
Epidemiological and Evolutionary Analysis of West Nile Virus Lineage 2 in Italy

 ,
,  , , , , , , , , ,
, , , , , , , , ,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample and Data Collection
2.2. Sample Analysis
Tissue Homogenisation, RNA Extraction, and Real-Time RT-PCR
2.3. Epidemiological Data Analysis
2.4. Sequence Data Preparation and Retrieval
2.4.1. Whole Genome Sequencing, Genome Assembly, and Sequence Processing at NCBI
2.4.2. Download of Reference Genomes
2.4.3. Sequence Metadata Collection and Curation
2.4.4. Sequence Data Cleaning and Formatting
2.5. Phylogenetic Analysis
2.5.1. Subsampling for Molecular Clock Analysis
2.5.2. Alignment and Recombination Detection
2.5.3. Model Selection
2.5.4. Maximum-Likelihood Phylogenies
2.5.5. Molecular Clock
2.5.6. Phylogeographic and Phylodynamic Analysis of the Italian Clade of WNV L2
2.6. Ecological and Epidemiological Modelling
3. Results
3.1. Epidemiological Scenario
3.2. Genetic Scenario
3.2.1. Genome Sequence Analysis
3.2.2. World Scale Phylogenomics of WNV
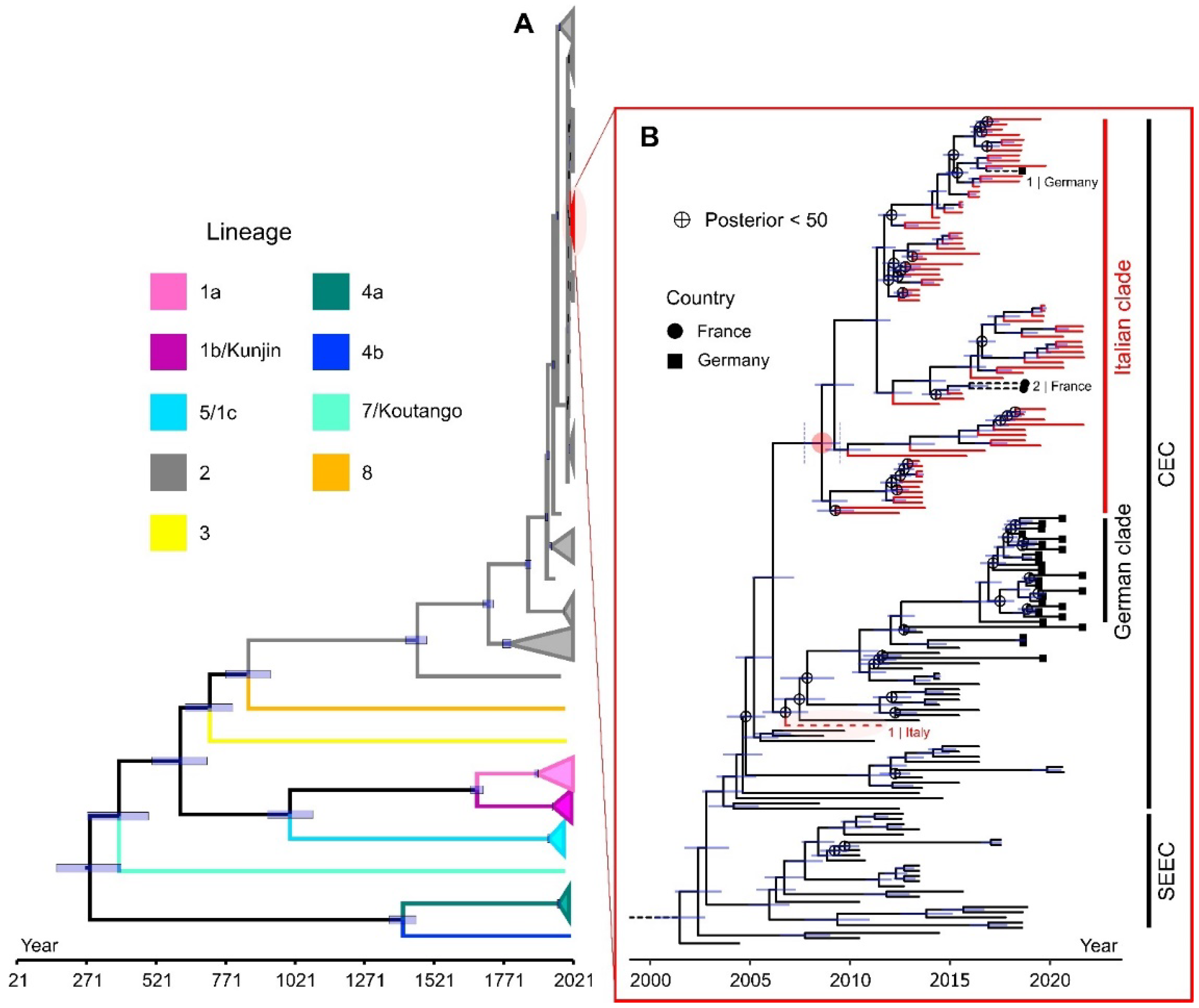
3.2.3. Phylodynamic and Phylogeographic Analysis of the WNV L2 Italian Clade
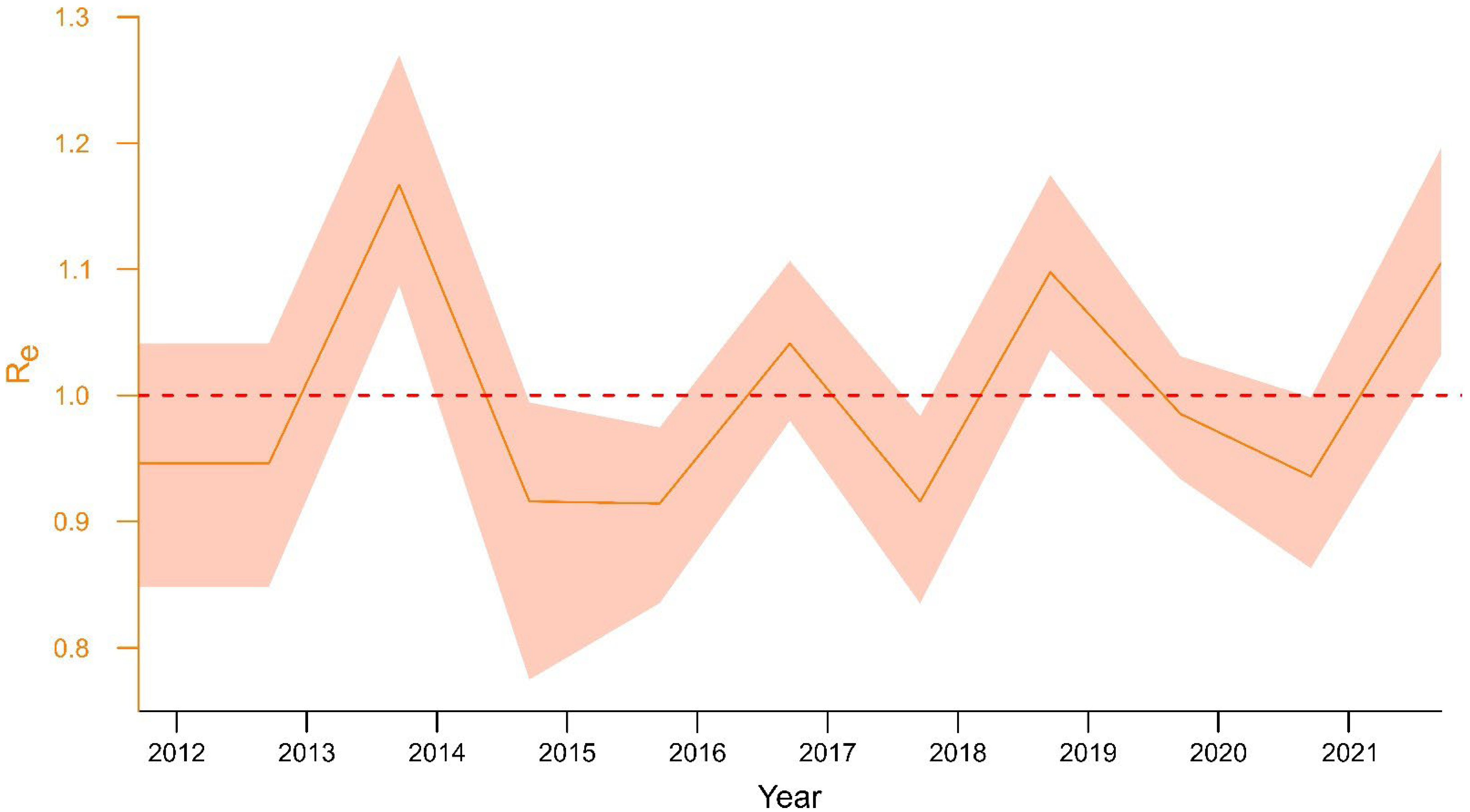
3.3. Epidemiological and Ecological Modelling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zannoli, S.; Sambri, V. West Nile Virus and Usutu Virus Co-Circulation in Europe: Epidemiology and Implications. Microorganisms 2019, 7, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chancey, C.; Grinev, A.; Volkova, E.; Rios, M. The Global Ecology and Epidemiology of West Nile Virus. BioMed Res. Int. 2015, 2015, 376230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzoli, A.; Jiménez-Clavero, M.A.; Barzon, L.; Cordioli, P.; Figuerola, J.; Koraka, P.; Martina, B.; Moreno, A.; Nowotny, N.; Pardigon, N.; et al. The challenge of West Nile virus in Europe: Knowledge gaps and research priorities. Eurosurveillance 2015, 20, 21135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mencattelli, G.; Ndione, M.H.D.; Rosà, R.; Marini, G.; Diagne, C.T.; Diagne, M.M.; Fall, G.; Faye, O.; Diallo, M.; Faye, O.; et al. Epidemiology of West Nile virus in Africa: An underestimated threat. PLoS Negl. Trop. Dis. 2022, 16, e0010075. [Google Scholar] [CrossRef]
