
Structural Basis for the Inhibition of Coronaviral Main Proteases by a Benzothiazole-Based Inhibitor
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Expression and Purification of Mpro Proteins from Human CoVs
2.2. Enzymatic Inhibition Assays
2.3. Crystallization of Mpro-YH-53 Complexes
2.4. Data Collection, Structure Determination, and Refinement
3. Results
3.1. Inhibitory Activities of YH-53 against Coronavirus Mpros
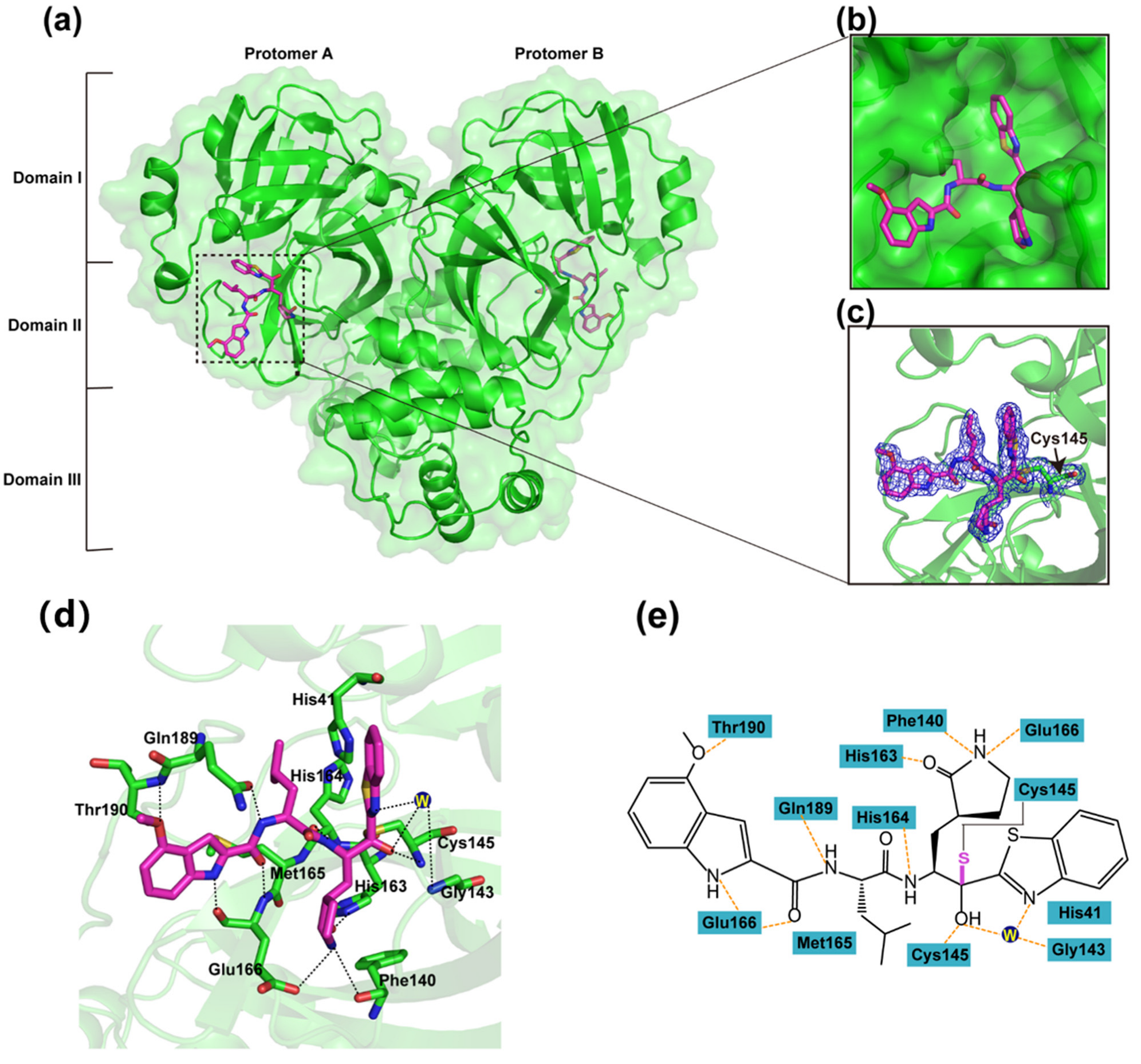
3.2. Inhibitory Mechanism of YH-53 against SARS-CoV-2 Mpro
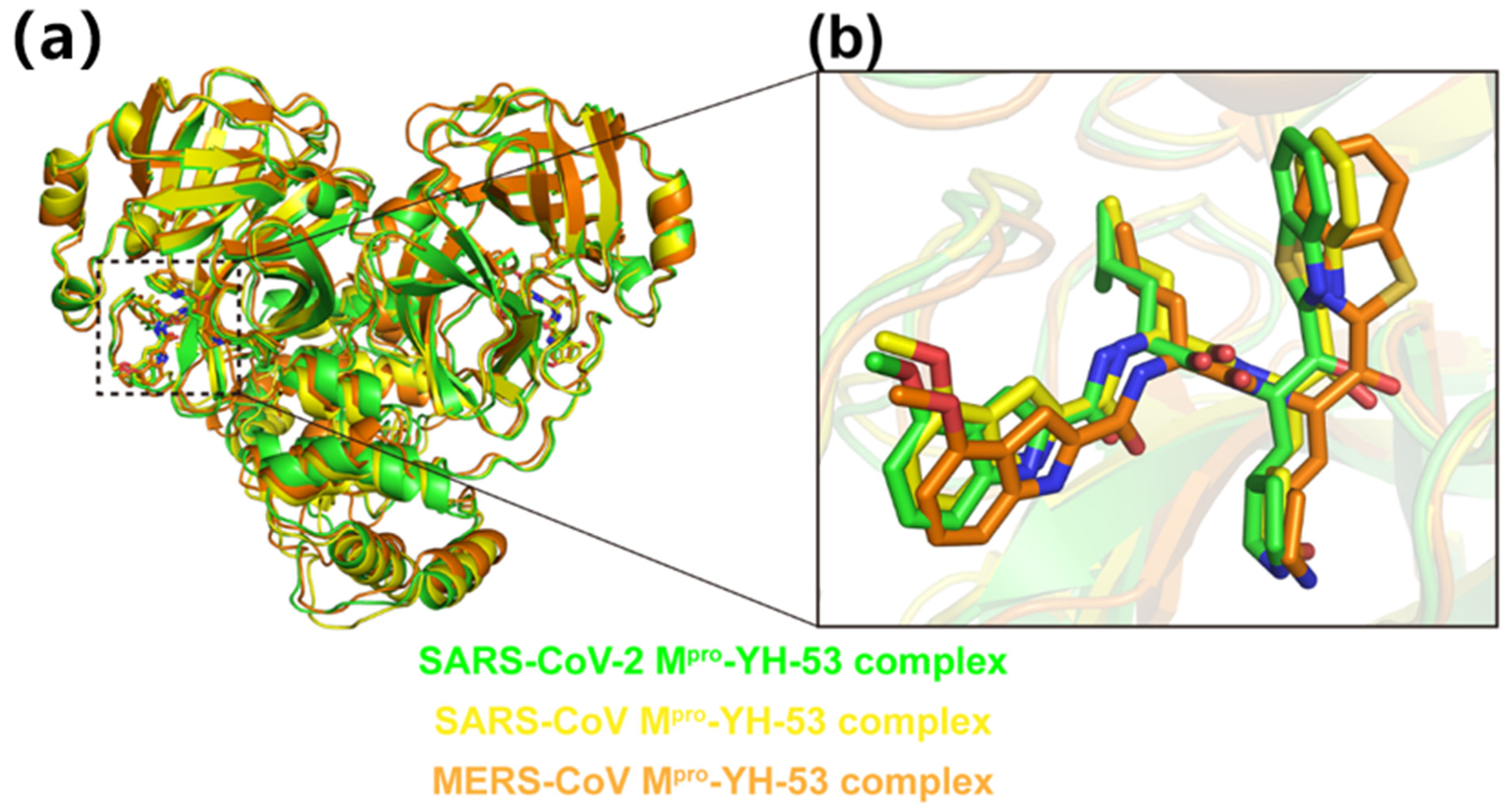
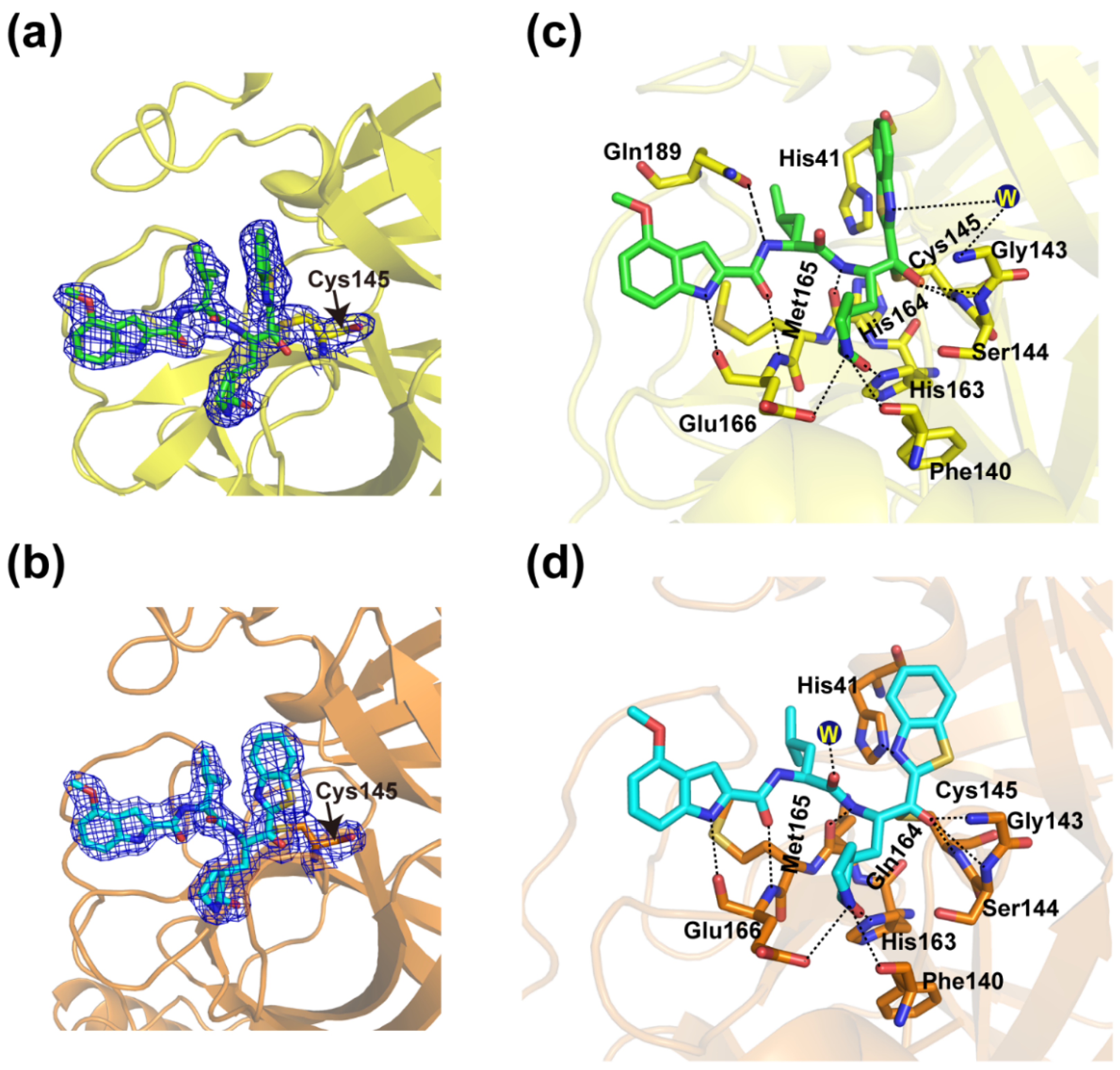
3.3. Crystal Structures of YH-53 in Complex with SARS-CoV and MERS-CoV Mpro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.T.; Leung, K.; Leung, G.M. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: A modelling study. Lancet 2020, 395, 689–697. [Google Scholar] [CrossRef]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef]
- Poutanen, S.M.; Low, D.E.; Henry, B.; Finkelstein, S.; Rose, D.; Green, K.; Tellier, R.; Draker, R.; Adachi, D.; Ayers, M.; et al. Identification of severe acute respiratory syndrome in Canada. N. Engl. J. Med. 2003, 348, 1995–2005. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, S.V.; Stoddard, S.D.; Oelkers, B.K.; Fitts, K.; Whalum, K.; Whalum, K.; Hemphill, A.D.; Manikonda, J.; Martinez, L.M.; Riley, E.G.; et al. Optimization Rules for SARS-CoV-2 Mpro Antivirals: Ensemble Docking and Exploration of the Coronavirus Protease Active Site. Viruses 2020, 12, 942. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Peng, F.; Wang, R.; Yange, M.; Guan, K.; Jiang, T.; Xu, G.; Sun, J.; Chang, C. The deadly coronaviruses: The 2003 SARS pandemic and the 2020 novel coronavirus epidemic in China. J. Autoimmun. 2020, 109, 102434. [Google Scholar] [CrossRef]
