
Morphological and Genetic Characterization of Eggerthella lenta Bacteriophage PMBT5

 ,
,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Media Preparation, Bacteria Strains and Cultivation
2.2. Bacteriophage Isolation, Propagation and Media
2.3. Transmission Electron Microscopy
2.4. Phage DNA Preparation
2.5. Genome Sequencing and Phylogenetic Analyses
2.6. Phage Structural Protein Analysis by UHPLC-MS-MS
3. Results and Discussion
3.1. Isolation Protocol of E. lenta Phage PMBT5 under Anaerobic Conditions
3.2. Morphology of Phage PMBT5 Particles and Phage Plaques
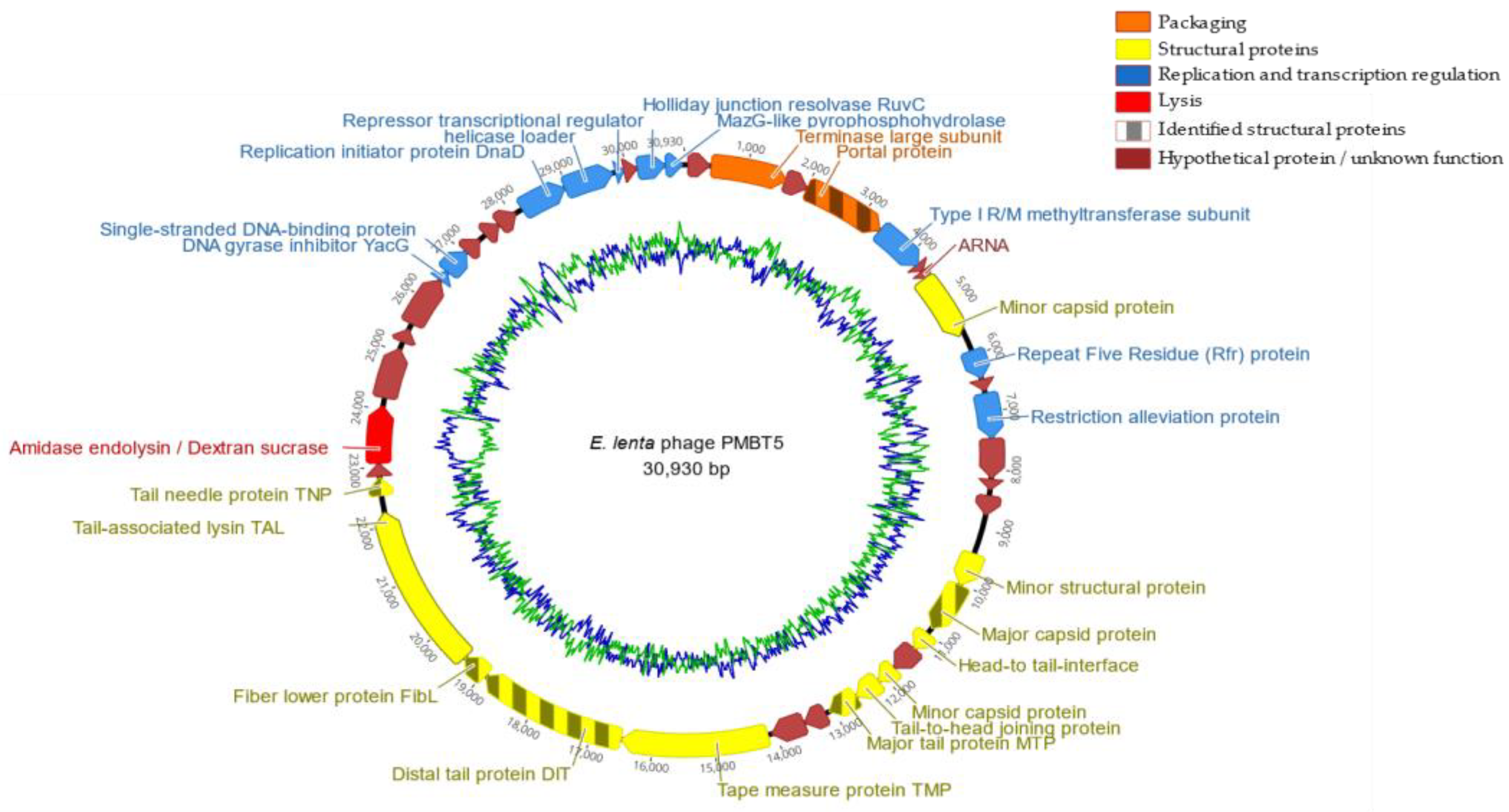
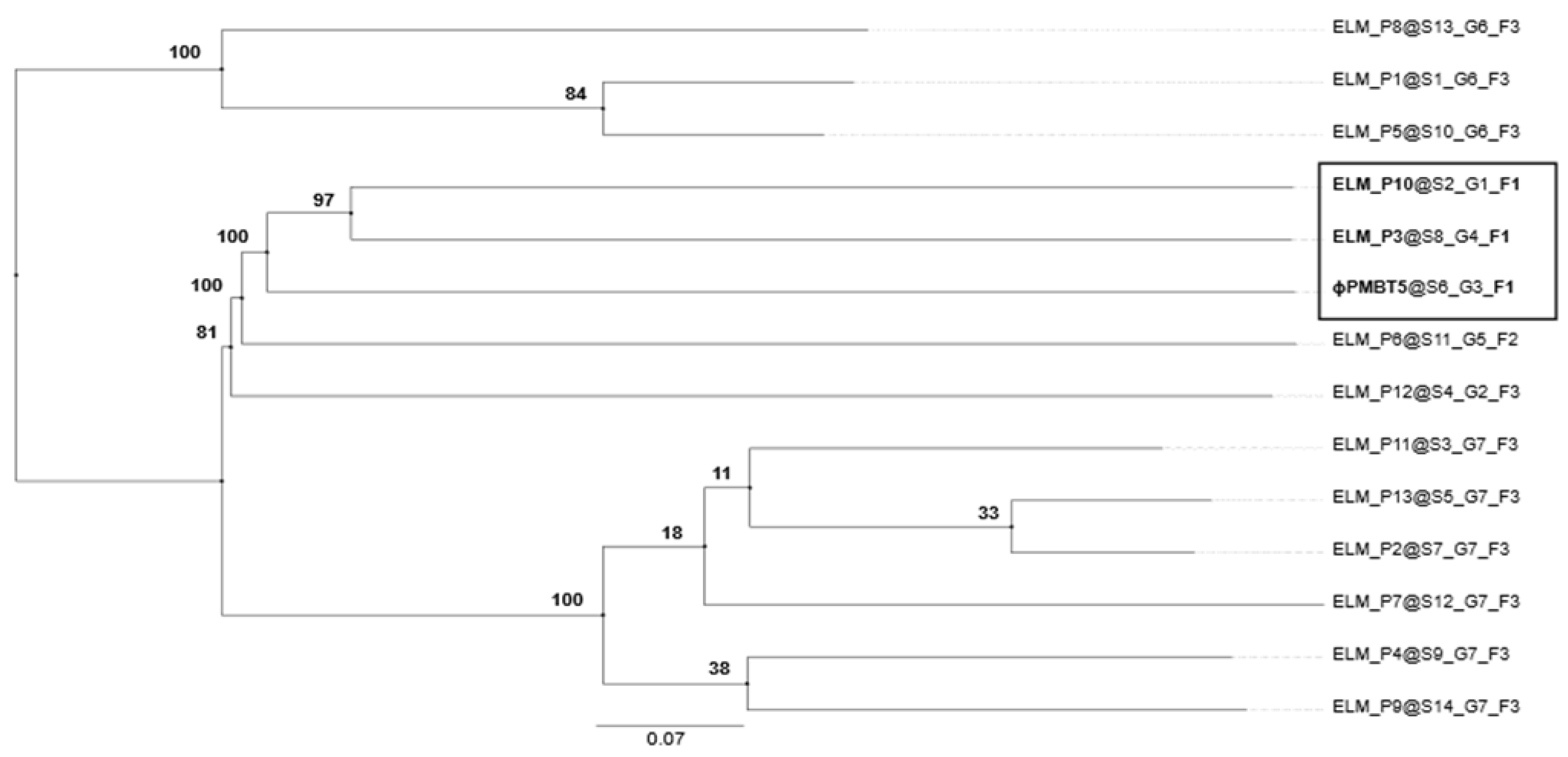
3.3. Phage PMBT5 Genome Sequence and Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cho, G.-S.; Ritzmann, F.; Eckstein, M.; Huch, M.; Briviba, K.; Behsnilian, D.; Neve, H.; Franz, C.M.A.P. Quantification of Slackia and Eggerthella spp. in human feces and adhesion of representatives strains to Caco-2 cells. Front. Microbiol. 2016, 7, 658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haiser, H.J.; Seim, K.L.; Balskus, E.P.; Turnbaugh, P.J. Mechanistic insight into digoxin inactivation by Eggerthella lenta augments our understanding of its pharmacokinetics. Gut Microbes 2014, 5, 233–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.S.; Chen, W.J.; Adeolu, M.; Chai, Y. Molecular signatures for the class Coriobacteriia and its different clades; proposal for division of the class Coriobacteriia into the emended order Coriobacteriales, containing the emended family Coriobacteriaceae and Atopobiaceae fam. nov., and Eggerthellales ord. nov., containing the family Eggerthellaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2013, 63, 3379–3397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppel, N.; Bisanz, J.E.; Pandelia, M.-E.; Turnbaugh, P.J.; Balskus, E.P. Discovery and characterization of a prevalent human gut bacterial enzyme sufficient for the inactivation of a family of plant toxins. eLife 2018, 7, e33953. [Google Scholar] [CrossRef] [PubMed]
- Claus, S.P.; Ellero, S.L.; Berger, B.; Krause, L.; Bruttin, A.; Molina, J.; Paris, A.; Want, E.J.; de Waziers, I.; Cloarec, O.; et al. Colonization-induced host-gut microbial metabolic interaction. mBio 2011, 2, e00271-10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bode, L.M.; Bunzel, D.; Huch, M.; Cho, G.-S.; Ruhland, D.; Bunzel, M.; Bub, A.; Franz, C.M.A.P.; Kulling, S.E. In vivo and in vitro metabolism of trans-resveratrol by human gut microbiota. Am. J. Clin. Nutr. 2013, 97, 295–309. [Google Scholar] [CrossRef]
