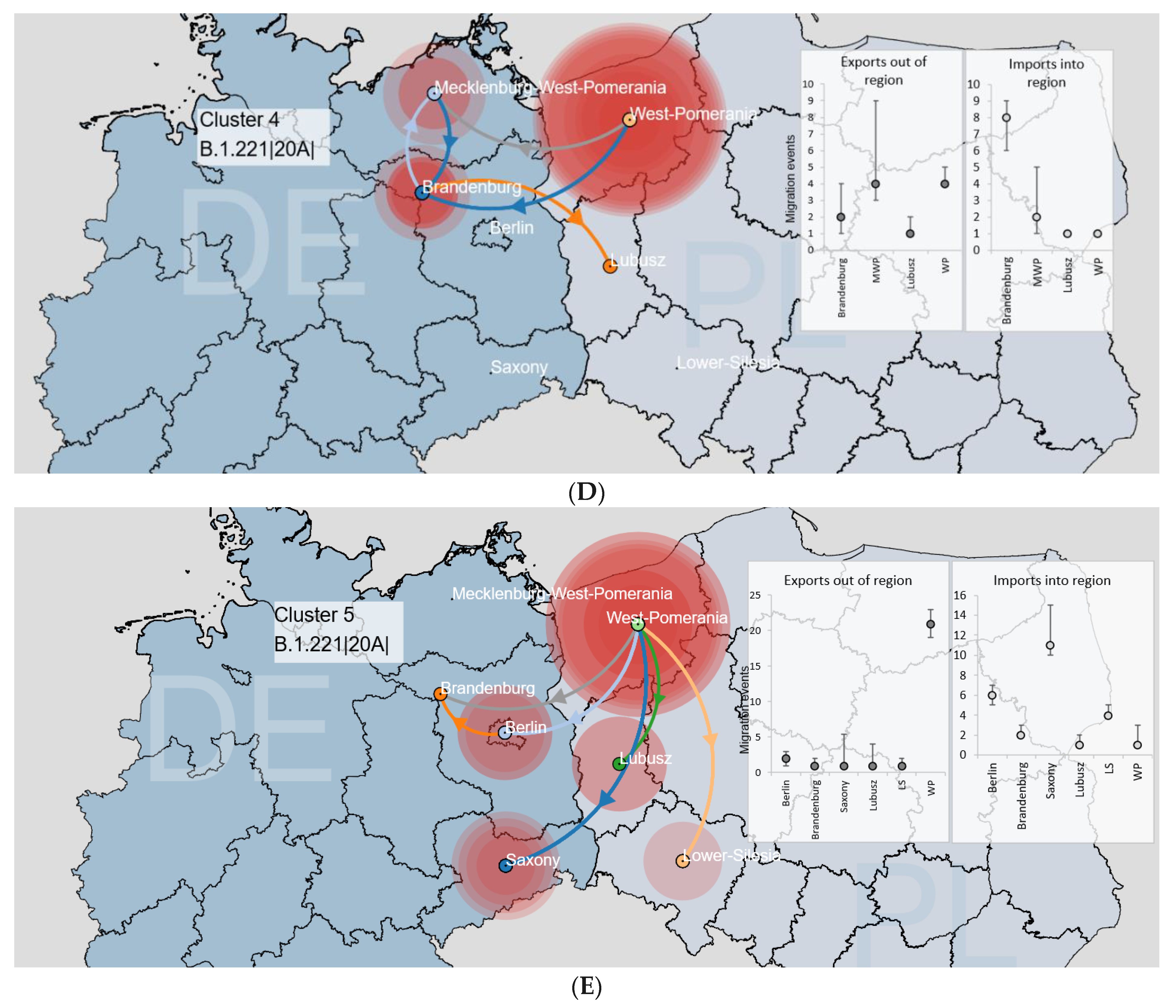
3.3.1. Binational Non-Alpha Clusters
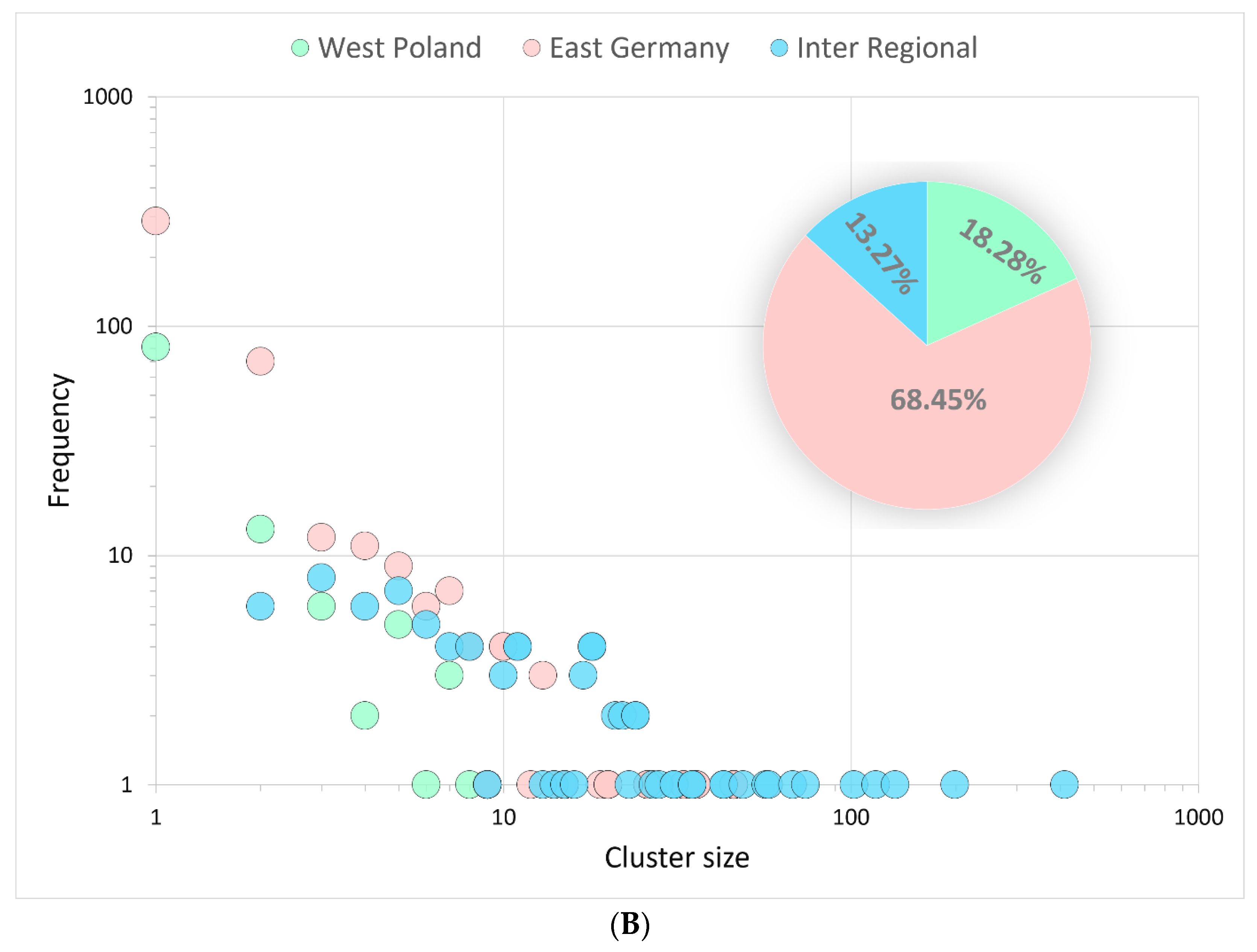
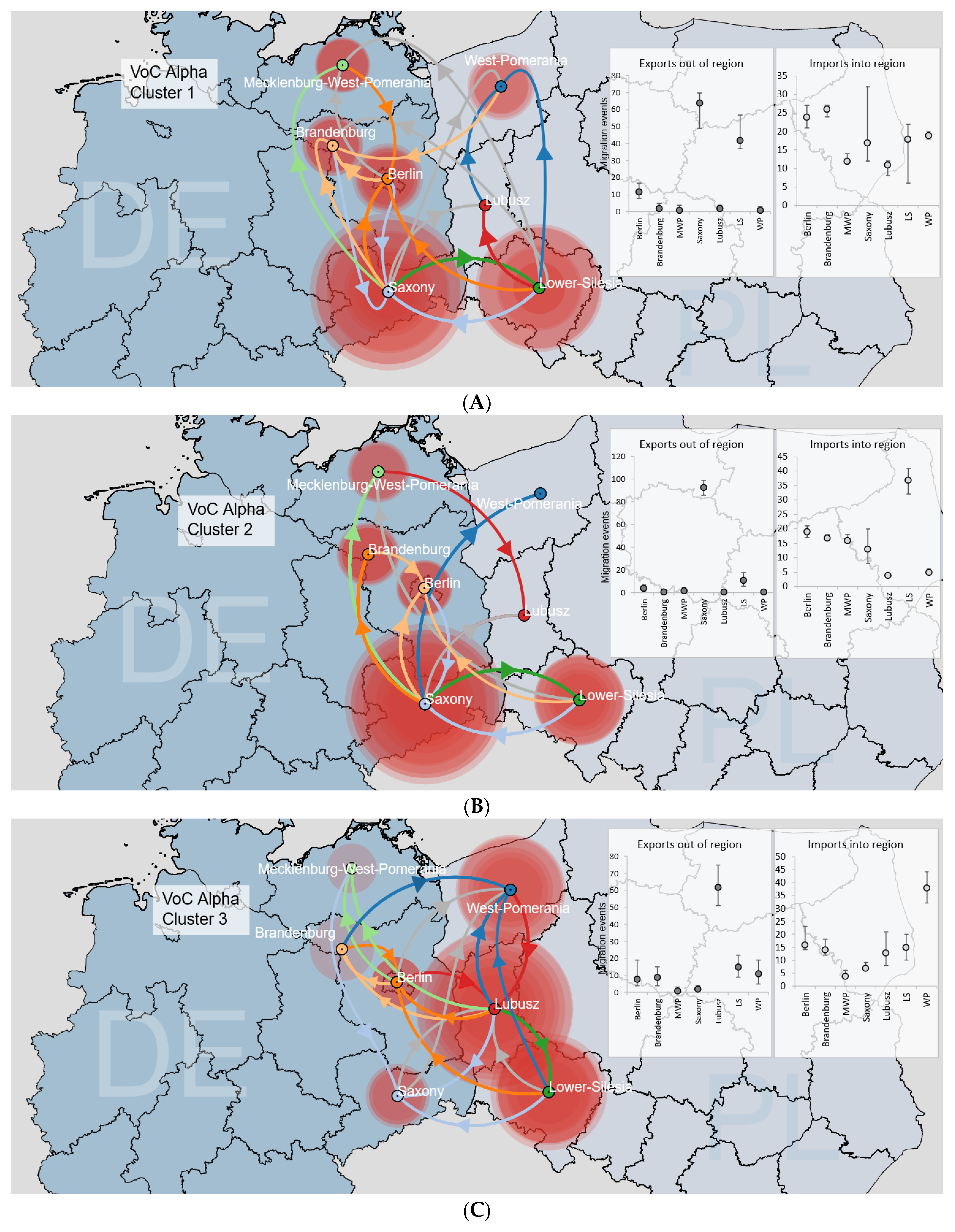
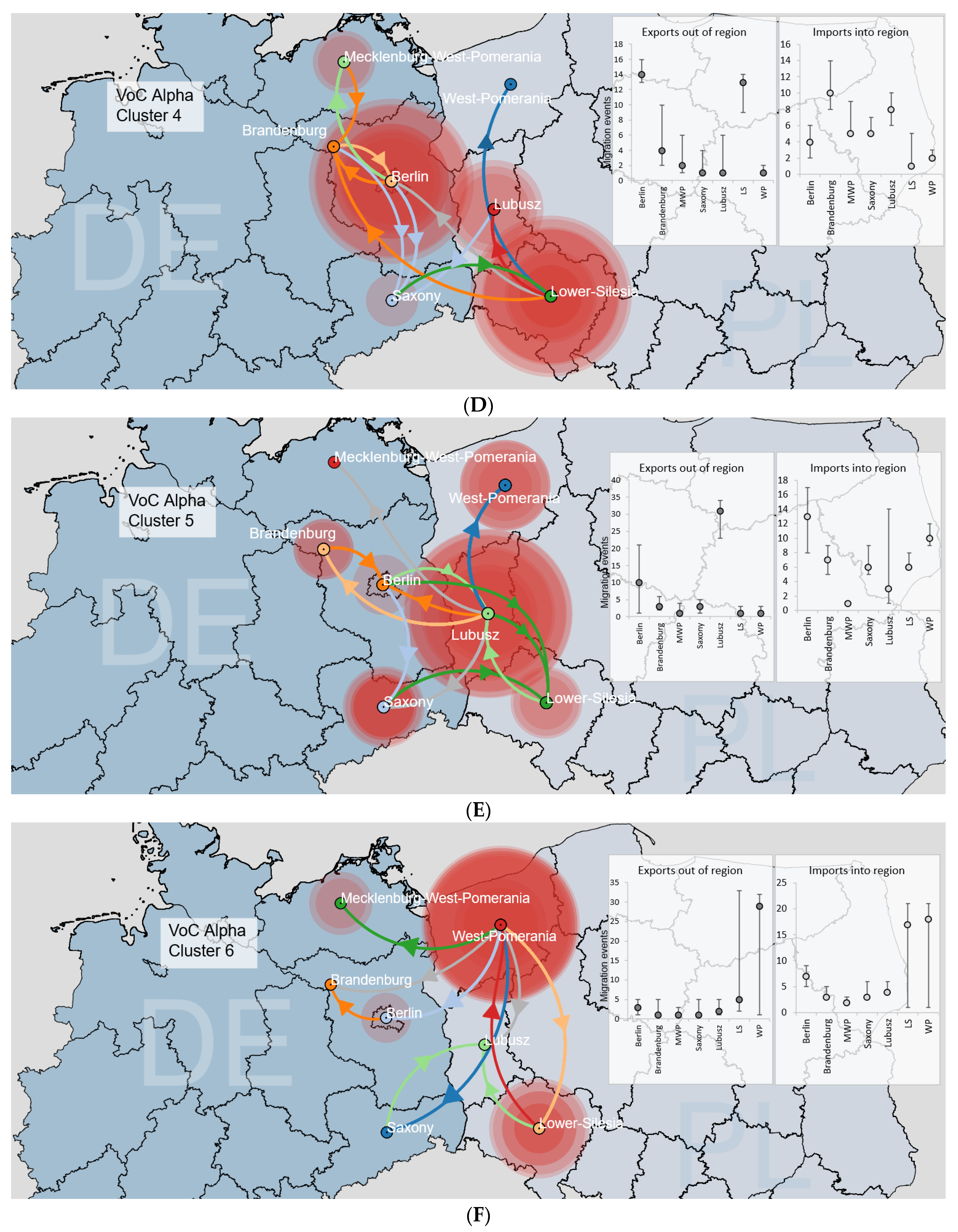
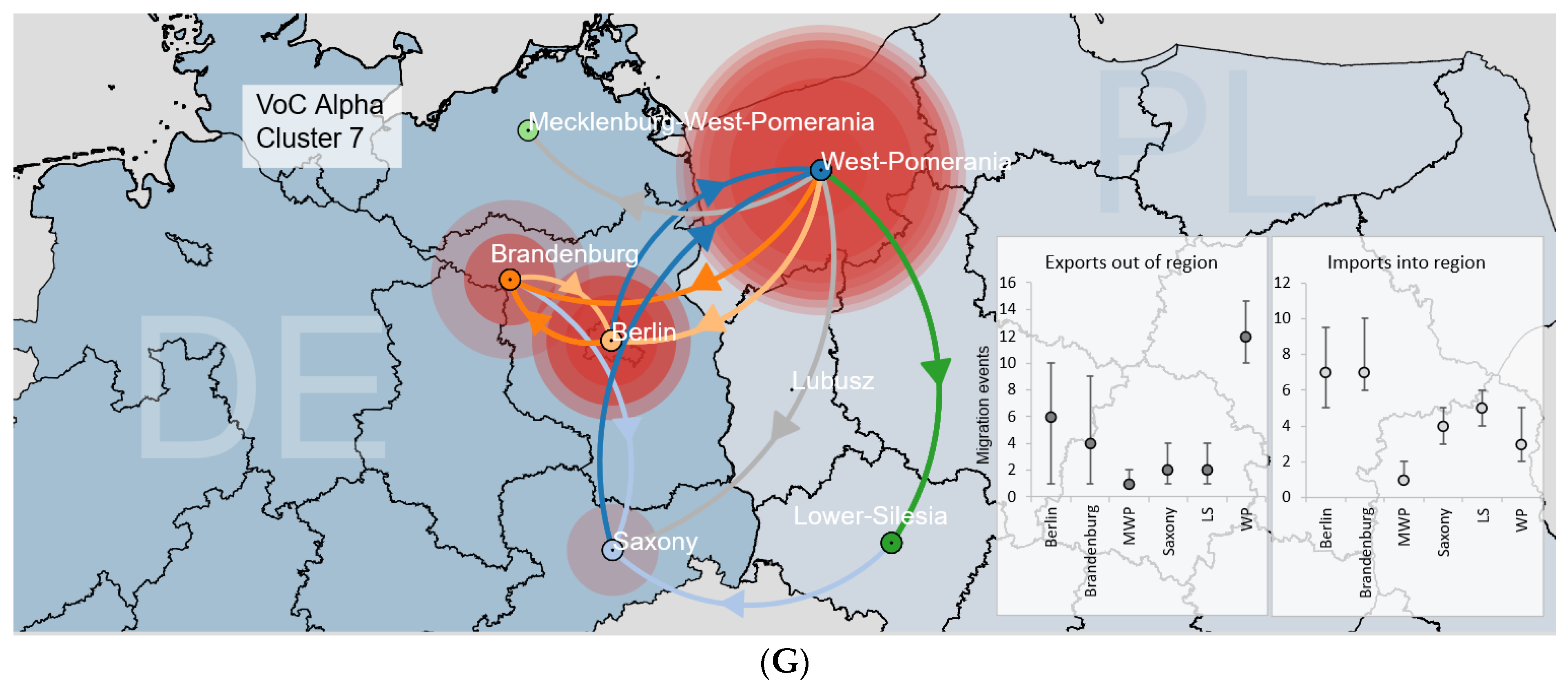
3.3.2. Binational B.1.1.7|Alpha Clusters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Pekar, J.; Larsen, B.; Nelson, M.; Hill, V.; Joy, J.; Rambaut, A.; Suchard, M.; Wertheim, J.; Lemey, P. The emergence of SARS-CoV-2 in Europe and North America. Science 2020, 370, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Oude Munnink, B.; Worp, N.; Nieuwenhuijse, D.; Sikkema, R.; Haagmans, B.; Fouchier, R.; Koopmans, M. The next phase of SARS-CoV-2 surveillance: Real-time molecular epidemiology. Nat. Med. 2021, 27, 1518–1524. [Google Scholar] [CrossRef]
- Alm, E.; Broberg, E.; Connor, T.; Hodcroft, E.; Komissarov, A.; Maurer-Stroh, S.; Melidou, A.; Neher, R.; O’Toole, Á.; Pereyaslov, D. Geographical and temporal distribution of SARS-CoV-2 clades in the WHO European Region, January to June 2020. Eurosurveillance 2020, 25, 2001410. [Google Scholar] [CrossRef]
