
Are Viral Infections Key Inducers of Autoimmune Diseases? Focus on Epstein–Barr Virus
 , and
, and
Abstract
:1. Introduction
2. Mechanism of Immune Tolerance Disruption by Viruses
3. EBV-Associated Diseases
4. EBV and Autoimmune Diseases
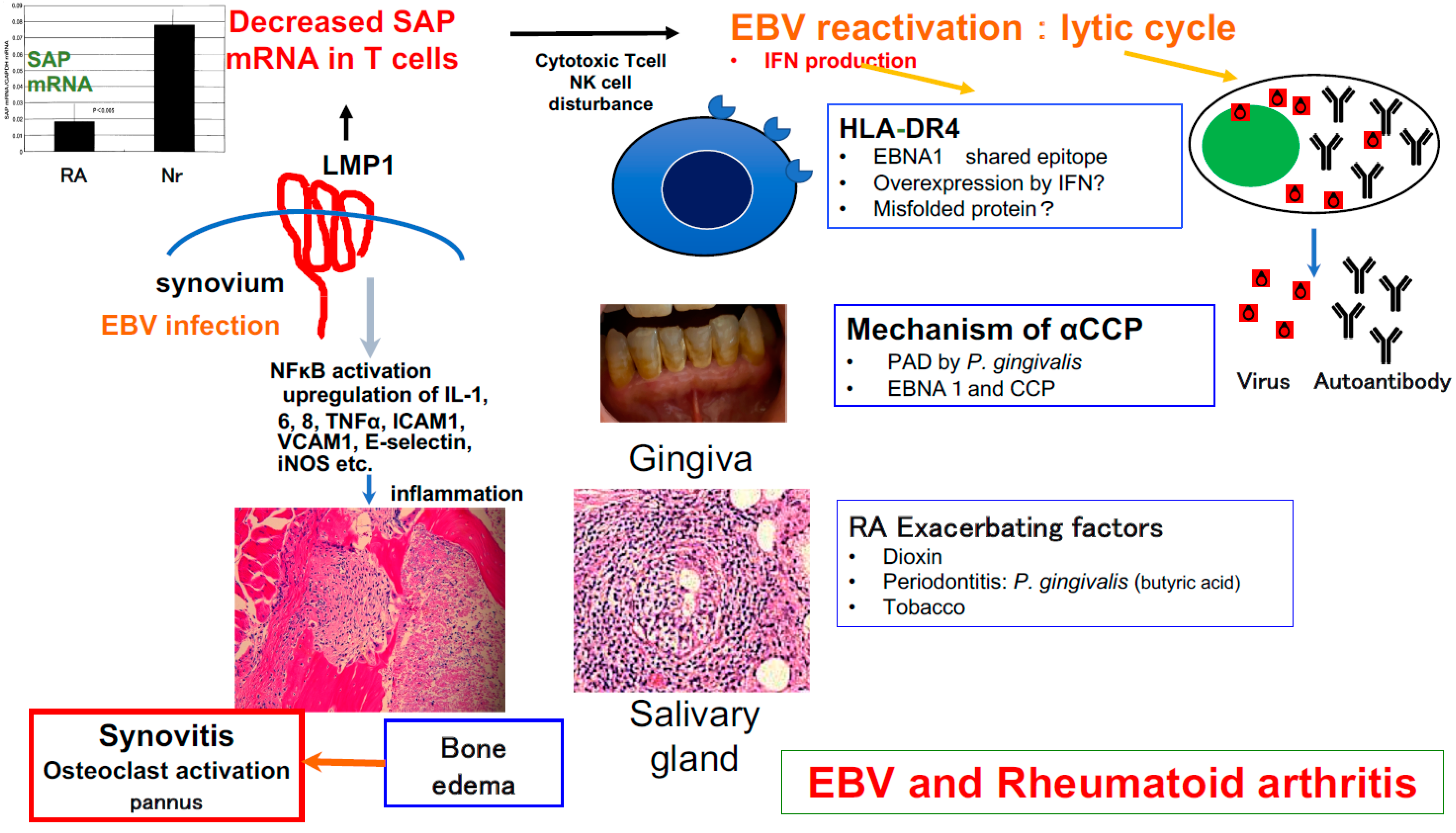
5. Involvement of EBV in RA Development
6. Signaling Lymphocytic Activation Molecule-Associated Protein (SAP)/SH2D1A and RA
7. Can EBV Directly Cause RA?
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wiertz, E.J.; Jones, T.R.; Sun, L.; Bogyo, M.; Geuze, H.J.; Ploegh, H.L. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 1996, 84, 769–779. [Google Scholar] [CrossRef]
- Schwartz, O.; Marechal, V.; Le Gall, S.; Lemonnier, F.; Heard, J.M. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 1996, 2, 338–342. [Google Scholar] [CrossRef]
- Le Gall, S.; Erdtmann, L.; Benichou, S.; Berlioz-Torrent, C.; Liu, L.; Benarous, R.; Heard, J.M.; Schwartz, O. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 1998, 8, 483–495. [Google Scholar] [CrossRef]
- Collins, K.L.; Chen, B.K.; Kalams, S.A.; Walker, B.D.; Baltimore, D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 1998, 391, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Albanese, M.; Tagawa, T.; Bouvet, M.; Maliqi, L.; Lutter, D.; Hoser, J.; Hastreiter, M.; Hayes, M.; Sugden, B.; Martin, L.; et al. Epstein-Barr virus microRNAs reduce immune surveillance by virus-specific CD8+ T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E6467–E6475. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, R.; Zamani, F.; Hajibaba, M.; Rasouli-Saravani, A.; Noroozbeygi, M.; Gorgani, M.; Hosseini-Fard, S.R.; Jalalifar, S.; Ajdarkosh, H.; Abedi, S.H.; et al. The pathogenic, therapeutic and diagnostic role of exosomal microRNA in the autoimmune diseases. J. Neuroimmunol. 2021, 358, 577640. [Google Scholar] [CrossRef]
- Hassani, A.; Khan, G. Epstein-Barr Virus and miRNAs: Partners in Crime in the Pathogenesis of Multiple Sclerosis? Front. Immunol. 2019, 10, 695. [Google Scholar] [CrossRef]
- Page, C.; François, C.; Goëb, V.; Duverlie, G. Human parvovirus B19 and autoimmune diseases. Review of the literature and pathophysiological hypotheses. J. Clin. Virol. 2015, 72, 69–74. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Duverlie, G. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef]
- Bar-Or, A.; Banwell, B.; Berger, J.R.; Lieberman, P.M. Guilty by association: Epstein-Barr virus in multiple sclerosis. Nat. Med. 2022, 28, 904–906. [Google Scholar] [CrossRef]
- Niller, H.H.; Wolf, H.; Ay, E.; Minarovits, J. Epigenetic dysregulation of epstein-barr virus latency and development of autoimmune disease. Adv. Exp. Med. Biol. 2011, 711, 82–102. [Google Scholar] [PubMed]
- Toussirot, E.; Roudier, J. Pathophysiological links between rheumatoid arthritis and the Epstein-Barr virus: An update. Joint Bone Spine. 2007, 74, 418–426. [Google Scholar] [PubMed]
- Balandraud, N.; Meynard, J.B.; Auger, I.; Sovran, H.; Mugnier, B.; Reviron, D.; Roudier, J.; Roudier, C. Epstein-Barr virus load in the peripheral blood of patients with rheumatoid arthritis: Accurate quantification using real-time polymerase chain reaction. Arthritis Rheum. 2003, 48, 1223–1228. [Google Scholar] [PubMed]
- Lossius, A.; Johansen, J.N.; Torkildsen, O.; Vartdal, F.; Holmøy, T. Epstein-Barr virus in systemic lupus erythematosus, rheumatoid arthritis and multiple sclerosis-association and causation. Viruses 2012, 4, 3701–3730. [Google Scholar]
- Rand, M.L.; Wright, J.F. Virus-associated idiopathic thrombocytopenic purpura. Transfus Sci. 1998, 19, 253–259. [Google Scholar]