- Kawada, Y.; Goshima, T.; Sawamura, R.; Yokoyama, S.; Yanase, E.; Niwa, T.; Ebihara, A.; Inagaki, M.; Yamaguchi, K.; Kuwata, K.; et al. Daidzein reductase of Eggerthella sp. YY7918, its octameric subunit structure containing FMN/FAD/4Fe-4S, and its enantioselective production of R-dihydroisoflavones. J. Biosci. Bioeng. 2018, 126, 301–309. [Google Scholar] [CrossRef]
- Gardiner, B.J.; Tai, A.Y.; Kotsanas, D.; Francis, M.J.; Roberts, S.A.; Ballard, S.A.; Junckerstorff, R.K.; Korman, T.M. Clinical and microbiological characteristics of Eggerthella lenta bacteremia. J. Clin. Microbiol. 2015, 53, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Ugarte-Torres, A.; Gillrie, M.R.; Griener, T.P.; Church, D.L. Eggerthella lenta bloodstream infections are associated with increased mortality following empiric piperacillin-tazobactam (TZP) Monotherapy: A Population-based Cohort Study. Clin. Infect. Dis. 2018, 67, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Koh, A.; Molinaro, A.; Ståhlman, M.; Khan, M.T.; Schmidt, C.; Mannerås-Holm, L.; Wu, H.; Carreras, A.; Jeong, H.; Olofsson, L.E.; et al. Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell 2018, 175, 947.e17–961.e17. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Castro-Mejía, J.L.; Muhammed, M.K.; Kot, W.; Neve, H.; Franz, C.M.A.P.; Hansen, L.H.; Vogensen, F.K.; Nielsen, D.S. Optimizing protocols for extraction of bacteriophages prior to metagenomic analyses of phage communities in the human gut. Microbiome 2015, 3, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, G.; Zhao, C.; Zhang, H.; Mattei, L.; Sherrill-Mix, S.; Bittinger, K.; Kessler, L.R.; Wu, G.D.; Baldassano, R.N.; DeRusso, P.; et al. The stepwise assembly of the neonatal virome is modulated by breastfeeding. Nature 2020, 581, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Hoyles, L.; McCartney, A.L.; Neve, H.; Gibson, G.R.; Sanderson, J.D.; Heller, K.J.; van Sinderen, D. Characterization of virus-like particles associated with the human faecal and caecal microbiota. Res. Microbiol. 