- Byas, A.D.; Ebel, G.D. Comparative Pathology of West Nile Virus in Humans and Non-Human Animals. Pathogens 2020, 9, 48. [Google Scholar] [CrossRef] [Green Version]
- Komar, N.; Langevin, S.; Hinten, S.; Nemeth, N.; Edwards, E.; Hettler, D.; Davis, B.; Bowen, R.; Bunning, M. Experimental Infection of North American Birds with the New York 1999 Strain of West Nile Virus. Emerg. Infect. Dis. 2003, 9, 311–322. [Google Scholar] [CrossRef]
- Reisen, W.K.; Padgett, K.; Fang, Y.; Woods, L.; Foss, L.; Anderson, J.; Kramer, V. Chronic Infections of West Nile Virus Detected in California Dead Birds. Vector-Borne Zoonotic Dis. 2013, 13, 401–405. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, S.S.; Langevin, S.A.; Brault, A.C.; Woods, L.; Carroll, B.D.; Reisen, W.K. Detection of Persistent West Nile Virus RNA in Ex-perimentally and Naturally Infected Avian Hosts. Am. J. Trop. Med. Hyg. 2012, 87, 559–564. [Google Scholar] [CrossRef] [Green Version]
- Giglia, G.; Mencattelli, G.; Lepri, E.; Agliani, G.; Gobbi, M.; Gröne, A.; van den Brand, J.M.; Savini, G.; Mandara, M.T. West Nile Virus and Usutu Virus: A Post-Mortem Moni-toring Study in Wild Birds from Rescue Centers, Central Italy. Viruses 2022, 14, 1994. [Google Scholar] [CrossRef]
- Hubálek, Z.; Halouzka, J. West Nile Fever–a Reemerging Mosquito-Borne Viral Disease in Europe. Emerg. Infect. Dis. 1999, 5, 643–650. [Google Scholar] [CrossRef]
- Mancuso, E.; Cecere, J.G.; Iapaolo, F.; Di Gennaro, A.; Sacchi, M.; Savini, G.; Spina, F.; Monaco, F. West Nile and Usutu Virus Introduction via Mi-gratory Birds: A Retrospective Analysis in Italy. Viruses 2022, 14, 416. [Google Scholar] [CrossRef] [PubMed]