- Islam, N.; Jdanov, D.; Shkolnikov, V.; Khunti, K.; Kawachi, I.; White, M.; Lewington, S.; Lacey, B. Effects of COVID-19 pandemic on life expectancy and premature mortality in 2020: Time series analysis in 37 countries. BMJ 2021, 375, e066768. [Google Scholar] [CrossRef]
- Islam, N.; Shkolnikov, V.; Acosta, R.; Klimkin, I.; Kawachi, I.; Irizarry, R.; Alicandro, G.; Khunti, K.; Yates, T.; Jdanov, D.; et al. Excess deaths associated with COVID-19 pandemic in 2020: Age and sex disaggregated time series analysis in 29 high income countries. BMJ 2021, 373, n1137. [Google Scholar] [CrossRef]
- Lai, A.; Bergna, A.; Caucci, S.; Clementi, N.; Vicenti, I.; Dragoni, F.; Cattelan, A.; Menzo, S.; Pan, A.; Callegaro, A.; et al. Molecular Tracing of SARS-CoV-2 in Italy in the First Three Months of the Epidemic. Viruses 2020, 12, 798. [Google Scholar] [CrossRef]
- López, M.; Chiner-Oms, Á.; García de Viedma, D.; Ruiz-Rodriguez, P.; Bracho, M.; Cancino-Muñoz, I.; D’Auria, G.; de Marco, G.; García-González, N.; Goig, G.; et al. The first wave of the COVID-19 epidemic in Spain was associated with early introductions and fast spread of a dominating genetic variant. Nat. Genet. 