
A Novel Class of HIV-1 Inhibitors Targeting the Vpr-Induced G2-Arrest in Macrophages by New Yeast- and Cell-Based High-Throughput Screening

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell and Yeast
2.2. Plasmids and Chemical Compounds
2.3. Protein Expression and Purification
2.4. Vpr Inhibitor Screening Using Overexpressing Yeast
2.5. Cell-Based Cytotoxicity Analysis
2.6. Cell-Based G2 Arrest Inhibition Assay Using CELAVIEW
2.7. Analysis of the Cell Cycle Using Flow Cytometry
2.8. HIV-1 Virus Preparation
2.9. Inhibition Assay of HIV Replication Treated by Hit Compounds
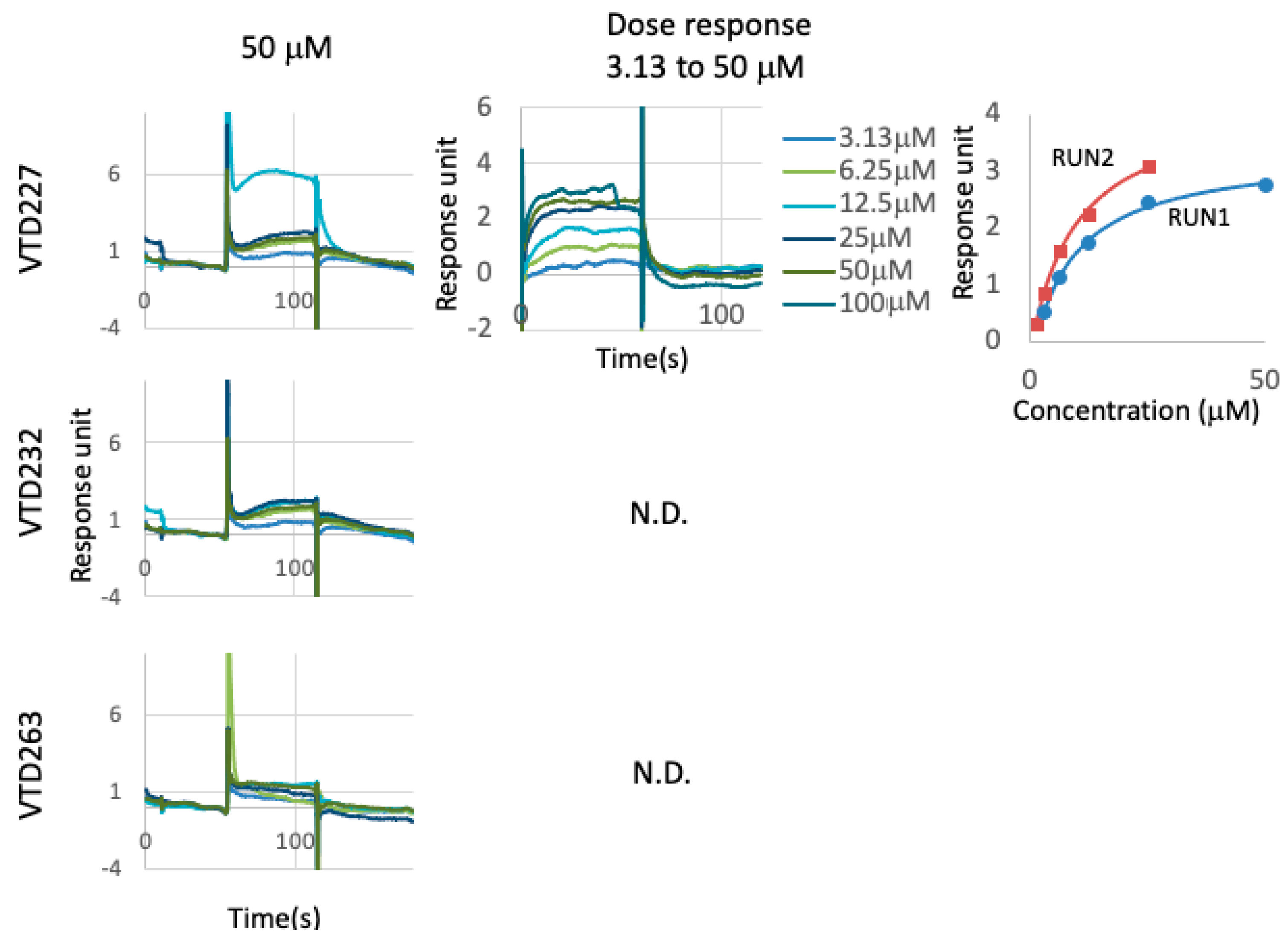
2.10. Surface Plasmon Resonance (SPR) Binding Analysis
2.11. Localization of Vpr Visualized by Confocal Microscopy
2.12. Expression Level of Vpr Assessed by Western Blotting Analysis
3. Results
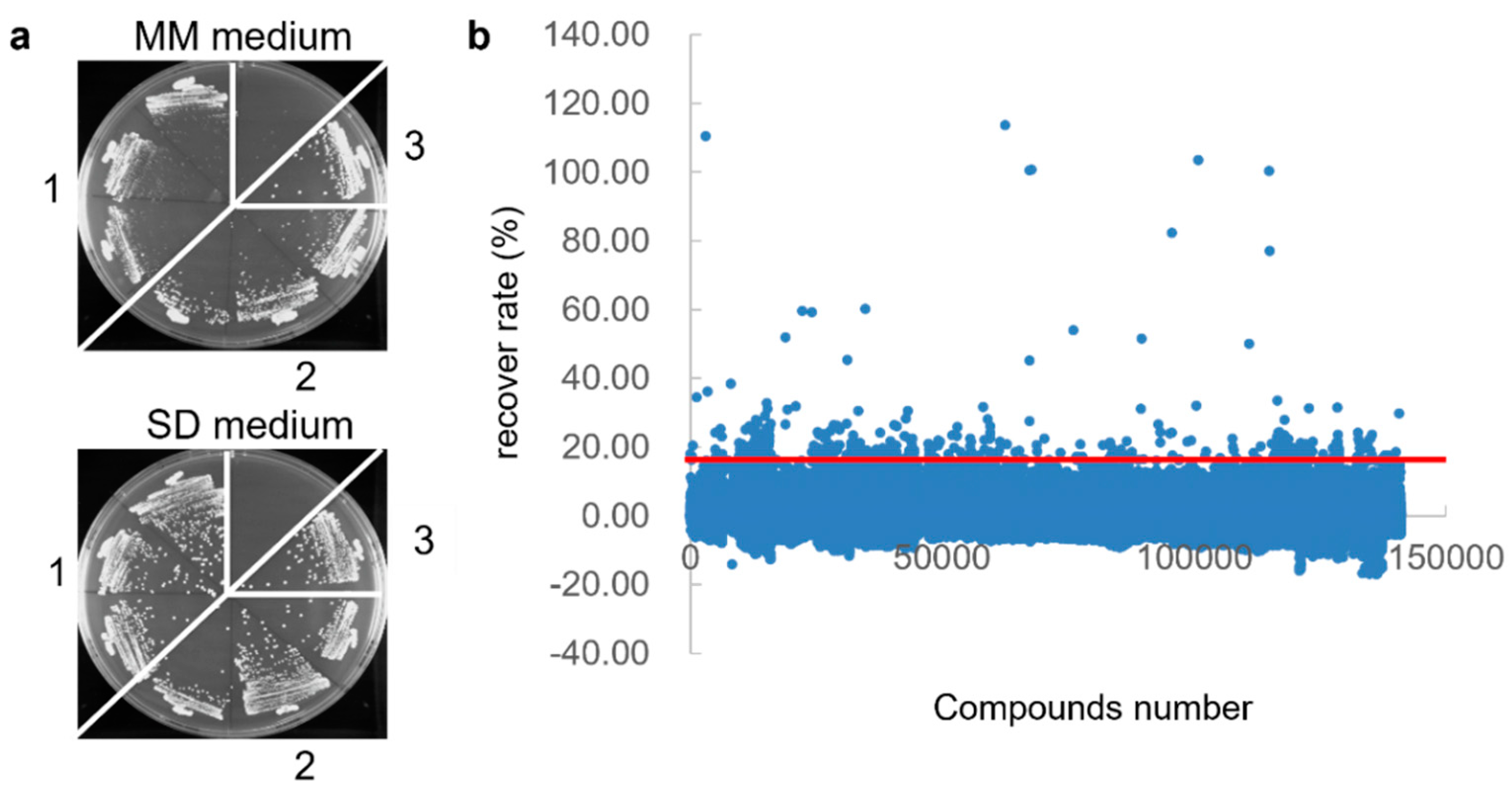
3.1. First Screening of Vpr Inhibitor Using Yeast-Based High-Throughput Screening
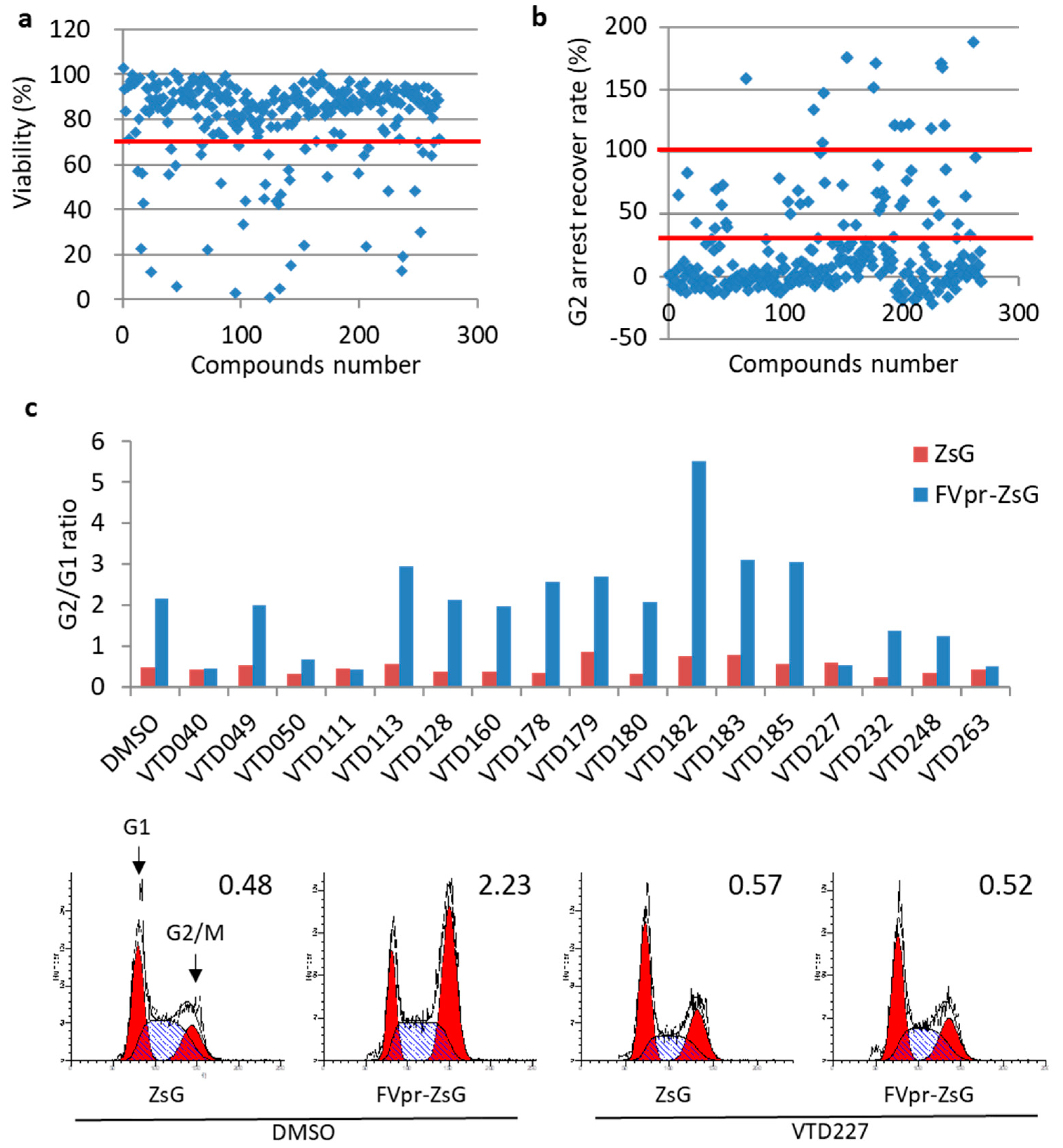
3.2. Second Screening of Vpr Inhibitor Using HeLa Cell-Based High-Throughput Screening
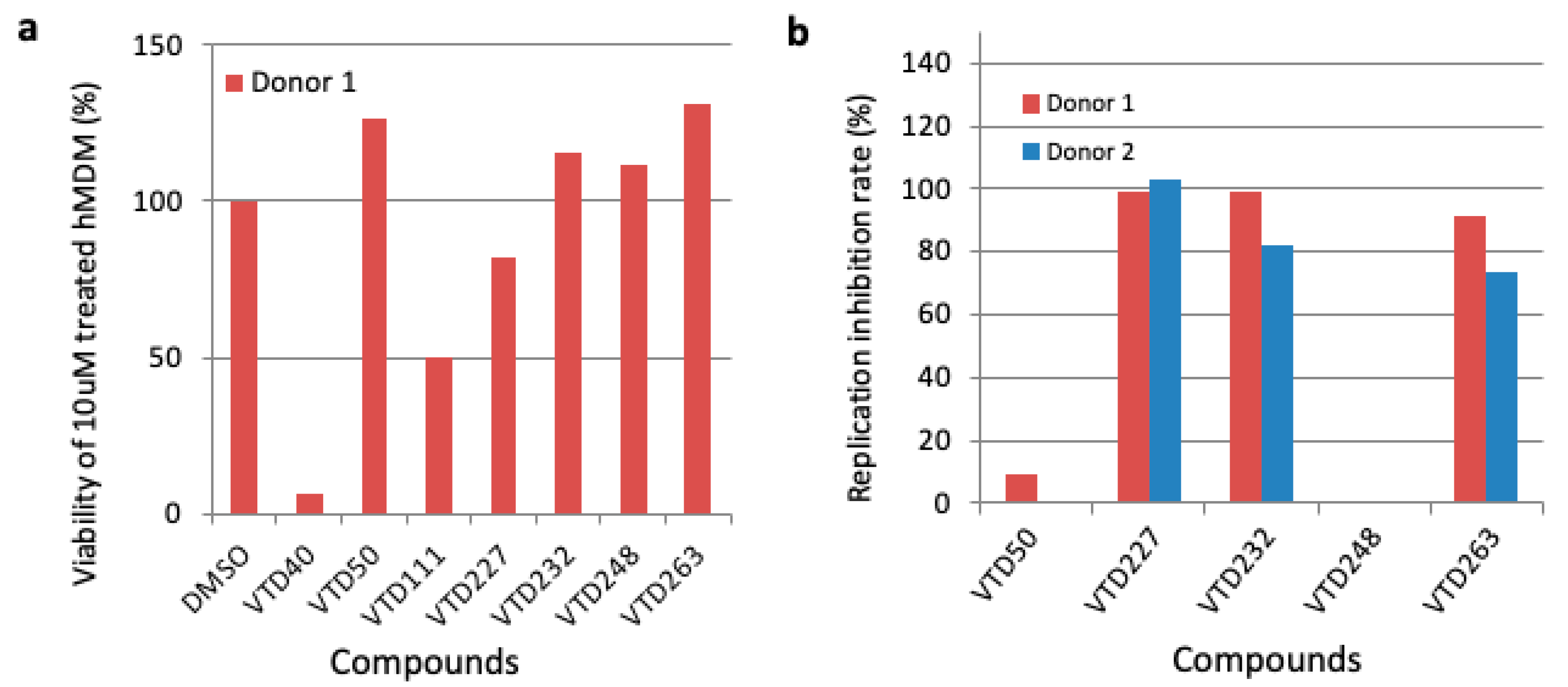
3.3. Third Screening of Vpr Inhibitor Using Human Monocyte-Derived Macrophages (MDM)
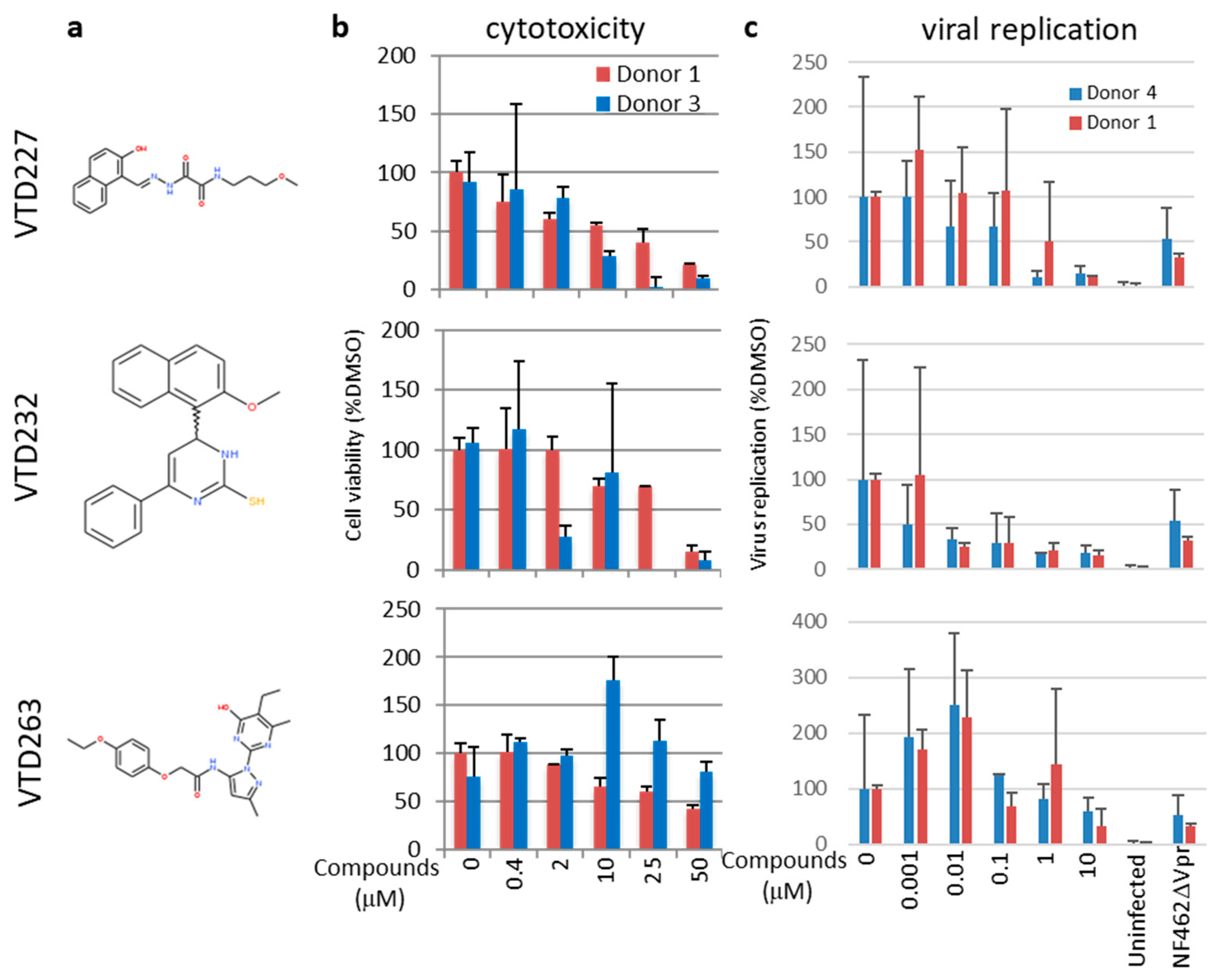
3.4. Final Selected Compounds Inhibited the Virus Replication in MDM in a Dose-Dependent Manner
3.5. Directly Binding Availability of Hit Compounds to Vpr Protein
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.; Singh, S.; Jung, H.Y.; Yang, G.; Jun, S.; Sastry, K.J.; Park, J.I. HIV-1 Vpr protein inhibits telomerase activity via the EDD-DDB1-VPRBP E3 ligase complex. J. Biol. Chem. 2013, 288, 15474–15480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guenzel, C.A.; Hérate, C.; Benichou, S. HIV-1 Vpr-a still “enigmatic multitasker”. Front. Microbiol. 2014, 5, 127. [Google Scholar] [CrossRef] [Green Version]
- Fabryova, H.; Strebel, K. Vpr and Its Cellular Interaction Partners: R We There Yet? Cells 2019, 8, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitahara-Kasahara, Y.; Kamata, M.; Yamamoto, T.; Zhang, X.; Miyamoto, Y.; Muneta, K.; Iijima, S.; Yoneda, Y.; Tsunetsugu-Yokota, Y.; Aida, Y. Novel Nuclear Import of Vpr Promoted by Importin Is Crucial for Human Immunodeficiency Virus Type 1 Replication in Macrophages. J. Virol. 2007, 81, 5284–5293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, D.R.; Lubow, J.; Lukic, Z.; Mashiba, M.; Collins, K.L. Vpr Promotes Macrophage-Dependent HIV-1 Infection of CD4+ T Lymphocytes. PLoS Pathog. 2015, 11, e1005054. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Dang, Y.; Baker, J.J.; Zhou, J.; Zheng, Y.-H. Evidence for Vpr-dependent HIV-1 replication in human CD4+ CEM.NKR T-cells. Retrovirology 2012, 9, 93. [Google Scholar] [CrossRef] [Green Version]