- Paré, J.; Moore, A. West Nile virus in horses—What do you need to know to diagnose the disease? Can. Vet. J. 2018, 59, 1119–1120. [Google Scholar] [PubMed]
- Silva, A.S.G.; Matos, A.C.D.; da Cunha, M.A.C.R.; Rehfeld, I.S.; Galinari, G.C.F.; Marcelino, S.A.C.; Saraiva, L.H.G.; Martins, N.R.D.S.; Maranhão, R.D.P.A.; Lobato, Z.I.P.; et al. West Nile virus associated with equid encephalitis in Brazil, 2018. Transbound. Emerg. Dis. 2018, 66, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riccò, M.; Peruzzi, S.; Balzarini, F. Epidemiology of West Nile Virus Infections in Humans, Italy, 2012-2020: A Summary of Available Evidences. Trop Med Infect Dis 2021, 6, 61. [Google Scholar] [CrossRef]
- Calistri, P.; Giovannini, A.; Hubalek, Z.; Ionescu, A.; Monaco, F.; Savini, G.; Lelli, R. Epidemiology of West Nile in Europe and in the Mediterranean Basin. Open Virol. J. 2010, 4, 29–37. [Google Scholar] [CrossRef] [Green Version]
- García-Carrasco, J.M.; Muñoz, A.R.; Olivero, J.; Segura, M.; Real, R. Predicting the spatio-temporal spread of West Nile virus in Eu-rope. PLoS Negl. Trop. Dis. 2021, 15, e0009022. [Google Scholar] [CrossRef]
- Fall, G.; Di Paola, N.; Faye, M.; Dia, M.; Freire, C.C.D.M.; Loucoubar, C.; Zanotto, P.M.D.A.; Faye, O.; Sall, A.A. Biological and phylogenetic characteristics of West African lineages of West Nile virus. PLoS Negl. Trop. Dis. 2017, 11, e0006078. [Google Scholar] [CrossRef] [Green Version]
- Mencattelli, G.; Iapaolo, F.; Monaco, F.; Fusco, G.; de Martinis, C.; Portanti, O.; Di Gennaro, A.; Curini, V.; Polci, A.; Berjaoui, S.; et al. West Nile Virus Lineage 1 in Italy: Newly In-troduced or a Re-Occurrence of a Previously Circulating Strain? Viruses 2022, 14, 64. [Google Scholar] [CrossRef]
- Bakonyi, T.; Ferenczi, E.; Erdélyi, K.; Kutasi, O.; Csörgő, T.; Seidel, B.; Weissenböck, H.; Brugger, K.; Bán, E.; Nowotny, N. Explosive spread of a neuroinvasive lineage 2 West Nile virus in Central Europe, 2008/2009. Veter-Microbiol. 2013, 165, 61–70. [Google Scholar] [CrossRef]