- da Costa, V.G.; Moreli, M.L.; Saivish, M.V. The emergence of SARS, MERS and novel SARS-2 coronaviruses in the 21st century. Arch. Virol. 2020, 165, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tenchov, R.; Smoot, J.; Liu, C.; Watkins, S.; Zhou, Q. A Comprehensive Review of the Global Efforts on COVID-19 Vaccine Development. ACS Cent. Sci. 2021, 7, 512–533. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Zheng, T.; Xu, K.; Han, Y.; Xu, L.; Huang, E.; An, Y.; Cheng, Y.; Li, S.; Liu, M.; et al. A Universal Design of Betacoronavirus Vaccines against COVID-19, MERS, and SARS. Cell 2020, 182, 722–733.e11. [Google Scholar] [CrossRef]
- Liu, L.; Iketani, S.; Guo, Y.; Chan, J.F.; Wang, M.; Liu, L.; Luo, Y.; Chu, H.; Huang, Y.; Nair, M.S.; et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature 2022, 602, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, L.; Li, Q.; Wang, B.; Liang, Z.; Sun, Y.; Nie, J.; Wu, J.; Su, X.; Qu, X.; et al. Reduced sensitivity of the SARS-CoV-2 Lambda variant to monoclonal antibodies and neutralizing antibodies induced by infection and vaccination. Emerg. Microbes Infect. 2022, 11, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Tuekprakhon, A.; Nutalai, R.; Dijokaite-Guraliuc, A.; Zhou, D.; Ginn, H.M.; Selvaraj, M.; Liu, C.; Mentzer, A.J.; Supasa, P.; Duyvesteyn, H.M.E.; et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell 2022, 185, 2422–2433.e13. [Google Scholar] [CrossRef]
- Koudelka, T.; Boger, J.; Henkel, A.; Schönherr, R.; Krantz, S.; Fuchs, S.; Rodríguez, E.; Redecke, L.; Tholey, A. N-Terminomics for the Identification of In Vitro Substrates and Cleavage Site Specificity of the SARS-CoV-2 Main Protease. Proteomics 2021, 21, e2000246. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Hegyi, A.; Ziebuhr, J. Conservation of substrate specificities among coronavirus main proteases. J. Gen. Virol. 2002, 83, 595–599. [Google Scholar] [CrossRef]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front. Chem. 2021, 9, 622898. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhou, X.; Zhang, Y.; Zhong, F.; Lin, C.; McCormick, P.J.; Jiang, F.; Luo, J.; Zhou, H.; Wang, Q.; et al. Crystal structure of SARS-CoV-2 main protease in complex with the natural product inhibitor shikonin illuminates a unique binding mode. Sci. Bull. 2021, 66, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors, I.I.; XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, N.; Sacco, M.D.; Ma, C.; Hu, Y.; Townsend, J.A.; Meng, X.; Zhang, F.; Zhang, X.; Ba, M.; Szeto, T.; et al. Expedited Approach toward the Rational Design of Noncovalent SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2022, 65, 2848–2865. [Google Scholar] [CrossRef]
- Drayman, N.; DeMarco, J.K.; Jones, K.A.; Azizi, S.A.; Froggatt, H.M.; Tan, K.; Maltseva, N.I.; Chen, S.; Nicolaescu, V.; Dvorkin, S.; et al. Masitinib is a broad coronavirus 3CL inhibitor that blocks replication of SARS-CoV-2. Science 2021, 373, 931–936. [Google Scholar] [CrossRef]
- Zhang, C.H.; Stone, E.A.; Deshmukh, M.; Ippolito, J.A.; Ghahremanpour, M.M.; Tirado-Rives, J.; Spasov, K.A.; Zhang, S.; Takeo, Y.; Kudalkar, S.N.; et al. Potent Noncovalent Inhibitors of the Main Protease of SARS-CoV-2 from Molecular Sculpting of the Drug Perampanel Guided by Free Energy Perturbation Calculations. ACS Cent. Sci. 2021, 7, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Du, X.; Duan, Y.; Pan, X.; Sun, Y.; You, T.; Han, L.; Jin, Z.; Shang, W.; Yu, J.; et al. High-throughput screening identifies established drugs as SARS-CoV-2 PLpro inhibitors. Protein Cell 2021, 12, 877–888. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 24, 1586–1593. [Google Scholar] [CrossRef]