- Mashiba, M.; Collins, D.R.; Terry, V.H.; Collins, K.L. Vpr overcomes macrophage-specific restriction of HIV-1 Env expression and virion production. Cell Host Microbe 2014, 16, 722–735. [Google Scholar] [CrossRef] [Green Version]
- Subbramanian, R.A.; Kessous-Elbaz, A.; Lodge, R.; Forget, J.; Yao, X.J.; Bergeron, D.; Cohen, E.A. Human immunodeficiency virus type 1 Vpr is a positive regulator of viral transcription and infectivity in primary human macrophages. J. Exp. Med. 1998, 187, 1103–1111. [Google Scholar] [CrossRef] [Green Version]
- Höhne, K.; Businger, R.; Van Nuffel, A.; Bolduan, S.; Koppensteiner, H.; Baeyens, A.; Vermeire, J.; Malatinkova, E.; Verhasselt, B.; Schindler, M. Virion encapsidated HIV-1 Vpr induces NFAT to prime non-activated T cells for productive infection. Open Biol. 2016, 6, 160046. [Google Scholar] [CrossRef] [Green Version]
- Kuramitsu, M.; Hashizume, C.; Yamamoto, N.; Azuma, A.; Kamata, M.; Yamamoto, N.; Tanaka, Y.; Aida, Y. A novel role for Vpr of human immunodeficiency virus type 1 as a regulator of the splicing of cellular pre-mRNA. Microbes Infect. 2005, 7, 1150–1160. [Google Scholar] [CrossRef]
- Hashizume, C.; Kuramitsu, M.; Zhang, X.; Kurosawa, T.; Kamata, M.; Aida, Y. Human immunodeficiency virus type 1 Vpr interacts with spliceosomal protein SAP145 to mediate cellular pre-mRNA splicing inhibition. Microbes Infect. 2007, 9, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Aida, Y. HIV-1 Vpr: A novel role in regulating RNA splicing. Curr. HIV Res. 2009, 7, 163–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamata, M.; Nitahara-Kasahara, Y.; Miyamoto, Y.; Yoneda, Y.; Aida, Y. Importin-alpha promotes passage through the nuclear pore complex of human immunodeficiency virus type 1 Vpr. J. Virol. 2005, 79, 3557–3564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aida, Y.; Matsuda, G. Role of Vpr in HIV-1 nuclear import: Therapeutic implications. Curr. HIV Res. 2009, 7, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, M.; Kamata, M.; Mojin, T.; Nakai, Y.; Aida, Y. Induction of apoptosis by the Vpr protein of human immunodeficiency virus type 1 occurs independently of G(2) arrest of the cell cycle. Virology 2000, 276, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Amini, S.; Saunders, M.; Kelley, K.; Khalili, K.; Sawaya, B.E. Interplay between HIV-1 Vpr and Sp1 modulates p21WAF1gene expression in human astrocytes. J. Biol. Chem. 2004, 279, 46046–46056. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Liu, R.; Lin, Y.; Liang, C.; Tan, J.; Qiao, W. HIV-1 Vpr protein activates the NF-κB pathway to promote G2/M cell cycle arrest. Virol. Sin. 2015, 30, 441–448. [Google Scholar] [CrossRef]
- Brégnard, C.; Benkirane, M.; Laguette, N. DNA damage repair machinery and HIV escape from innate immune sensing. Front. Microbiol. 2014, 5, 176. [Google Scholar] [CrossRef]
- Zahoor, M.A.; Xue, G.; Sato, H.; Aida, Y. Genome-wide transcriptional profiling reveals that HIV-1 Vpr differentially regulates interferon-stimulated genes in human monocyte-derived dendritic cells. Virus Res. 2015, 208, 156–163. [Google Scholar] [CrossRef]
- Zahoor, M.A.; Xue, G.; Sato, H.; Murakami, T.; Takeshima, S.N.; Aida, Y. HIV-1 Vpr induces interferon-stimulated genes in human monocyte-derived macrophages. PLoS ONE 2014, 9, e106418. [Google Scholar] [CrossRef] [Green Version]
- Ayyavoo, V.; Mahboubi, A.; Mahalingam, S.; Ramalingam, R.; Kudchodkar, S.; Williams, W.V.; Green, D.R.; Weiner, D.B. HIV-1 Vpr suppresses immune activation and apoptosis through regulation of nuclear factor κB. Nat. Med. 1997, 3, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Lin, Y.; Jia, R.; Geng, Y.; Liang, C.; Tan, J.; Qiao, W. HIV-1 Vpr stimulates NF-kappaB and AP-1 signaling by activating TAK1. Retrovirology 2014, 11, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jowett, J.B.; Planelles, V.; Poon, B.; Shah, N.P.; Chen, M.L.; Chen, I.S. The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2 + M phase of the cell cycle. J. Virol. 1995, 69, 6304–6313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogel, M.E.; Wu, L.I.; Emerman, M. The human immunodeficiency virus type 1 vpr gene prevents cell proliferation during chronic infection. J. Virol. 1995, 69, 882–888. [Google Scholar] [CrossRef] [Green Version]
- Blondot, M.-L.L.; Dragin, L.; Lahouassa, H.; Margottin-Goguet, F. How SLX4 cuts through the mystery of HIV-1 Vpr-mediated cell cycle arrest. Retrovirology 2014, 11, 117. [Google Scholar] [CrossRef]