- Beck, C.; Goffart, I.L.; Franke, F.; Gonzalez, G.; Dumarest, M.; Lowenski, S.; Blanchard, Y.; Lucas, P.; Lamballerie, X.; Grard, G.; et al. Contrasted Epidemiological Patterns of West Nile Virus Lineages 1 and 2 Infections in France from 2015 to 2019. Pathogens 2020, 9, 908. [Google Scholar] [CrossRef]
- Veo, C.; della Ventura, C.; Moreno, A.; Rovida, F.; Percivalle, E.; Canziani, S.; Torri, D.; Calzolari, M.; Baldanti, F.; Galli, M.; et al. Evolutionary Dynamics of the Lineage 2 West Nile Virus That Caused the Largest European Epidemic: Italy 2011–2018. Viruses 2019, 11, 814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autorino, G.L.; Battisti, A.; Deubel, V.; Ferrari, G.; Forletta, R.; Giovannini, A.; Lelli, R.; Murri, S.; Scicluna, M.T. West Nile virus Epidemic in Horses, Tuscany Region, Italy. Emerg. Infect. Dis. 2002, 8, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Rizzoli, A.; Rosà, R.; Rosso, F.; Buckley, A.; Gould, E. West Nile Virus Circulation Detected in Northern Italy in Sentinel Chickens. Vector-Borne Zoonotic Dis. 2007, 7, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Monaco, F.; Lelli, R.; Teodori, L.; Pinoni, C.; Di Gennaro, A.; Polci, A.; Calistri, P.; Savini, G. Re-Emergence of West Nile Virus in Italy. Zoonoses Public Health 2009, 57, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Bagnarelli, P.; Marinelli, K.; Trotta, D.; Monachetti, A.; Tavio, M.; Del Gobbo, R.; Capobianchi, M.R.; Menzo, S.; Nicoletti, L.; Magurano, F.; et al. case of autochthonous West Nile virus lineage 2 infection in Italy, September 2011. Eurosurveillance 2011, 16, 20002. [Google Scholar] [CrossRef]
- Savini, G.; Capelli, G.; Monaco, F.; Polci, A.; Russo, F.; Di Gennaro, A.; Marini, V.; Teodori, L.; Montarsi, F.; Pinoni, C.; et al. Evidence of West Nile virus lineage 2 circulation in Northern Italy. Veter-Microbiol. 2012, 158, 267–273. [Google Scholar] [CrossRef]