2021, 53, 1405–1414. [Google Scholar] [CrossRef]
- Oude Munnink, B.; Nieuwenhuijse, D.; Stein, M.; O’Toole, Á.; Haverkate, M.; Mollers, M.; Kamga, S.; Schapendonk, C.; Pronk, M.; Lexmond, P.; et al. Rapid SARS-CoV-2 whole-genome sequencing and analysis for informed public health decision-making in the Netherlands. Nat. Med. 2020, 26, 1405–1410. [Google Scholar] [CrossRef]
- Serwin, K.; Ossowski, A.; Szargut, M.; Cytacka, S.; Urbańska, A.; Majchrzak, A.; Niedźwiedź, A.; Czerska, E.; Pawińska-Matecka, A.; Gołąb, J.; et al. Molecular Evolution and Epidemiological Characteristics of SARS-COV-2 in (Northwestern) Poland. Viruses 2021, 13, 1295. [Google Scholar] [CrossRef] [PubMed]
- Plümper, T.; Neumayer, E. Lockdown policies and the dynamics of the first wave of the SARS-CoV-2 pandemic in Europe. J. Eur. Public Policy 2020, 29, 321–341. [Google Scholar] [CrossRef]
- Hodcroft, E.; Zuber, M.; Nadeau, S.; Vaughan, T.; Crawford, K.; Althaus, C.; Reichmuth, M.; Bowen, J.; Walls, A.; Corti, D.; et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Ruktanonchai, N.; Hong, S.; Colizza, V.; Poletto, C.; Van den Broeck, F.; Gill, M.; Ji, X.; Levasseur, A.; Oude Munnink, B.; et al. Untangling introductions and persistence in COVID-19 resurgence in Europe. Nature 2021, 595, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, M.; Hill, V.; Ruis, C.; Dellicour, S.; Bajaj, S.; McCrone, J.; Baele, G.; Parag, K.; Battle, A.; Gutierrez, B.; et al. Spatiotemporal invasion dynamics of SARS-CoV-2 lineage B.1.1.7 emergence. Science 2021, 373, 889–895. [Google Scholar] [CrossRef]
- Mlcochova, P.; Kemp, S.; Dhar, M.; Papa, G.; Meng, B.; Ferreira, I.; Datir, R.; Collier, D.; Albecka, A.; Singh, S.; et al. SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef]
- Davies, N.G.; Jarvis, C.I.; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature 2021, 593, 270–274. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Dellicour, S.; Durkin, K.; Hong, S.; Vanmechelen, B.; Martí-Carreras, J.; Gill, M.; Meex, C.; Bontems, S.; André, E.; Gilbert, M.; et al. A Phylodynamic Workflow to Rapidly Gain Insights into the Dispersal History and Dynamics of SARS-CoV-2 Lineages. Mol. Biol. Evol. 2021, 38, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.; Schmidt, H.; Chernomor, O.; Schrempf, D.; Woodhams, M.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavare, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
- Yang, Z. A space-time process model for the evolution of DNA sequences. Genetics 1995, 139, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.; Max Carvalho, L.; Pybus, O. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Sagulenko, P.; Puller, V.; Neher, R. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.; Suchard, M. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.; Lemey, P.; Baele, G.; Ayres, D.; Drummond, A.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Lemey, P.; Rambaut, A.; Bedford, T.; Faria, N.; Bielejec, F.; Baele, G.; Russell, C.; Smith, D.; Pybus, O.; Brockmann, D.; et al. Unifying viral genetics and human transportation data to predict the global transmission dynamics of human influenza H3N2. PLoS Pathog. 2014, 10, e1003932. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.; Xie, D.; Baele, G.; Suchard, M. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Bollen, N.; Artesi, M.; Durkin, K.; Hong, S.; Potter, B.; Boujemla, B.; Vanmechelen, B.; Martí-Carreras, J.; Wawina-Bokalanga, T.; Meex, C.; et al. Exploiting genomic surveillance to map the spatio-temporal dispersal of SARS-CoV-2 spike mutations in Belgium across 2020. Sci. Rep. 2021, 11, 18580. [Google Scholar] [CrossRef]
- Dellicour, S.; Rose, R.; Faria, N.; Lemey, P.; Pybus, O. SERAPHIM: Studying environmental rasters and phylogenetically informed movements. Bioinformatics 2016, 32, 3204–3206. [Google Scholar] [CrossRef]
- Faria, N.; Suchard, M.; Rambaut, A.; Lemey, P. Toward a quantitative understanding of viral phylogeography. Curr. Opin. Virol. 2011, 1, 423–429. [Google Scholar] [CrossRef] [Green Version]