- Boras, B.; Jones, R.M.; Anson, B.J.; Arenson, D.; Aschenbrenner, L.; Bakowski, M.A.; Beutle, N.; Binder, J.; Chen, E.; Eng, H.; et al. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun. 2021, 12, 6055. [Google Scholar] [CrossRef]
- Vuong, W.; Khan, M.B.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.A.; McKay, R.T.; van Belkum, M.J.; Joyc, M.A.; et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020, 11, 4282. [Google Scholar] [CrossRef]
- Abe, K.; Kabe, Y.; Uchiyama, S.; Iwasaki, Y.W.; Ishizu, H.; Uwamino, Y.; Takenouch, T.; Uno, S.; Ishii, M.; Maruno, T.; et al. Pro108Ser mutation of SARS-CoV-2 3CLpro reduces the enzyme activity and ameliorates the clinical severity of COVID-19. Sci. Rep. 2022, 12, 1299. [Google Scholar] [CrossRef]
- Cross, T.J.; Takahashi, G.R.; Diessner, E.M.; Crosby, M.G.; Farahmand, V.; Zhuang, S.; Butts, C.T.; Martin, R.M. Sequence Characterization and Molecular Modeling of Clinically Relevant Variants of the SARS-CoV-2 Main Protease. Biochemistry 2020, 59, 3741–3756. [Google Scholar] [CrossRef] [PubMed]
- Thanigaimalai, P.; Konno, S.; Yamamoto, T.; Koiwai, Y.; Taguchi, A.; Takayama, K.; Yakushiji, F.; Akaji, T.; Chen, S.-N.; Naser-Tavakolian, A.; et al. Development of potent dipeptide-type SARS-CoV 3CL protease inhibitors with novel P3 scaffolds: Design, synthesis, biological evaluation, and docking studies. Eur. J. Med. Chem. 2013, 68, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Konno, S.; Kobayashi, K.; Senda, M.; Funai, Y.; Seki, Y.; Tamai, I.; Schäkel, L.; Sakata, K.; Pillaiyar, T.; Taguchi, A.; et al. 3CL Protease Inhibitors with an Electrophilic Arylketone Moiety as Anti-SARS-CoV-2 Agents. J. Med. Chem. 2022, 65, 2926–2939. [Google Scholar] [CrossRef] [PubMed]