2014, 165, 803–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shkoporov, A.N.; Clooney, A.G.; Sutton, T.D.S.; Ryan, F.J.; Daly, K.M.; Nolan, J.A.; McDonnell, S.A.; Khokhlova, E.V.; Draper, L.A.; Forde, A.; et al. The human gut virome is highly diverse, stable, and individual specific. Cell Host Microbe 2019, 26, 527.e5–541.e5. [Google Scholar] [CrossRef]
- Manrique, P.; Dills, M.; Young, M.J. The human gut phage community and its implications for health and disease. Viruses 2017, 9, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sausset, R.; Petit, M.A.; Gaboriau-Routhiau, V.; de Paepe, M. New insights into intestinal phages. Mucosal Immunol. 2020, 13, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.M.; Lin, H.C. A theoretical model of temperate phages as mediators of gut microbiome dysbiosis. F1000Research 2019, 8, F1000 Faculty Rev-997. [Google Scholar] [CrossRef] [Green Version]
- D’Herelle, F.H. Sur un microbe invisible antagoniste des bacilles dysentériques. C. R. Acad. Sci. 1917, 165, 373–375. [Google Scholar]
- Twort, F.W. An Investigation on the nature of ultra-microscopic viruses. Lancet 1915, 186, 1241–1243. [Google Scholar] [CrossRef] [Green Version]
- Stern, A.; Mick, E.; Tirosh, I.; Sagy, O.; Sorek, R. CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Res. 2012, 22, 1985–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the human gut: The “known unknown” of the microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yutin, N.; Makarova, K.S.; Gussow, A.B.; Krupovic, M.; Segall, A.; Edwards, R.A.; Koonin, E.V. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat. Microbiol. 2018, 3, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; You, X.; Mai, G.; Tokuyasu, T.; Liu, C. A human gut phage catalog correlates the gut phageome with type 2 diabetes. Microbiome 2018, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Soto-Perez, P.; Bisanz, J.E.; Berry, J.D.; Lam, K.N.; Bondy-Denomy, J.; Turnbaugh, P.J. CRISPR-Cas system of a prevalent human gut bacterium reveals hyper-targeting against phages in a human virome catalog. Cell Host Microbe 2019, 26, 325.e5–335.e5. [Google Scholar] [CrossRef]
