
Oncolytic Measles Virus Encoding MicroRNA for Targeted RNA Interference †
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
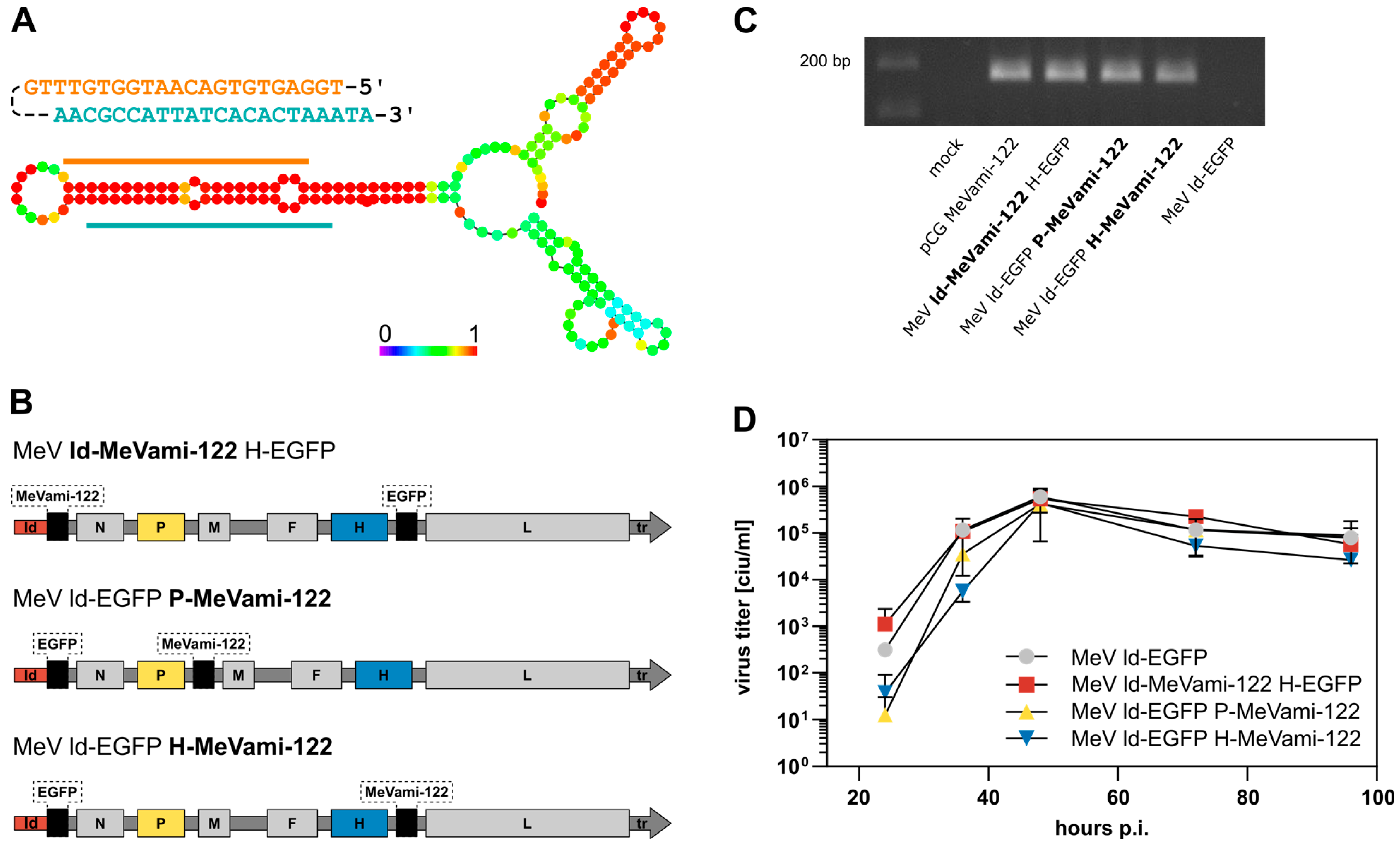