- Faria, N.; Suchard, M.; Rambaut, A.; Streicker, D.; Lemey, P. Simultaneously reconstructing viral cross-species transmission history and identifying the underlying constraints. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2013, 368, 20120196. [Google Scholar] [CrossRef] [Green Version]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [Green Version]
- Kass, R.; Raftery, A. Bayes Factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- Du Plessis, L.; McCrone, J.; Zarebski, A.; Hill, V.; Ruis, C.; Gutierrez, B.; Raghwani, J.; Ashworth, J.; Colquhoun, R.; Connor, T.; et al. Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK. Science 2021, 371, 708–712. [Google Scholar] [CrossRef]
- Bittorf, M. Will Economic Relations between Germany and Poland Thrive Post-Coronavirus? Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&ved=2ahUKEwiEqIWpk__1AhWOzYsKHbdmAeMQFnoECAIQAQ&url=https%3A%2F%2Fwww.kfw.de%2FPDF%2FDownload-Center%2FKonzernthemen%2FResearch%2FPDF-Dokumente-Fokus-Volkswirtschaft%2FFokus-englische-Dateien%2FFokus-2020-EN%2FFocus-No.-296-July-2020-Germany-Poland.pdf&usg=AOvVaw2rVjibO6dO5QROm2T7WYxy (accessed on 1 March 2022).