- Aggarwala, V.; Liang, G.; Bushman, F.D. Viral communities of the human gut: Metagenomic analysis of composition and dynamics. Mob. DNA 2017, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Cornuault, J.K.; Petit, M.-A.; Mariadassou, M.; Benevides, L.; Moncaut, E.; Langella, P.; Sokol, H.; de Paepe, M. Phages infecting Faecalibacterium prausnitzii belong to novel viral genera that help to decipher intestinal viromes. Microbiome 2018, 6, 65. [Google Scholar] [CrossRef]
- d’Humières, C.; Touchon, M.; Dion, S.; Cury, J.; Ghozlane, A.; Garcia-Garcera, M.; Bouchier, C.; Ma, L.; Denamur, E.; Rocha, E.P.C. A simple, reproducible and cost-effective procedure to analyse gut phageome: From phage isolation to bioinformatic approach. Sci. Rep. 2019, 9, 11331. [Google Scholar] [CrossRef] [Green Version]
- García-Aljaro, C.; Muniesa, M.; Jofre, J. Isolation of bacteriophages of the anaerobic bacteria Bacteroides. Methods Mol. Biol. 2018, 1693, 11–22. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Khokhlova, E.V.; Fitzgerald, C.B.; Stockdale, S.R.; Draper, L.A.; Ross, R.P.; Hill, C. ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 2018, 9, 4781. [Google Scholar] [CrossRef] [Green Version]
- Fortier, L.-C. Bacteriophages contribute to shaping Clostridioides (Clostridium) difficile species. Front. Microbiol. 2018, 9, 2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, T.D.; Chalgren, S. Medium for use in antibiotic susceptibility testing of anaerobic bacteria. Antimicrob. Agents Chemother. 1976, 10, 926–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambrook, J.; Russel, D. (Eds.) Molecular Cloning—A Laboratory Manual; Cold Spring Harbor Laboratory: New York, NY, USA, 2001. [Google Scholar]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.; Korobeynikov, A.; Lapidus, A.; Prjibelsky, A.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling genomes and mini-metagenomes from highly chimeric reads. In Research in Computational Molecular Biology; Deng, M., Jiang, R., Sun, F., Zhang, X., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 158–170. ISBN 978-3-642-37195-0. [Google Scholar]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [Green Version]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [Green Version]
- Farris, J.S. Estimating phylogenetic trees from distance matrices. Am. Nat. 1972, 106, 645–668. [Google Scholar] [CrossRef]
- FigTree v. 1.4.3. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 17 March 2020).