- Bressy, C.; Droby, G.N.; Maldonado, B.D.; Steuerwald, N.; Grdzelishvili, V.Z. Cell Cycle Arrest in G2/M Phase Enhances Replication of Interferon-Sensitive Cytoplasmic RNA Viruses via Inhibition of Antiviral Gene Expression. J. Virol. 2018, 93, e01885-18. [Google Scholar] [CrossRef] [Green Version]
- Hakata, Y.; Miyazawa, M.; Landau, N.R. Interactions with DCAF1 and DDB1 in the CRL4 E3 ubiquitin ligase are required for Vpr-mediated G2 arrest. Virol. J. 2014, 11, 108. [Google Scholar] [CrossRef] [Green Version]
- Laguette, N.; Brégnard, C.; Hue, P.; Basbous, J.; Yatim, A.; Larroque, M.; Kirchhoff, F.; Constantinou, A.; Sobhian, B.; Benkirane, M. Premature activation of the slx4 complex by vpr promotes g2/m arrest and escape from innate immune sensing. Cell 2014, 156, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.; Siarot, L.; Matsuura, R.; Lo, C.-W.; Sato, H.; Otsuki, H.; Aida, Y. Distinct MCM10 Proteasomal Degradation Profiles by Primate Lentiviruses Vpr Proteins. Viruses 2020, 12, 98. [Google Scholar] [CrossRef] [Green Version]
- Hammer, S.M.; Squires, K.E.; Hughes, M.D.; Grimes, J.M.; Demeter, L.M.; Currier, J.S.; Eron, J.J., Jr.; Feinberg, J.E.; Balfour, H.H., Jr.; Deyton, L.R.; et al. A Controlled Trial of Two Nucleoside Analogues plus Indinavir in Persons with Human Immunodeficiency Virus Infection and CD4 Cell Counts of 200 per Cubic Millimeter or Less. N. Engl. J. Med. 1997, 337, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Collier, D.A.; Monit, C.; Gupta, R.K. The Impact of HIV-1 Drug Escape on the Global Treatment Landscape. Cell Host Microbe 2019, 26, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Hezareh, M.; Gunthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.; Balkin, A. Risk factors for gastrointestinal adverse events in HIV treated and untreated patients. AIDS Rev. 2009, 11, 30–38. [Google Scholar]
- Gupta, A.; Juneja, S.; Vitoria, M.; Habiyambere, V.; Dongmo Nguimfack, B.; Doherty, M.; Low-Beer, D. Projected uptake of new antiretroviral (ARV) medicines in adults in low- and middle-income countries: A forecast analysis 2015–2025. PLoS ONE 2016, 11, e0164619. [Google Scholar] [CrossRef]
- Vitoria, M.; Rangaraj, A.; Ford, N.; Doherty, M. Current and future priorities for the development of optimal HIV drugs. Curr. Opin. HIV AIDS 2019, 14, 143–149. [Google Scholar] [CrossRef]
- Pierson, T.; McArthur, J.; Siliciano, R.F. Reservoirs for HIV-1: Mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu. Rev. Immunol. 2000, 18, 665–708. [Google Scholar] [CrossRef]