- Jakhmola, S.; Upadhyay, A.; Jain, K.; Mishra, A.; Jha, H.C. Herpesviruses and the hidden links to Multiple Sclerosis neuropathology. J. Neuroimmunol. 2021, 358, 577636. [Google Scholar]
- Smatti, M.K.; Cyprian, F.S.; Nasrallah, G.K.; Al Thani, A.A.; Almishal, R.O.; Yassine, H.M. Viruses and Autoimmunity: A Review on the Potential Interaction and Molecular Mechanisms. Viruses 2019, 11, 762. [Google Scholar]
- Lee, K.H.; Choi, S.; Kwon, J.S.; Kim, S.H.; Park, S.Y. Varicella zoster virus (VZV)-specific immunity and subclinical VZV reactivation in patients with autoimmune diseases. Korean J. Intern. Med. 2021, 36, 992–1000. [Google Scholar]
- Schreiner, P.; Mueller, N.J.; Fehr, J.; Maillard, M.H.; Brand, S.; Michetti, P.; Schoepfer, A.; Restellini, S.; Vulliemoz, M.; Vavricka, S.R.; et al. Varicella zoster virus in inflammatory bowel disease patients: What every gastroenterologist should know. J. Crohns Colitis. 2021, 15, 316–325. [Google Scholar]
- Nishioka, Y.; Noda, T.; Okada, S.; Myojin, T.; Kubo, S.; Higashino, T.; Nakajima, H.; Sugiyama, T.; Ishii, H.; Imamura, T. Association between influenza and the incidence rate of new-onset type 1 diabetes in Japan. J. Diabetes Investig. 2021, 12, 1797–1804. [Google Scholar]
- Allen, D.W.; Kim, K.W.; Rawlinson, W.D.; Craig, M.E. Maternal virus infections in pregnancy and type 1 diabetes in their offspring: Systematic review and meta-analysis of observational studies. Rev. Med. Virol. 2018, 28, e1974. [Google Scholar] [CrossRef] [PubMed]
- Quaresma, J.A.; Yoshikawa, G.T.; Koyama, R.V.; Dias, G.A.; Fujihara, S.; Fuzii, H.T. HTLV-1, Immune Response and Autoimmunity. Viruses 2015, 8, 5. [Google Scholar] [CrossRef]
- Giardullo, L.; Corrado, A.; Maruotti, N.; Rotondo, C.; Cantatore, F.P. Rheumatological Diseases in HIV Infection. Curr. Rheumatol Rev. 2021, 17, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Vega, L.E.; Espinoza, L.R. HIV infection and its effects on the development of autoimmune disorders. Pharmacol. Res. 2018, 129, 1–9. [Google Scholar] [PubMed]
- Wang, C.R.; Tsai, H.W. Human hepatitis viruses-associated cutaneous and systemic vasculitis. World J. Gastroenterol. 2021, 27, 19–36. [Google Scholar] [CrossRef]
- Geravandi, S.; Liu, H.; Maedler, K. Enteroviruses and T1D: Is It the Virus, the Genes or Both which Cause T1D. Microorganisms 2020, 8, 1017. [Google Scholar] [CrossRef]
- Gómez-Rial, J.; Rivero-Calle, I.; Salas, A.; Martinón-Torres, F. Rotavirus and autoimmunity. J. Infect. 2020, 81, 183–189. [Google Scholar] [CrossRef]
- Firestein, G.S.; Budd, R.C.; Gabriel, S.E.; McInnes, I.B.; O’Dell, J.R. Firestein & Kelley’s Textbook of Rheumatology, 11th ed.; Koretzky, G., Ed.; Elsevier: Philadelphia, PA, USA, 2020. [Google Scholar]
- van Regenmortel, M.H.V.; Mahy, B.W.J. (Eds.) Encyclopedia of Virology, 3rd ed.; Academic Press (Elsevier): Amsterdam, The Netherlands, 2008. [Google Scholar]
- Sandhu, H.S.; Kaplan, H.J. (Eds.) Clinical Cases in Uveitis. Differential Diagnosis and Management, 1st ed.; Elsevier: Philadelphia, PA, USA, 2020. [Google Scholar]
- Batterbury, M.; Murphy, C. (Eds.) Ophthalmology, 4th ed.; Elsevier: London, UK, 2018. [Google Scholar]
- Lee, J.H.; Agarwal, A.; Mahendradas, P.; Lee, C.S.; Gupta, V.; Pavesio, C.E.; Agrawal, R. Viral posterior uveitis. Surv. Ophthalmol. 2017, 62, 404–445. [Google Scholar] [CrossRef]
- Lynn, W.A.; Lightman, S. The eye in systemic infection. Lancet 2004, 364, 1439–1450. [Google Scholar]
- Calonje, J.E.; Brenn, T.; Lazar, A.J.; Billings, S.D. (Eds.) McKee’s Pathology of the Skin with Clinical Correlations, 5th ed.; Elsevier: London, UK, 2018. [Google Scholar]