- Göker, M.; García-Blázquez, G.; Voglmayr, H.; Tellería, M.T.; Martín, M.P. Molecular taxonomy of phytopathogenic fungi: A case study in Peronospora. PLoS ONE 2009, 4, e6319. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; Goodwin, L.A.; et al. Complete genome sequence of DSM 30083(T), the type strain (U5/41(T)) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genom. Sci. 2014, 9, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carstens, A.B.; Kot, W.; Lametsch, R.; Neve, H.; Hansen, L.H. Characterisation of a novel enterobacteria phage, CAjan, isolated from rat faeces. Arch. Virol. 2016, 161, 2219–2226. [Google Scholar] [CrossRef] [PubMed]
- Ewert, D.L.; Paynter, M.J. Enumeration of bacteriophages and host bacteria in sewage and the activated-sludge treatment process. Appl. Environ. Microbiol. 1980, 39, 576–583. [Google Scholar] [CrossRef] [Green Version]
- Muniesa, M.; Imamovic, L.; Jofre, J. Bacteriophages and genetic mobilization in sewage and faecally polluted environments. Microb. Biotechnol. 2011, 4, 725–734. [Google Scholar] [CrossRef]
- Deng, L.; Silins, R.; Castro-Mejía, J.L.; Kot, W.; Jessen, L.; Thorsen, J.; Shah, S.; Stokholm, J.; Bisgaard, H.; Moineau, S.; et al. A protocol for extraction of infective viromes suitable for metagenomics sequencing from low volume fecal samples. Viruses 2019, 11, 667. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, A.; Dion, M.; Deng, L.; Tremblay, D.; Moncaut, E.; Shah, S.A.; Stokholm, J.; Krogfelt, K.A.; Schjørring, S.; Bisgaard, H.; et al. Virulent coliphages in 1-year-old children fecal samples are fewer, but more infectious than temperate coliphages. Nat. Commun. 2020, 11, 378. [Google Scholar] [CrossRef]
- Duncan, S.H.; Hold, G.L.; Harmsen, H.J.M.; Stewart, C.S.; Flint, H.J. Growth requirements and fermentation products of Fusobacterium prausnitzii, and a proposal to reclassify it as Faecalibacterium prausnitzii gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 2002, 52, 2141–2146. [Google Scholar] [CrossRef] [Green Version]
- Jurczak-Kurek, A.; Gąsior, T.; Nejman-Faleńczyk, B.; Bloch, S.; Dydecka, A.; Topka, G.; Necel, A.; Jakubowska-Deredas, M.; Narajczyk, M.; Richert, M.; et al. Biodiversity of bacteriophages: Morphological and biological properties of a large group of phages isolated from urban sewage. Sci. Rep. 2016, 6, 34338. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Huang, J.; Yan, G.; Lei, L.; Wang, S.; Yu, L.; Zhou, L.; Gao, A.; Feng, X.; Han, W.; et al. Identification and characterization of Dpo42, a novel depolymerase derived from the Escherichia coli phage vB_EcoM_ECOO78. Front. Microbiol. 2017, 8, 1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latka, A.; Maciejewska, B.; Majkowska-Skrobek, G.; Briers, Y.; Drulis-Kawa, Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 2017, 101, 3103–3119. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef]