- Reece, M.D.; Taylor, R.R.; Song, C.; Gavegnano, C. Targeting Macrophage Dysregulation for Viral Infections: Novel Targets for Immunomodulators. Front. Immunol. 2021, 12, 768695. [Google Scholar] [CrossRef]
- Lubow, J.; Collins, K.L. Vpr Is a VIP: HIV Vpr and Infected Macrophages Promote Viral Pathogenesis. Viruses 2020, 12, 809. [Google Scholar] [CrossRef]
- Kogan, M.; Rappaport, J. HIV-1 Accessory Protein Vpr: Relevance in the pathogenesis of HIV and potential for therapeutic intervention. Retrovirology 2011, 8, 25. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, M.E. The HIV-1 vpr protein: A multifaceted target for therapeutic intervention. Int. J. Mol. Sci. 2017, 18, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, E.B.B.; Watanabe, N.; Saito, A.; Futamura, Y.; Abd El Galil, K.H.; Koito, A.; Najimudin, N.; Osada, H. Vipirinin, a coumarin-based HIV-1 Vpr inhibitor, interacts with a hydrophobic region of Vpr. J. Biol. Chem. 2011, 286, 14049–14056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, N.; Nishihara, Y.; Yamaguchi, T.; Koito, A.; Miyoshi, H.; Kakeya, H.; Osada, H. Fumagillin suppresses HIV-1 infection of macrophages through the inhibition of Vpr activity. FEBS Lett. 2006, 580, 2598–2602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Yamamoto, N.; Nonaka, M.; Hashimoto, Y.; Matsuda, G.; Takeshima, S.n.; Matsuyama, M.; Igarashi, T.; Miura, T.; Tanaka, R.; et al. Inhibition of human immunodeficiency virus type 1 (HIV-1) nuclear import via Vpr-Importin α interactions as a novel HIV-1 therapy. Biochem. Biophys. Res. Commun. 2009, 380, 838–843. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, K.; Murakami, T.; Xue, G.; Shimizu, Y.; Takeda, E.; Hashimoto, Y.; Honda, K.; Kondoh, Y.; Osada, H.; Tsunetsugu-Yokota, Y.; et al. Identification of a novel Vpr-binding compound that inhibits HIV-1 multiplication in macrophages by chemical array. Biochem. Biophys. Res. Commun. 2010, 403, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Koyama, H.; Hagiwara, K.; Miura, T.; Xue, G.; Hashimoto, Y.; Kitahara, G.; Aida, Y.; Suzuki, M. Synthesis and biological evaluation of deoxy-hematoxylin derivatives as a novel class of anti-HIV-1 agents. Bioorg. Med. Chem. Lett. 2012, 22, 1469–1474. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, K.; Ishii, H.; Murakami, T.; Takeshima, S.N.; Chutiwitoonchai, N.; Kodama, E.N.; Kawaji, K.; Kondoh, Y.; Honda, K.; Osada, H.; et al. Synthesis of a Vpr-Binding derivative for use as a novel HIV-1 Inhibitor. PLoS ONE 2015, 10, e0145573. [Google Scholar] [CrossRef]