- MSD MANUAL Professional Version; Merck Sharp & Dohme Corp.: Kenilworth, NJ, USA, 2021.
- Fujiwara, S.; Takei, M. Epstein-Barr virus and autoimmune diseases. Clin. Exp. Neuroimmunol. 2015, 6 (Suppl. 1), 38–48. [Google Scholar]
- Pak, C.Y.; Eun, H.M.; McArthur, R.G.; Yoon, J.W. Association of cytomegalovirus infection with autoimmune type1 diabetes. Lancet 1988, 2, 1–4. [Google Scholar] [CrossRef]
- Eng, M.A.; Kallemuchikkal, U.; Gorevic, P.D. Hepatitis C virus, autoimmunity and lymphoproliferation. Mt. Sinai. J. Med. 2000, 67, 120–132. [Google Scholar]
- Kukreja, A.; Maclaren, N.K. Current cases in which epitope mimicry is considered as a component cause of autoimmune disease: Immune-mediated (type 1) diabetes. Cell. Mol. Life Sci. 2000, 57, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Rose, N.R. Critical Cytokine Pathways to Cardiac Inflammation. J. Interferon Cytokine Res. 2011, 31, 705–710. [Google Scholar] [CrossRef]
- Jiang, Y.; Arase, N.; Kohyama, M.; Hirayasu, K.; Suenaga, T.; Jin, H.; Matsumoto, M.; Shida, K.; Lanier, L.L.; Saito, T.; et al. Transport of misfolded endoplasmic reticulum proteins to the cell surface by MHC class II molecules. Int. Immunol. 2013, 25, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Gangaplara, A.; Martens, C.; Dahlstrom, E.; Metidji, A.; Gokhale, A.S.; Glass, D.D.; Lopez-Ocasio, M.; Baur, R.; Kanakabandi, K.; Porcella, S.F.; et al. Type I interferon signaling attenuates regulatory T cell function in viral infection and in the tumor microenvironment. PLoS Pathog. 2018, 14, e1006985. [Google Scholar] [CrossRef]
- Qingyong, J.; Perchellet, A.; Goverman, J.M. Viral Infection Triggers Central Nervous System Autoimmunity Via Activation of Dual TCR-Expressing CD8+ T Cells. Nat. Immunol. 2010, 11, 628–634. [Google Scholar]
- Harley, J.B.; Chen, X.; Pujato, M.; Miller, D.; Maddox, A.; Forney, C.; Magnusen, A.F.; Lynch, A.; Chetal, K.; Yukawa, M.; et al. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat. Genet. 2018, 50, 699–707. [Google Scholar]
- Alspaugh, M.A.; Jensen, F.C.; Rabin, H.; Tan, E.M. Lymphocyte transformed by Epstein-Barr virus: Induction of nuclear antigen reactive with antibody in rheumatoid arthritis. J. Exp. Med. 1978, 147, 1018–1027. [Google Scholar] [CrossRef]
- Billings, P.B.; Hoch, S.O.; White, P.J.; Carson, D.A.; Vaughan, J.H. Antibodies to the Epstein-Barr virus nuclear antigen and to rheumatoid arthritis nuclear antigen identify the same polypeptide. Proc. Natl. Acad. Sci. USA 1983, 80, 7104–7108. [Google Scholar]
- Rhodes, G.; Carson, D.A.; Valbracht, J.; Houghten, R.; Vaughan, J.H. Human immune response to synthetic peptides from the Epstein-Barr virus nuclear antigen. J. Immunol. 1985, 134, 211–216. [Google Scholar] [PubMed]
- Rumpold, H.; Rhodes, G.H.; Bloch, P.L.; Carson, D.A.; Vaughan, J.H. The glycine-alanine repeating region is the major epitope of the Epstein-Barr nuclear antigen-1 (EBNA-1). J. Immunol. 1987, 138, 593–599. [Google Scholar] [PubMed]
- Fox, R.; Sportsman, R.; Rhodes, G.; Luka, J.; Pearson, G.; Vaughan, J.H. Rheumatoid arthritis synovial membrane contains a 62000-molecular-weight protein that shares an antigenic epitope with the Epstein Barr virus encoded associated nuclear antigen. J. Clin. Inv. 1986, 77, 1539–1547. [Google Scholar] [CrossRef]