- Isidro, A.; Henriques, A.O.; Tavares, P. The portal protein plays essential roles at different steps of the SPP1 DNA packaging process. Virology 2004, 322, 253–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, H.E.; Dawson, M.T.; Devine, K.M.; McConnell, D.J. Characterization of PBSX, a defective prophage of Bacillus subtilis. J. Bacteriol. 1990, 172, 2667–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krogh, S.; O’Reilly, M.; Nolan, N.; Devine, K.M. The phage-like element PBSX and part of the skin element, which are resident at different locations on the Bacillus subtilis chromosome, are highly homologous. Microbiology 1996, 142, 2031–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godinho, L.M.; El Sadek Fadel, M.; Monniot, C.; Jakutyte, L.; Auzat, I.; Labarde, A.; Djacem, K.; Oliveira, L.; Carballido-Lopez, R.; Ayora, S.; et al. The revisited genome of Bacillus subtilis bacteriophage SPP1. Viruses 2018, 10, 705. [Google Scholar] [CrossRef] [Green Version]
- Mc Grath, S.; Neve, H.; Seegers, J.F.M.L.; Eijlander, R.; Vegge, C.S.; Brøndsted, L.; Heller, K.J.; Fitzgerald, G.F.; Vogensen, F.K.; van Sinderen, D. Anatomy of a lactococcal phage tail. J. Bacteriol. 2006, 188, 3972–3982. [Google Scholar] [CrossRef] [Green Version]
- Levin, M.E.; Hendrix, R.W.; Casjens, S.R. A programmed translational frameshift is required for the synthesis of a bacteriophage lambda tail assembly protein. J. Mol. Biol. 1993, 234, 124–139. [Google Scholar] [CrossRef]
- Murray, N.E. Type I restriction systems: Sophisticated molecular machines (a legacy of Bertani and Weigle). Microbiol. Mol. Biol. Rev. 2000, 64, 412–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, J.; Mahony, J.; Ainsworth, S.; Nauta, A.; van Sinderen, D. Bacteriophage orphan DNA methyltransferases: Insights from their bacterial origin, function, and occurrence. Appl. Environ. Microbiol. 2013, 79, 7547–7555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Kennedy, M.A. Current understanding of the structure and function of pentapeptide repeat proteins. Biomolecules 2021, 11, 638. [Google Scholar] [CrossRef] [PubMed]
- Pell, L.G.; Kanelis, V.; Donaldson, L.W.; Howell, P.L.; Davidson, A.R. The phage lambda major tail protein structure reveals a common evolution for long-tailed phages and the type VI bacterial secretion system. Proc. Natl. Acad. Sci. USA 2009, 106, 4160–4165. [Google Scholar] [CrossRef] [Green Version]
- Vegge, C.S.; Brøndsted, L.; Neve, H.; Mc Grath, S.; van Sinderen, D.; Vogensen, F.K. Structural characterization and assembly of the distal tail structure of the temperate lactococcal bacteriophage TP901-1. J. Bacteriol. 2005, 187, 4187–4197. [Google Scholar] [CrossRef] [Green Version]
- Christie, G.E.; Matthews, A.M.; King, D.G.; Lane, K.D.; Olivarez, N.P.; Tallent, S.M.; Gill, S.R.; Novick, R.P. The complete genomes of Staphylococcus aureus bacteriophages 80 and 80α-implications for the specificity of SaPI mobilization. Virology 2010, 407, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Auzat, I.; Dröge, A.; Weise, F.; Lurz, R.; Tavares, P. Origin and function of the two major tail proteins of bacteriophage SPP1. Mol. Microbiol. 2008, 70, 557–569. [Google Scholar] [CrossRef]
- Zinke, M.; Schröder, G.F.; Lange, A. Major tail proteins of bacteriophages of the order Caudovirales. J. Biol. Chem. 2022, 298, 101472. [Google Scholar] [CrossRef]
- Goulet, A.; Spinelli, S.; Mahony, J.; Cambillau, C. Conserved and diverse traits of adhesion devices from Siphoviridae recognizing proteinaceous or saccharidic receptors. Viruses 2020, 12, 512. [Google Scholar] [CrossRef]