- Win, N.N.; Ngwe, H.; Abe, I.; Morita, H. Naturally occurring Vpr inhibitors from medicinal plants of Myanmar. J. Nat. Med. 2017, 71, 579–589. [Google Scholar] [CrossRef] [Green Version]
- Win, N.N.; Ito, T.; Win, Y.Y.; Ngwe, H.; Kodama, T.; Abe, I.; Morita, H. Quassinoids: Viral protein R inhibitors from Picrasma javanica bark collected in Myanmar for HIV infection. Bioorg. Med. Chem. Lett. 2016, 26, 4620–4624. [Google Scholar] [CrossRef]
- Woo, S.-Y.; Win, N.N.; Noe Oo, W.M.; Ngwe, H.; Ito, T.; Abe, I.; Morita, H. Viral protein R inhibitors from Swertia chirata of Myanmar. J. Biosci. Bioeng. 2019, 128, 445–449. [Google Scholar] [CrossRef]
- Nakamura, Y.; Arai, A.; Takebe, Y.; Masuda, M. A chemical compound for controlled expression of nmt1-driven gene in the fission yeast Schizosaccharomyces pombe. Anal. Biochem. 2011, 412, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Yashiroda, Y.; Okamoto, R.; Hatsugai, K.; Takemoto, Y.; Goshima, N.; Saito, T.; Hamamoto, M.; Sugimoto, Y.; Osada, H.; Seimiya, H.; et al. A novel yeast cell-based screen identifies flavone as a tankyrase inhibitor. Biochem. Biophys. Res. Commun. 2010, 394, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, A.; Shirai, A.; Yashiroda, Y.; Kamata, A.; Horinouchi, S.; Yoshida, M. pDUAL, a multipurpose, multicopy vector capable of chromosomal integration in fission yeast. Yeast 2004, 21, 1289–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, T.; Aida, Y. Visualizing Vpr-induced G2 arrest and apoptosis. PLoS ONE 2014, 9, e86840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, E.; Murakami, T.; Matsuda, G.; Murakami, H.; Zako, T.; Maeda, M.; Aida, Y. Nuclear exportin receptor cas regulates the NPI-1-mediated nuclear import of HIV-1 vpr. PLoS ONE 2011, 6, e27815. [Google Scholar] [CrossRef] [Green Version]
- Matsuyama, A.; Arai, R.; Yashiroda, Y.; Shirai, A.; Kamata, A.; Sekido, S.; Kobayashi, Y.; Hashimoto, A.; Hamamoto, M.; Hiraoka, Y.; et al. ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 2006, 24, 841–847. [Google Scholar] [CrossRef]
- Maruyama, Y.; Kawamura, Y.; Nishikawa, T.; Isogai, T.; Nomura, N.; Goshima, N. HGPD: Human Gene and Protein Database, 2012 update. Nucleic Acids Res. 2012, 40, D924–D929. [Google Scholar] [CrossRef]