- Hattori, S.I.; Higashi-Kuwata, N.; Hayashi, H.; Allu, S.R.; Raghavaiah, J.; Bulut, H.; Das, D.; Anson, B.J.; Lendy, E.K.; Takamatsu, Y.; et al. A small molecule compound with an indole moiety inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2021, 12, 668. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Sivaraman, J.; Song, J. Mechanism for controlling the dimer-monomer switch and coupling dimerization to catalysis of the severe acute respiratory syndrome coronavirus 3C-like protease. J. Virol. 2008, 82, 4620–4629. [Google Scholar] [CrossRef]
- Li, J.; Lin, C.; Zhou, X.; Zhong, F.; Zeng, P.; Yang, Y.; Zhang, Y.; Yu, B.; Fan, X.; McCormick, P.J.; et al. Structural Basis of the Main Proteases of Coronavirus Bound to Drug Candidate PF-07321332. J. Virol. 2022, 96, e0201321. [Google Scholar] [CrossRef]
- Li, J.; Lin, C.; Zhou, X.; Zhong, F.; Zeng, P.; McCormick, P.J.; Jiang, H.; Zhang, J. Structural Basis of Main Proteases of Coronavirus Bound to Drug Candidate PF-07304814. J. Mol. Biol. 2022, 434, 167706. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SARS-CoV-2 Mpro-YH-53 | SARS-CoV Mpro-YH-53 | MERS-CoV Mpro-YH-53 | |
|---|---|---|---|
| PDB code | 7XRS | 7YGQ | 7XRY |
| Data collection | |||
| Synchrotron | SSRF | SSRF | SSRF |
| Beam line | BL02U1 | BL02U1 | BL02U1 |
| Wavelength (Å) | 0.97918 | 0.97919 | 0.97918 |
| Space group | P1211 | P1 | P212121 |
| a,b,c (Å) | 55.56, 99.44, 59.67 | 55.06, 60.59, 68.30 | 80.02, 93.8, 102.16 |
| α,β,γ (°) | 90.00, 107.93, 90.00 | 90.20, 120.72, 108.43 | 90.00, 90.00, 90.00 |
| Total reflections | 252,466 | 174,371 | 518,857 |
| Unique reflections | 46,180 | 50,175 | 53,633 |