- Roudier, J.; Rhodes, G.; Petersen, J.; Vaughan, J.H.; Carson, D.A. The Epstein-Barr virus glycoprotein gp110, a molecular link between HLA DR4, HLA DR1, and rheumatoid arthritis. Scand. J. Immunol. 1988, 27, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Pratesi, F.; Tommasi, C.; Anzilotti, C.; Chimenti, D.; Migliorini, P. Deiminated Epstein-Barr virus nuclear antigen 1 is a target of anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheum. 2006, 54, 733–741. [Google Scholar]
- Croia, C.; Serafini, B.; Bombardieri, M.; Kelly, S.; Humby, F.; Severa, M.; Rizzo, F.; Coccia, E.M.; Migliorini, P.; Aloisi, F.; et al. Epstein-Barr virus persistence and infection of autoreactive plasma cells in synovial lymphoid structures in rheumatoid arthritis. Ann. Rheum. Dis. 2013, 72, 1559–1568. [Google Scholar] [CrossRef]
- Niedobitek, G.; Lisner, R.; Swoboda, B.; Rooney, N.; Fassbender, H.G.; Kirchner, T.; Aigner, T.; Herbst, H. Lack of evidence for an involvement of Epstein-Barr virus infection of synovial membranes in the pathogenesis of rheumatoid arthritis. Arthritis Rheum. 2000, 43, 151–154. [Google Scholar]
- Fox, R.I.; Chilton, T.; Rhodes, G.; Vaughan, J.H. Lack of reactivity of rheumatoid arthritis synovial membrane DNA with cloned Epstein-Barr virus DNA probes. J. Immunol. 1986, 137, 498–501. [Google Scholar]
- Koide, J.; Takada, K.; Sugiura, M.; Sekine, H.; Ito, T.; Saito, K.; Mori, S.; Takeuchi, T.; Uchida, S.; Abe, T. Spontaneous establishment of an Epstein-Barr virus-infected fibroblast line from the synovial tissue of a rheumatoid arthritis patient. J. Virol. 1990, 71, 2478–7481. [Google Scholar] [CrossRef]
- Takei, M.; Mitamura, K.; Fujiwara, S.; Fujiwara, S.; Sasaki, K.; Nishi, T.; Kuga, T.; Ookubo, T.; Horie, T.; Ryu, J.; et al. Detection of Epstein-Barr virus-encoded small RNA 1 and latent membrane protein 1 in synovial lining cells from rheumatoid arthritis patients. Int. Immunol. 1997, 9, 739–743. [Google Scholar] [CrossRef]
- Edinger, J.W.; Bonneville, M.; Scotet, E.; Houssaint, E.; Schumacher, H.R.; Posnett, D.N. EBV gene expression not altered in rheumatoid synovia despite the presence of EBV antigen-specific T cell clones. J. Immunol. 1999, 162, 3694–3701. [Google Scholar] [PubMed]
- Takeda, T.; Mizugaki, Y.; Matsubara, L.; Imai, S.; Koike, T.; Takada, K. Lytic Epstein-Barr virus infection in the synovial tissue of patients with rheumatoid arthritis. Arthritis Rheum. 2000, 43, 1218–1225. [Google Scholar] [CrossRef]
- Mehraein, Y.; Lennerz, C.; Ehlhardt, S.; Remberger, K.; Ojak, A.; Zang, K.D. Latent Epstein-Barr virus (EBV) infection and cytomegalovirus (CMV) infection in synovial tissue of autoimmune chronic arthritis determined by RNA- and DNA-in situ hybridization. Mod. Pathol. 2004, 17, 781–789. [Google Scholar] [PubMed]
- Saal, J.G.; Krimmel, M.; Steidle, M.; Gerneth, F.; Wagner, S.; Fritz, P.; Koch, S.; Zacher, J.; Sell, S.; Einsele, H.; et al. Epstein-Barr virus infection increases the risk of rheumatoid arthritis in individuals with the shared HLA-DR4 epitope. Arthritis Rheum. 1999, 42, 1485–1496. [Google Scholar]
- Brousset, P. Lack of Epstein-Barr virus infection of synovial membranes in patients with rheumatoid arthritis: Comment on the article by Niedobitek et al. Arthritis Rheum. 2000, 43, 2614. [Google Scholar] [PubMed]
- Brousset, P.; Caulier, M.; Cantagrel, A.; Dromer, C.; Mazières, B.; Delsol, G. Absence of Epstein-Barr virus carrying cells in synovial membranes and subcutaneous nodules of patients with rheumatoid arthritis. Ann. Rheum. Dis. 1993, 52, 608–609. [Google Scholar]
- Tosato, G.; Steinberg, A.D.; Blaese, R.M. Defective EBV-specific suppressor T-cell function in rheumatoid arthritis. N. Engl. J. Med. 1981, 19, 1238–1243. [Google Scholar]