3.1. Genetic Design, Transgene Expression, and Replication Kinetics of miR-122-Encoding MeV
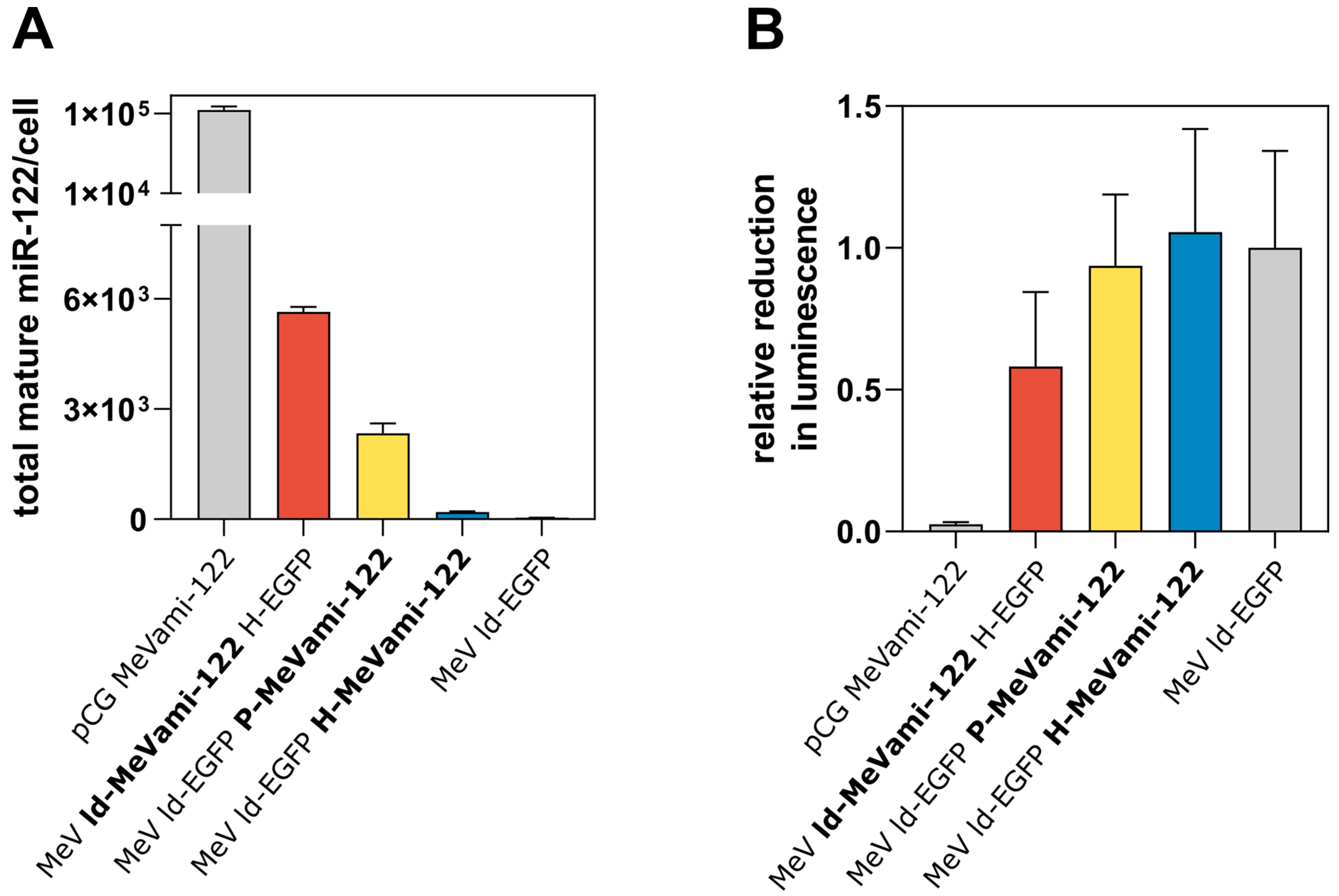
3.2. Cells Infected with