- Moreno, S.; Klar, A.; Nurse, P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzym. 1991, 194, 795–823. [Google Scholar] [CrossRef]
- Masuda, M.; Nagai, Y.; Oshima, N.; Tanaka, K.; Murakami, H.; Igarashi, H.; Okayama, H. Genetic Studies with the Fission Yeast Schizosaccharomyces pombe Suggest Involvement of Wee1, Ppa2, and Rad24 in Induction of Cell Cycle Arrest by Human Immunodeficiency Virus Type 1 Vpr. J. Virol. 2000, 74, 2636–2646. [Google Scholar] [CrossRef] [Green Version]
- Nishi, K.; Yoshida, M.; Nishimura, M.; Nishikawa, M.; Nishiyama, M.; Horinouchi, S.; Beppu, T. A leptomycin B resistance gene of Schizosaccharomyces pombe encodes a protein similar to the mammalian P-glycoproteins. Mol. Microbiol. 1992, 6, 761–769. [Google Scholar] [CrossRef]
- Nagao, K.; Taguchi, Y.; Arioka, M.; Kadokura, H.; Takatsuki, A.; Yoda, K.; Yamasaki, M. bfr1+, a novel gene of Schizosaccharomyces pombe which confers brefeldin A resistance, is structurally related to the ATP-binding cassette superfamily. J. Bacteriol. 1995, 177, 1536–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Orenstein, J.M.; Fox, C.; Wahl, S.M. Macrophages as a source of HIV during opportunistic infections. Science 1997, 276, 1857–1861. [Google Scholar] [CrossRef] [PubMed]
- Sattentau, Q.J.; Stevenson, M. Macrophages and HIV-1: An Unhealthy Constellation. Cell Host Microbe 2016, 19, 304–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Bieniasz, P.D. HIV-1 Vpr induces cell cycle arrest and enhances viral gene expression by depleting CCDC137. eLife 2020, 9, e55806. [Google Scholar] [CrossRef]
- Murakami, T.; Matsuura, R.; Chutiwitoonchai, N.; Takei, M.; Aida, Y. Huntingtin-Interacting Protein 1 Promotes Vpr-Induced G2 Arrest and HIV-1 Infection in Macrophages. Viruses 2021, 13, 2308. [Google Scholar] [CrossRef]
- Yan, J.; Shun, M.-C.; Zhang, Y.; Hao, C.; Skowronski, J. HIV-1 Vpr counteracts HLTF-mediated restriction of HIV-1 infection in T cells. Proc. Natl. Acad. Sci. USA 2019, 116, 9568–9577. [Google Scholar] [CrossRef] [Green Version]
- Goh, W.C.; Rogel, M.E.; Kinsey, C.M.; Michael, S.F.; Fultz, P.N.; Nowak, M.A.; Hahn, B.; Emerman, M. HIV-1 Vpr increases viral expression by manipulation of the cell cycle: A mechanism for selection of Vpr in vivo. Nat. Med. 1998, 4, 65–71. [Google Scholar] [CrossRef]