- Hammarskjold, M.-L.; Simurda, M.C. Epstein-Barr virus latent membrane protein transactivates the human immunodeficiency virus type 1 long terminal repeat through induction of NF-kB activity. J. Virol. 1992, 66, 6496–6501. [Google Scholar] [PubMed]
- Henderson, S.; Rowe, M.; Gregory, C.; Croom-Carter, D.; Wang, F.; Longnecker, R.; Kieff, E.; Rickinson, A. Induction of bcl-2 expression by Epstein-Barr virus latent membrane protein 1 protects infected B cells from programmed cell death. Cell 1991, 65, 1107–1115. [Google Scholar]
- Wang, D.; Liebowitz, D.; Kieff, E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell 1985, 43, 831–840. [Google Scholar]
- Fahaeus, R.; Rymo, L.; Rhim, J.S. Morphological transformation of human keratinocytes expressing the LMP gene of Epstein-Barr virus. Nature 1990, 345, 447–449. [Google Scholar]
- Sayos, J.; Wu, C.; Morra, M.; Wang, N.; Zhang, X.; Allen, D.; van Schaik, S.; Notarangelo, L.; Geha, R.; Roncarolo, M.G.; et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature 1998, 395, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Nichols, K.E.; Koretzky, G.A.; June, C.H. SAP natural inhibitor or grand SLAM of T cell activation? Nat. Immunol. 2001, 2, 665–666. [Google Scholar] [CrossRef]
- Chan, B.; Lanyi, A.; Song, H.K.; Griesbach, J.; Simarro-Grande, M.; Poy, F.; Howie, D.; Sumegi, J.; Terhorst, C.; Eck, M.J. SAP couples Fyn to SLAM immune receptors. Nat. Cell Biol. 2003, 5, 155–160. [Google Scholar] [CrossRef]
- Tangye, S.G.; Lazetic, S.; Woollatt, E.; Sutherland, G.R.; Lanier, L.L.; Phillips, J.H. Cutting edge: Human 2B4, an activating NK cell receptor, recruits the protein tyrosine phosphatase SHP-2 and the adaptor signaling protein SAP. J. Immunol. 1999, 162, 6981–6985. [Google Scholar]
- Brown, M.H.; Boles, K.; van der Merwe, P.A.; Sutherland, G.R.; Lanier, L.L.; Phillips, J.H. 2B4, the natural killer and T cell immunoglobulin superfamily surface protein, is a ligand for CD48. J. Exp. Med. 1998, 188, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Sayos, J.; Martín, M.; Chen, A.; Simarro, M.; Howie, D.; Morra, M.; Engel, P.; Terhorst, C. Links Cell surface receptors Ly-9 and CD84 recruit the X-linked lymphoproliferative disease gene product SAP. Blood 2001, 97, 3867–3874. [Google Scholar] [CrossRef]
- Bottino, C.; Falco, M.; Parolini, S.; Marcenaro, E.; Augugliaro, R.; Sivori, S.; Landi, E.; Biassoni, R.; Notarangelo, L.D.; Moretta, L.; et al. NTB-A [correction of GNTB-A], a novel SH2D1A-associated surface molecule contributing to the inability of natural killer cells to kill Epstein-Barr virus-infected B cells in X-linked lymphoproliferative disease. J. Exp. Med. 2001, 194, 235–246. [Google Scholar] [CrossRef]
- Li, C.; Iosef, C.; Jia, C.Y.; Han, V.K.; Li, S.S. Dual functional roles for the X-linked lymphoproliferative syndrome gene product SAP/SH2D1A in signaling through the signaling lymphocyte activation molecule (SLAM) family of immune receptors. J. Biol. Chem. 2003, 278, 3852–3859. [Google Scholar] [CrossRef]
- Latour, S.; Roncagalli, R.; Chen, R.; Bakinowski, M.; Shi, X.; Schwartzberg, P.L.; Davidson, D.; Veillette, A. Binding of SAP SH2 domain to FynT SH3 domain reveals a novel mechanism of receptor signaling in immune regulation. Nat. Cell Biol. 2003, 5, 149–154. [Google Scholar] [CrossRef]
- Engel, P.; Eck, M.J.; Terhorst, C. The SAP and SLAM families in immune responses and X-linked lymphoproliferative disease. Nat. Rev. Immunol. 2003, 3, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Davidson, D.; Shi, X.; Zhang, S.; Wang, H.; Nemer, M.; Ono, N.; Ohno, S.; Yanagi, Y.; Veillette, A. Genetic evidence linking SAP, the X-linked lymphoproliferative gene product, to Src-related kinase FynT in T(H)2 cytokine regulation. Immunity 2004, 21, 707–717. [Google Scholar] [CrossRef]