- Barzon, L.; Montarsi, F.; Quaranta, E.; Monne, I.; Pacenti, M.; Michelutti, A.; Toniolo, F.; Danesi, P.; Marchetti, G.; Gobbo, F.; et al. Early start of seasonal transmission and co-circulation of West Nile virus lineage 2 and a newly introduced lineage 1 strain, northern Italy, June 2022. Eurosurveillance 2022, 27, 2200548. [Google Scholar] [CrossRef]
- Candeloro, L.; Ippoliti, C.; Iapaolo, F.; Monaco, F.; Morelli, D.; Cuccu, R.; Fronte, P.; Calderara, S.; Vincenzi, S.; Porrello, A.; et al. Predicting WNV Circulation in Italy Using Earth Ob-servation Data and Extreme Gradient Boosting Model. Remote Sensing 2020, 12, 3064. [Google Scholar] [CrossRef]
- Rizzo, C.; Napoli, C.; Venturi, G.; Pupella, S.; Lombardini, L.; Calistri, P.; Monaco, F.; Cagarelli, R.; Angelini, P.; Bellini, R.; et al. West Nile virus transmission: Results from the integrated surveillance system in Italy, 2008 to 2015. Eurosurveillance 2016, 21, 30340. [Google Scholar] [CrossRef] [Green Version]
- Colangeli, P.; Iannetti, S.; Cerella, A.; Ippoliti, C.; Di Lorenzo, A.; Santucci, U.; Simonetti, P.; Calistri, P.; Lelli, R. The national information system for the notification of animal diseases in Italy. Veter-Ital. 2011, 47, 303–312. [Google Scholar]
- Del Amo, J.; Sotelo, E.; Fernández-Pinero, J.; Gallardo, C.; Llorente, F.; Agüero, M.; Jiménez-Clavero, M.A. A novel quantitative multiplex real-time RT-PCR for the simultaneous detection and differentiation of West Nile virus lineages 1 and 2, and of Usutu virus. J. Virol. Methods 2013, 189, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Calc|LibreOffice—Free Office Suite—Based on OpenOffice—Compatible with Microsoft. Available online: https://www.libreoffice.org/discover/calc/ (accessed on 2 November 2022).
- Microsoft Excel, Software di Foglio di Calcolo|Microsoft 365. Available online: https://www.microsoft.com/it-it/microsoft-365/excel (accessed on 2 November 2022).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; Available online: https://www.R-project.org/ (accessed on 2 November 2022).