- Vermeire, J.; Roesch, F.; Sauter, D.; Rua, R.; Hotter, D.; Van Nuffel, A.; Vanderstraeten, H.; Naessens, E.; Iannucci, V.; Landi, A.; et al. HIV Triggers a cGAS-Dependent, Vpu- and Vpr-Regulated Type I Interferon Response in CD4+ T Cells. Cell Rep. 2016, 17, 413–424. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Screening Stage | Screening Methods | Compounds’ Number |
|---|---|---|
| 1st screening | Vpr inhibition on the overexpressing yeast | 140,000 to 560 |
| Reproductivity, dose, and selectivity assay | 560 to 268 | |
| 2nd screening | WST-1 assay (HeLa cell) | 268 to 224 |
| G2 arrest inhibition on the Vpr transfected HeLa cells (CELAVIEW) | 224 to 17 | |
| G2 arrest inhibition on the Vpr transfected HeLa cells (flow cytometry) | 17 to 7 | |
| 3rd screening | WST-1 assay (MDM cell) | 7 to 5 |
| HIV replication inhibition on the HIV-infected MDM | 5 to 3 |
| Compound | IUPAC Name | HeLa 1 CC50 (µM) | MDM 2 CC50 (µM) | MDM IC50 (µM) | MDM 3 SI | Vpr Binding 4 Kd (µM) |
|---|---|---|---|---|---|---|
| VTD227 | N-[(1E)-2-(2-hydroxynaphthyl)-1-azavinyl]-N′-(3-methoxypropyl)ethane-1,2-diamide | 99.0 ± 22.0 | 17.9 ± 5.3 | 0.78 ± 0.59 | 22.9 | 8.8 ± 0.48 |
| VTD232 | 4-(2-methoxy-1-naphthyl)-6-phenyl-3,4-dihydro-2(1H)-pyrimidinethione | 57.1 ± 30.9 | 30.5 ± 5.0 | 0.0039 ± 0.0046 | 7820 | Not binding |
| VTD263 | 2-(4-ethoxyphenoxy)-N-[1-(5-ethyl-6-methyl-4-oxo(3-hydropyrimidin-2-yl))-3-methylpyrazol-5-yl]acetamide | 100< | 38.8 ± 0.76 | 11.4 ± 4.0 | 3.4 | Not binding |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, H.; Murakami, T.; Matsuura, R.; Abe, M.; Matsuoka, S.; Yashiroda, Y.; Yoshida, M.; Akari, H.; Nagasawa, Y.; Takei, M.; et al. A Novel Class of HIV-1 Inhibitors Targeting the Vpr-Induced G2-Arrest in Macrophages by New Yeast- and Cell-Based High-Throughput Screening. Viruses 2022, 14, 1321. https://doi.org/10.3390/v14061321
Sato H, Murakami T, Matsuura R, Abe M, Matsuoka S, Yashiroda Y, Yoshida M, Akari H, Nagasawa Y, Takei M, et al. A Novel Class of HIV-1 Inhibitors Targeting the Vpr-Induced G2-Arrest in Macrophages by New Yeast- and Cell-Based High-Throughput Screening. Viruses. 2022; 14(6):1321. https://doi.org/10.3390/v14061321
Chicago/Turabian StyleSato, Hirotaka, Tomoyuki Murakami, Ryosuke Matsuura, Masako Abe, Seiji Matsuoka, Yoko Yashiroda, Minoru Yoshida, Hirofumi Akari, Yosuke Nagasawa, Masami Takei, and et al. 2022. "A Novel Class of HIV-1 Inhibitors Targeting the Vpr-Induced G2-Arrest in Macrophages by New Yeast- and Cell-Based High-Throughput Screening" Viruses 14, no. 6: 1321. https://doi.org/10.3390/v14061321