- Wu, C.; Nguyen, K.B.; Pien, G.C.; Wang, N.; Gullo, C.; Howie, D.; Sosa, M.R.; Edwards, M.J.; Borrow, P.; Satoskar, A.R.; et al. SAP controls T cell responses to virus and terminal differentiation of TH2 cells. Nat. Immunol. 2001, 2, 410–414. [Google Scholar] [CrossRef]
- Chen, R.; Relouzat, F.; Roncagalli, R.; Aoukaty, A.; Tan, R.; Latour, S.; Veillette, A. Molecular dissection of 2B4 signaling: Implications for signal transduction by SLAM-related receptors. Mol. Cell. Biol. 2004, 24, 5144–5156. [Google Scholar] [CrossRef] [PubMed]
- Roncagalli, R.; Taylor, J.E.; Zhang, S.; Shi, X.; Chen, R.; Cruz-Munoz, M.E.; Yin, L.; Latour, S.; Veillette, A. Negative regulation of natural killer cell function by EAT-2, a SAP-related adaptor. Nat. Immunol. 2005, 6, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Cannons, J.L.; Yu, L.J.; Hill, B.; Mijares, L.A.; Dombroski, D.; Nichols, K.E.; Antonellis, A.; Koretzky, G.A.; Gardner, K.; Schwartzberg, P.L. SAP regulates T(H)2 differentiation and PKC-theta-mediated activation of NF-kappaB1. Immunity 2004, 21, 693–706. [Google Scholar] [CrossRef]
- Gu, C.; Tangye, S.G.; Sun, X.; Luo, Y.; Lin, Z.; Wu, J. The X-linked lymphoproliferative disease gene product SAP associates with PAK-interacting exchange factor and participates in T cell activation. Proc. Natl. Acad. Sci. USA 2006, 103, 14447–14452. [Google Scholar] [CrossRef]
- Lo, K.Y.; Chin, W.H.; Ng, Y.P.; Cheng, A.W.; Cheung, Z.H.; Ip, N.Y. SLAM-associated protein as a potential negative regulator in Trk signaling. J. Biol. Chem. 2005, 280, 41744–41752. [Google Scholar] [CrossRef]
- Takei, M.; Ishiwata, T.; Mitamura, K.; Fujiwara, S.; Sasaki, K.; Nishi, T.; Kuga, T.; Ookubo, T.; Horie, T.; Ryu, J.; et al. Decreased expression of signaling lymphocytic-activation molecule associated protein (SAP) transcripts in T cells from patients with rheumatoid arthritis. Int. Immunol. 1997, 9, 739–743. [Google Scholar] [CrossRef]
- Ito, M.; Hiramatsu, H.; Kobayashi, K.; Suzue, K.; Kawahata, M.; Hioki, K.; Ueyama, Y.; Koyanagi, Y.; Sugamura, K.; Tsuji, K.; et al. NOD/SCID/gamma(c) mouse: An excellent recipient mouse model for engraftment of human cells. Blood 2002, 100, 3175–3182. [Google Scholar] [CrossRef]
- Traggiai, E.; Chicha, L.; Mazzucchelli, L.; Bronz, L.; Piffaretti, J.C.; Lanzavecchia, A.; Manz, M.G. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science 2004, 304, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef] [PubMed]
- Melkus, M.W.; Estes, J.D.; Padgett-Thomas, A.; Denton, P.W.; Othieno, F.A.; Wege, A.K.; Haase, A.T.; Garcia, J.V. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat. Med. 2006, 12, 1316–1322. [Google Scholar] [CrossRef]
- Yajima, M.; Imadome, K.; Nakagawa, A.; Watanabe, S.; Terashima, K.; Nakamura, H.; Ito, M.; Shimizu, N.; Honda, M.; Yamamoto, N.; et al. A new humanized mouse model of Epstein-Barr virus infection that reproduces persistent infection, lymphoproliferative disorder, and cell-mediated and humoral immune responses. J. Infect. Dis. 2008, 198, 673–682. [Google Scholar] [CrossRef]