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; Plessis, L.d.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Schuppert, A.; Polotzek, K.; Schmitt, J.; Busse, R.; Karschau, J.; Karagiannidis, C. Different spreading dynamics throughout Germany during the second wave of the COVID-19 pandemic: A time series study based on national surveillance data. Lancet Reg. Health—Eur. 2021, 6, 100151. [Google Scholar] [CrossRef]
- Buqing, Y.; Poetsch, A.; Stadtmüller, M.; Rost, F.; Winkler, S.; Dalpke, A. Phylogenetic analysis of SARS-CoV-2 lineage development across the first and second waves in Eastern Germany, 2020. bioRxiv 2021. [Google Scholar] [CrossRef]
- Frampton, D.; Rampling, T.; Cross, A.; Bailey, H.; Heaney, J.; Byott, M.; Scott, R.; Sconza, R.; Price, J.; Margaritis, M.; et al. Genomic characteristics and clinical effect of the emergent SARS-CoV-2 B.1.1.7 lineage in London, UK: A whole-genome sequencing and hospital-based cohort study. Lancet Infect. Dis. 2021, 21, 1246–1256. [Google Scholar] [CrossRef]
- Vrancken, B.; Mehta, S.; Ávila-Ríos, S.; García-Morales, C.; Tapia-Trejo, D.; Reyes-Terán, G.; Navarro-Álvarez, S.; Little, S.; Hoenigl, M.; Pines, H.; et al. Dynamics and Dispersal of Local Human Immunodeficiency Virus Epidemics within San Diego and across the San Diego-Tijuana Border. Clin. Infect. Dis. 2021, 73, e2018–e2025. [Google Scholar] [CrossRef]
- Candido, D.; Claro, I.; de Jesus, J.; Souza, W.; Moreira, F.; Dellicour, S.; Mellan, T.; du Plessis, L.; Pereira, R.; Sales, F.; et al. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science 2020, 369, 1255–1260. [Google Scholar] [CrossRef]
- Dellicour, S.; Hong, S.; Vrancken, B.; Chaillon, A.; Gill, M.; Maurano, M.; Ramaswami, S.; Zappile, P.; Marier, C.; Harkins, G.; et al. Dispersal dynamics of SARS-CoV-2 lineages during the first epidemic wave in New York City. PLoS Pathog. 2021, 17, e1009571. [Google Scholar] [CrossRef]
- Nguyen, P.; Kant, R.; Broeck, F.; Suvanto, M.; Alburkat, H.; Virtanen, J.; Ahvenainen, E.; Castren, R.; Hong, S.; Baele, G.; et al. The phylodynamics of SARS-CoV-2 during 2020 in Finland—Disappearance and re-emergence of introduced strains; Research Square. 2021; manuscript in preparation. [Google Scholar] [CrossRef]
- Duchene, S.; Featherstone, L.; Freiesleben de Blasio, B.; Holmes, E.; Bohlin, J.; Pettersson, J. The impact of public health interventions in the Nordic countries during the first year of SARS-CoV-2 transmission and evolution. Eurosurveillance 2021, 26, 2001996. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serwin, K.; Aksak-Wąs, B.; Parczewski, M. Phylodynamic Dispersal of SARS-CoV-2 Lineages Circulating across Polish–German Border Provinces. Viruses 2022, 14, 884. https://doi.org/10.3390/v14050884
Serwin K, Aksak-Wąs B, Parczewski M. Phylodynamic Dispersal of SARS-CoV-2 Lineages Circulating across Polish–German Border Provinces. Viruses. 2022; 14(5):884. https://doi.org/10.3390/v14050884
Chicago/Turabian StyleSerwin, Karol, Bogusz Aksak-Wąs, and Miłosz Parczewski. 2022. "Phylodynamic Dispersal of SARS-CoV-2 Lineages Circulating across Polish–German Border Provinces" Viruses 14, no. 5: 884. https://doi.org/10.3390/v14050884
APA StyleSerwin, K., Aksak-Wąs, B., & Parczewski, M. (2022). Phylodynamic Dispersal of SARS-CoV-2 Lineages Circulating across Polish–German Border Provinces. Viruses, 14(5), 884. https://doi.org/10.3390/v14050884