- Kizziah, J.L.; Manning, K.A.; Dearborn, A.D.; Dokland, T. Structure of the host cell recognition and penetration machinery of a Staphylococcus aureus bacteriophage. PLoS Pathog. 2020, 16, e1008314. [Google Scholar] [CrossRef] [Green Version]
- Veesler, D.; Robin, G.; Lichière, J.; Auzat, I.; Tavares, P.; Bron, P.; Campanacci, V.; Cambillau, C. Crystal structure of bacteriophage SPP1 distal tail protein (gp19.1): A baseplate hub paradigm in gram-positive infecting phages. J. Biol. Chem. 2010, 285, 36666–36673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinelli, S.; Tremblay, D.; Moineau, S.; Cambillau, C.; Goulet, A. Structural insights into lactococcal Siphophage p2 baseplate activation mechanism. Viruses 2020, 12, 878. [Google Scholar] [CrossRef] [PubMed]
- Veesler, D.; Spinelli, S.; Mahony, J.; Lichière, J.; Blangy, S.; Bricogne, G.; Legrand, P.; Ortiz-Lombardia, M.; Campanacci, V.; van Sinderen, D.; et al. Structure of the phage TP901-1 1.8 MDa baseplate suggests an alternative host adhesion mechanism. Proc. Natl. Acad. Sci. USA 2012, 109, 8954–8958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olia, A.S.; Casjens, S.; Cingolani, G. Structure of phage P22 cell envelope–penetrating needle. Nat. Struct. Mol. Biol. 2007, 14, 1221–1226. [Google Scholar] [CrossRef]
- Leavitt, J.C.; Gogokhia, L.; Gilcrease, E.B.; Bhardwaj, A.; Cingolani, G.; Casjens, S.R. The tip of the tail needle affects the rate of DNA delivery by bacteriophage P22. PLoS ONE 2013, 8, e70936. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, A.; Molineux, I.J.; Casjens, S.R.; Cingolani, G. Atomic structure of bacteriophage Sf6 tail needle knob. J. Biol. Chem. 2011, 286, 30867–30877. [Google Scholar] [CrossRef] [Green Version]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage endolysins as novel antimicrobials. Future Microbiol. 2012, 7, 1147–1171. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Xu, W.; Lei, L.; Huang, J.; Feng, X.; Sun, C.; Du, C.; Zuo, J.; Li, Y.; Du, T.; et al. LysGH15, a novel bacteriophage lysin, protects a murine bacteremia model efficiently against lethal methicillin-resistant Staphylococcus aureus infection. J. Clin. Microbiol. 2011, 49, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Montes, E. Dextran: Sources, structures, and properties. Polysaccharides 2021, 2, 554–565. [Google Scholar] [CrossRef]
- Wang, I.N.; Smith, D.L.; Young, R. Holins: The protein clocks of bacteriophage infections. Annu. Rev. Microbiol. 2000, 54, 799–825. [Google Scholar] [CrossRef]
- Weigel, C.; Seitz, H. Bacteriophage replication modules. FEMS Microbiol. Rev. 2006, 30, 321–381. [Google Scholar] [CrossRef] [PubMed]
- Curtis, F.A.; Reed, P.; Sharples, G.J. Evolution of a phage RuvC endonuclease for resolution of both Holliday and branched DNA junctions. Mol. Microbiol. 2005, 55, 1332–1345. [Google Scholar] [CrossRef] [PubMed]