| Resolution (Å) | 1.93(2.03–1.93) | 2.04(2.15–2.04) | 1.99(2.09–1.99) |
| R-merge (%) | 5.7(49.4) | 2.8(31.6) | 6.2(92.6) |
| Mean I/σ (I) | 8.1/2.5 | 9.3/2.5 | 11.0/2.6 |
| Completeness (%) | 98.9(92.9) | 96.4(96.3) | 99.9(99.5) |
| Redundancy | 5.5(3.6) | 3.5(3.1) | 9.7(7.8) |
| Refinement | |||
| Resolution (Å) | 52.86–1.93 | 31.40–2.04 | 44.86–1.99 |
| Rwork/Rfree(%) | 19.96/24.31 | 20.54/23.50 | 22.36/25.35 |
| Atoms | 4667 | 4552 | 4800 |
| Mean temperature factor (Å2) | 33.9 | 46.8 | 29.0 |
| Bond lengths (Å) | 0.007 | 0.007 | 0.007 |
| Bond angles (°) | 0.952 | 1.034 | 0.92 |
| Preferred | 97.98 | 98.14 | 97.82 |
| Allowed | 2.02 | 1.86 | 2.18 |
| Outliers | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, X.; Lin, C.; Xu, Q.; Zhou, X.; Zeng, P.; McCormick, P.J.; Jiang, H.; Li, J.; Zhang, J. Structural Basis for the Inhibition of Coronaviral Main Proteases by a Benzothiazole-Based Inhibitor. Viruses 2022, 14, 2075. https://doi.org/10.3390/v14092075
Hu X, Lin C, Xu Q, Zhou X, Zeng P, McCormick PJ, Jiang H, Li J, Zhang J. Structural Basis for the Inhibition of Coronaviral Main Proteases by a Benzothiazole-Based Inhibitor. Viruses. 2022; 14(9):2075. https://doi.org/10.3390/v14092075
Chicago/Turabian StyleHu, Xiaohui, Cheng Lin, Qin Xu, Xuelan Zhou, Pei Zeng, Peter J. McCormick, Haihai Jiang, Jian Li, and Jin Zhang. 2022. "Structural Basis for the Inhibition of Coronaviral Main Proteases by a Benzothiazole-Based Inhibitor" Viruses 14, no. 9: 2075. https://doi.org/10.3390/v14092075
APA StyleHu, X., Lin, C., Xu, Q., Zhou, X., Zeng, P., McCormick, P. J., Jiang, H., Li, J., & Zhang, J. (2022). Structural Basis for the Inhibition of Coronaviral Main Proteases by a Benzothiazole-Based Inhibitor. Viruses, 14(9), 2075. https://doi.org/10.3390/v14092075