- Mencattelli, G.; Iapaolo, F.; Polci, A.; Marcacci, M.; Di Gennaro, A.; Teodori, L.; Curini, V.; Di Lollo, V.; Secondini, B.; Scialabba, S.; et al. West Nile Virus Lineage 2 Overwintering in Italy. Trop. Med. Infect. Dis. 2022, 7, 160. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Bolyen, E.; Dillon, M.R.; Bokulich, N.A.; Ladner, J.T.; Larsen, B.B.; Hepp, C.M.; Lemmer, D.; Sahl, J.W.; Sanchez, A.; Holdgraf, C.; et al. Reproducibly sampling SARS-CoV-2 genomes across time, geography, and viral diversity. F1000Research 2020, 9, 657. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phyloge-netic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus ge-nomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phy-logenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Ef-ficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data inte-gration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, U.; Santos, P.D.; Groschup, M.H.; Hattendorf, C.; Eiden, M.; Höper, D.; Eisermann, P.; Keller, M.; Michel, F.; Klopfleisch, R.; et al. West Nile Virus Epidemic in Germany Triggered by Epizootic Emergence, 2019. Viruses 2020, 12, 448. [Google Scholar] [CrossRef] [Green Version]
- Reisen, W.K.; Wheeler, S.S. Overwintering of West Nile Virus in the United States. J. Med. Èntomol. 2019, 56, 1498–1507. [Google Scholar] [CrossRef]
- Marini, G.; Rosà, R.; Pugliese, A.; Rizzoli, A.; Rizzo, C.; Russo, F.; Montarsi, F.; Capelli, G. West Nile virus transmission and human infection risk in Veneto (Italy): A modelling analysis. Sci. Rep. 2018, 8, 14005. [Google Scholar] [CrossRef] [Green Version]
- Bakonyi, T.; Ivanics, É.; Erdélyi, K.; Ursu, K.; Ferenczi, E.; Weissenböck, H.; Nowotny, N. Lineage 1 and 2 Strains of Encephalitic West Nile Virus, Central Europe. Emerg. Infect. Dis. 2006, 12, 618–623. [Google Scholar] [CrossRef]
- Marini, G.; Calzolari, M.; Angelini, P.; Bellini, R.; Bellini, S.; Bolzoni, L.; Torri, D.; Defilippo, F.; Dorigatti, I.; Nikolay, B.; et al. A quantitative comparison of West Nile virus incidence from 2013 to 2018 in Emilia-Romagna, Italy. PLoS Neglected Trop. Dis. 2020, 14, e0007953. [Google Scholar] [CrossRef] [Green Version]
- Marini, G.; Manica, M.; Delucchi, L.; Pugliese, A.; Rosà, R. Spring temperature shapes West Nile virus transmission in Europe. Acta Trop. 2020, 215, 105796. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mencattelli, G.; Silverj, A.; Iapaolo, F.; Ippoliti, C.; Teodori, L.; Di Gennaro, A.; Curini, V.; Candeloro, L.; Conte, A.; Polci, A.; et al. Epidemiological and Evolutionary Analysis of West Nile Virus Lineage 2 in Italy. Viruses 2023, 15, 35. https://doi.org/10.3390/v15010035
Mencattelli G, Silverj A, Iapaolo F, Ippoliti C, Teodori L, Di Gennaro A, Curini V, Candeloro L, Conte A, Polci A, et al. Epidemiological and Evolutionary Analysis of West Nile Virus Lineage 2 in Italy. Viruses. 2023; 15(1):35. https://doi.org/10.3390/v15010035
Chicago/Turabian StyleMencattelli, Giulia, Andrea Silverj, Federica Iapaolo, Carla Ippoliti, Liana Teodori, Annapia Di Gennaro, Valentina Curini, Luca Candeloro, Annamaria Conte, Andrea Polci, and et al. 2023. "Epidemiological and Evolutionary Analysis of West Nile Virus Lineage 2 in Italy" Viruses 15, no. 1: 35. https://doi.org/10.3390/v15010035