- Fujiwara, S.; Imadome, K.; Takei, M. Modeling EBV infection and pathogenesis in new-generation humanized mice. Exp. Mol. Med. 2015, 47, e135. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, Y.; Takei, M.; Yajima, M.; Imadome, K.; Inomata, H.; Shiozaki, M.; Ikumi, N.; Nozaki, T.; Shiraiwa, H.; Kitamura, N.; et al. Epstein-Barr virus induces erosive arthritis in humanized mice. PLoS ONE 2011, 6, e26630. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, Y.; Takei, M.; Iwata, M.; Nagatsuka, Y.; Tsuzuki, H.; Imai, K.; Imadome, K.I.; Fujiwara, S.; Kitamura, N. Human osteoclastogenesis in Epstein-Barr virus-induced erosive arthritis in humanized NOD/Shi-scid/IL-2Rγnull mice. PLoS ONE 2021, 16, e0249340. [Google Scholar] [CrossRef]
- Sawada, S.; Takei, M. Epstein-Barr virus etiology in rheumatoid synovitis. Autoimmun. Rev. 2005, 4, 106–110. [Google Scholar] [CrossRef]
- Sawada, S.; Takei, M.; Inomata, H.; Nozaki, T.; Shiraiwa, H. What is after cytokine blocking therapy, a novel therapeutic target-synovial Epstein-Barr virus for rheumatoid arthritis. Autoimmun. Rev. 2007, 6, 126–130. [Google Scholar] [CrossRef]
- Sawada, S.; Takei, M.; Ishiwata, T.; Shiraiwa, H.; Inomata, H.; Nozaki, T. SLAM-associated protein solves a mystery of autoimmunity. Ann. N. Y. Acad. Sci. 2007, 1109, 19–30. [Google Scholar] [CrossRef]
- Sawada, S.; Takei, M.; Ishiwata, T. SAP discovery: The sword edges--beneficial and harmful. Autoimmun. Rev. 2007, 6, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Takei, M.; Nozaki, T.; Inomata, H.; Kuwana, Y.; Kitamura, N.; Shiraiwa, H.; Ikumi, N.; Nagasawa, Y.; Sawada, S.; Yajima, M.; et al. Virus and Autoimmune Diseases: EBV is a Possible Cause of Rheumatoid Arthritis. J. Nihon. Univ. Med. Ass. 2012, 71, 302–310. [Google Scholar]
- Takei, M.; Kitamura, N.; Shiraiwa, H.; Inomata, H.; Nozaki, T.; Kuwana, Y.; Shiozaki, M.; Sawada, S.; Ishiwata, T. The possible curative therapy for rheumatoid arthritis―EBV infection control gene SAP and its application. Jpn. J. Clin. Immunol. 2008, 31, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Takei, M.; Kitamura, N.; Shiraiwa, H.; Inomata, H.; Nozaki, T.; Kuwana, Y.; Siozaki, M.; Masuda, H.; Matsubara, T.; Sasaki, A.; et al. Is Epstein-Barr virus a key conductor in some intractable disease?—Is SAP/SH2D1A a double-edged sword? Nihon Uni. J. Med. 2008, 50, 105–121. [Google Scholar]
- Sokal, E.M.; Hoppenbrouwers, K.; Vandermeulen, C.; Moutschen, M.; Léonard, P.; Moreels, A.; Haumont, M.; Bollen, A.; Smets, F.; Denis, M. Recombinant gp350 vaccine for infectious mononucleosis: A phase 2, randomized, double-blind, placebo-controlled a trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J. Infect. Dis. 2007, 196, 1749–1753. [Google Scholar] [CrossRef]
- Lucas, K.G.; Sun, Q.; Burton, R.L.; Tilden, A.; Vaughan, W.P.; Carabasi, M.; Salzman, D.; Ship, A. A phase I-II trial to examine the toxicity of CMV- and EBV-specific cytotoxic T lymphocytes when used for prophylaxis against EBV and CMV disease in recipients of CD34-selected/T cell-depleted stem cell transplants. Hum. Gene Ther. 2000, 11, 1453–1463. [Google Scholar]
- Tsumiyama, K.; Miyazaki, Y.; Shiozawa, S. Self-organized criticality theory of autoimmunity. PLoS ONE 2009, 4, e8382. [Google Scholar] [CrossRef]
- Chuang, H.C.; Lay, J.D.; Hsieh, W.C.; Wang, H.C.; Chang, Y.; Chuang, S.E.; Su, I.J. Epstein-Barr virus LMP1 inhibits the expression of SAP gene and upregulates Th1 cytokines in the pathogenesis of hemophagocytic syndrome. Blood 2005, 106, 3090–3096. [Google Scholar] [CrossRef]
- Rosenstein, E.D.; Greenwald, R.A.; Kushner, L.J.; Weissmann, G. Hypothesis: The humoral immune response to oral bacteria provides a stimulus for the development of rheumatoid arthritis. Inflammation 2004, 28, 311–318. [Google Scholar] [CrossRef]