- Bidnenko, E.; Ehrlich, S.D.; Chopin, M.C. Lactococcus lactis phage operon coding for an endonuclease homologous to RuvC. Mol. Microbiol. 1998, 28, 823–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, M.; Marianovsky, I.; Glaser, G. MazG—A regulator of programmed cell death in Escherichia coli. Mol. Microbiol. 2006, 59, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Rihtman, B.; Bowman-Grahl, S.; Millard, A.; Corrigan, R.M.; Clokie, M.R.J.; Scanlan, D.J. Cyanophage MazG is a pyrophosphohydrolase but unable to hydrolyse magic spot nucleotides. Environ. Microbiol. Rep. 2019, 11, 448–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- McNair, K.; Bailey, B.A.; Edwards, R.A. PHACTS, a computational approach to classifying the lifestyle of phages. Bioinformatics 2012, 28, 614–618. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Song, W.; Sun, H.-X.; Zhang, C.; Cheng, L.; Peng, Y.; Deng, Z.; Wang, D.; Wang, Y.; Hu, M.; Liu, W.; et al. Prophage Hunter: An integrative hunting tool for active prophages. Nucleic Acids Res. 2019, 47, W74–W80. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession * | Description | Coverage [%] | No. of Distinct Peptides Identified | No. of Amino Acids | Molecular Weight (kDa) |
|---|---|---|---|---|---|
| lcl_gp04 ORF4 Portal protein | [locus_tag = HOT76_gp04] [db_xref = GeneID:54998161] [protein = portal protein] [protein_id = YP_009807283.1] [location = 2003..3385] [gbkey = CDS] | 2 | 1 | 460 | 51.3 |
| lcl_gp16 ORF16 Major capsid protein | [locus_tag = HOT76_gp16] [db_xref = GeneID:54998173] [protein = main capsid protein] [protein_id = YP_009807295.1] [location = 10024..10899] [gbkey = CDS] | 31 | 10 | 291 | 31.6 |
| lcl_gp21 ORF21 Major tail protein | [locus_tag = HOT76_gp21] [db_xref = GeneID:54998178] [protein = hypothetical protein] [protein_id = YP_009807300.1] [location = 12569..13075] [gbkey = CDS] | 23 | 3 | 168 | 18.1 |
| lcl_gp25 ORF25 Distal tail protein | [locus_tag = HOT76_gp25] [db_xref = GeneID:54998182] [protein = hypothetical protein] [protein_id = YP_009807304.1] [location = 16515..18968] [gbkey = CDS] | 1 | 1 | 817 | 89.8 |
| lcl_gp26 ORF26 Fiber lower protein | [locus_tag = HOT76_gp26] [db_xref = GeneID:54998183] [protein = hypothetical protein] [protein_id = YP_009807305.1] [location = 18968..19426] [gbkey = CDS] | 7 | 1 | 152 | 16.3 |
| lcl_gp28 ORF28 Tail needle protein | [locus_tag = HOT76_gp28] [db_xref = GeneID:54998185] [protein = hypothetical protein] [protein_id = YP_009807307.1] [location = 22521..22841] [gbkey = CDS] | 9 | 1 | 106 | 11.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sprotte, S.; Rasmussen, T.S.; Cho, G.-S.; Brinks, E.; Lametsch, R.; Neve, H.; Vogensen, F.K.; Nielsen, D.S.; Franz, C.M.A.P. Morphological and Genetic Characterization of Eggerthella lenta Bacteriophage PMBT5. Viruses 2022, 14, 1598. https://doi.org/10.3390/v14081598
Sprotte S, Rasmussen TS, Cho G-S, Brinks E, Lametsch R, Neve H, Vogensen FK, Nielsen DS, Franz CMAP. Morphological and Genetic Characterization of Eggerthella lenta Bacteriophage PMBT5. Viruses. 2022; 14(8):1598. https://doi.org/10.3390/v14081598
Chicago/Turabian StyleSprotte, Sabrina, Torben S. Rasmussen, Gyu-Sung Cho, Erik Brinks, René Lametsch, Horst Neve, Finn K. Vogensen, Dennis S. Nielsen, and Charles M. A. P. Franz. 2022. "Morphological and Genetic Characterization of Eggerthella lenta Bacteriophage PMBT5" Viruses 14, no. 8: 1598. https://doi.org/10.3390/v14081598
APA StyleSprotte, S., Rasmussen, T. S., Cho, G.-S., Brinks, E., Lametsch, R., Neve, H., Vogensen, F. K., Nielsen, D. S., & Franz, C. M. A. P. (2022). Morphological and Genetic Characterization of Eggerthella lenta Bacteriophage PMBT5. Viruses, 14(8), 1598. https://doi.org/10.3390/v14081598