- Imai, K.; Inoue, H.; Tamura, M.; Cueno, M.E.; Inoue, H.; Takeichi, O.; Kusama, K.; Saito, I.; Ochiai, K. The periodontal pathogen Porphyromonas gingivalis induces the Epstein-Barr virus lytic switch transactivator ZEBRA by histone modification. Biochimie 2012, 94, 839–846. [Google Scholar] [CrossRef]
- Inoue, H.; Mishima, K.; Yamamoto-Yoshida, S.; Ushikoshi-Nakayama, R.; Nakagawa, Y.; Yamamoto, K.; Ryo, K.; Ide, F.; Saito, I. Aryl hydrocarbon receptor-mediated induction of EBV reactivation as a risk factor for Sjögren’s syndrome. J. Immunol. 2012, 188, 4654–4662. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.H.; Xiong, D.; Xu, Y.F.; Cao, S.M.; Xue, W.Q.; Qin, H.D.; Liu, W.S.; Cao, J.Y.; Zhang, Y.; Feng, Q.S.; et al. An epidemiological and molecular study of the relationship between smoking, risk of nasopharyngeal carcinoma, and Epstein-Barr virus activation. J. Natl. Cancer Inst. 2012, 104, 1396–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Virus | Autoimmune Disease | Reference |
|---|---|---|
| Parvovirus B19 | SLE, adult-onset Still’s disease, RA | [8] |
| Epstein–Barr virus | RA, SLE, SS, PN, IgA nephropathy, MS, autoimmune thyroiditis, G-B syndrome, ITP | [5,6,7,9,10,11,12,13,14,15,16,36] |
| Human cytomegalovirus | SLE, PN, G-B syndrome | [17,37] |
| Varicella-zoster virus | ITP, SLE, RA, inflammatory bowel disease, autoimmune hepatitis | [15,16,18,19] |
| Influenza virus | ITP, type I DM | [15,20] |
| Rubella virus | ITP, type I DM | [15,21] |
| Human T-cell lymphotropic virus type 1 | HAM, SS, RA, PM, SSc, SLE | [22] |
| Human immunodeficiency virus type 1 | SS, SLE, APS, PBC, PM, autoimmune hepatitis, vasculitis, ITP | [23,24] |
| Hepatitis B virus | PN, cryoglobulin vasculitis | [25] |
| Hepatitis C virus | SS, RA, PN, cryoglobulin vasculitis | [17,38] |
| Coxsackie B virus | Type I DM | [17,26] |
| Rotavirus | Type I DM | [16,27] |
| Frequency | Manifestation | ||
|---|---|---|---|
| Arthritis [28,29] | Uveitis [30,31,32,33] | Erythema Nodosum [34] | |
| Often | HBV, HCV, rubella (adult female), parvovirus B19, HTLV-1, HIV | Herpesvirus family (HSV, HCMV, VZV) in AIDS | |
| Sometimes | Macacine alphaherpesvirus 1, Mayaro virus, mumps virus, VZV | HTLV-1, rubella virus | HBV, EBV, HIV, cytomegalovirus |
| Rare | Adenovirus type 7, herpesvirus family (EBV, HCMV, VZV) | Filovirus (Ebola), arbovirus (West Nile) | |
| Polyclonal B cell activation |
| Dual TCR-expressing T cells |
| Molecular mimicry |
| Antigen spreading |
| Bystander activation |
| HLA dysfunction |
| Regulatory T cell dysfunction |
| Aberrant function of signal transduction; SAP etc. |
| Pathogenic cytokine secretion from virus-infected cells -especially Interferon-. Misfolded proteins complexed with HLA, Attenuation of regulatory T cell function etc. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takei, M.; Kitamura, N.; Nagasawa, Y.; Tsuzuki, H.; Iwata, M.; Nagatsuka, Y.; Nakamura, H.; Imai, K.; Fujiwara, S. Are Viral Infections Key Inducers of Autoimmune Diseases? Focus on Epstein–Barr Virus. Viruses 2022, 14, 1900. https://doi.org/10.3390/v14091900
Takei M, Kitamura N, Nagasawa Y, Tsuzuki H, Iwata M, Nagatsuka Y, Nakamura H, Imai K, Fujiwara S. Are Viral Infections Key Inducers of Autoimmune Diseases? Focus on Epstein–Barr Virus. Viruses. 2022; 14(9):1900. https://doi.org/10.3390/v14091900
Chicago/Turabian StyleTakei, Masami, Noboru Kitamura, Yosuke Nagasawa, Hiroshi Tsuzuki, Mitsuhiro Iwata, Yasuko Nagatsuka, Hideki Nakamura, Kenichi Imai, and Shigeyoshi Fujiwara. 2022. "Are Viral Infections Key Inducers of Autoimmune Diseases? Focus on Epstein–Barr Virus" Viruses 14, no. 9: 1900. https://doi.org/10.3390/v14091900