miRNA-Encoding MeV Produce Functional miRNAs, Albeit at Comparatively Low Copy Numbers
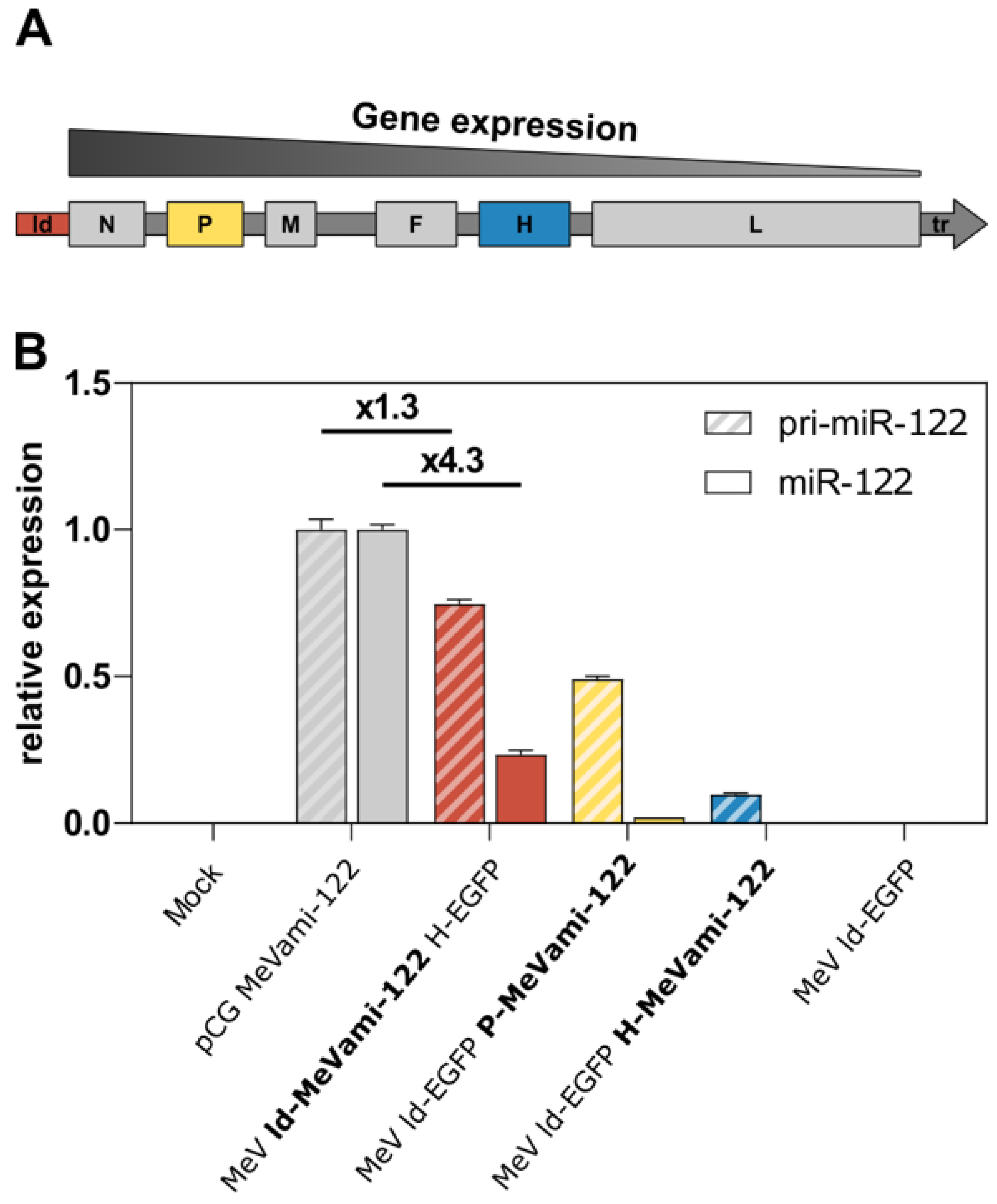
3.3. Limited Processing of MeV-Encoded miRNA
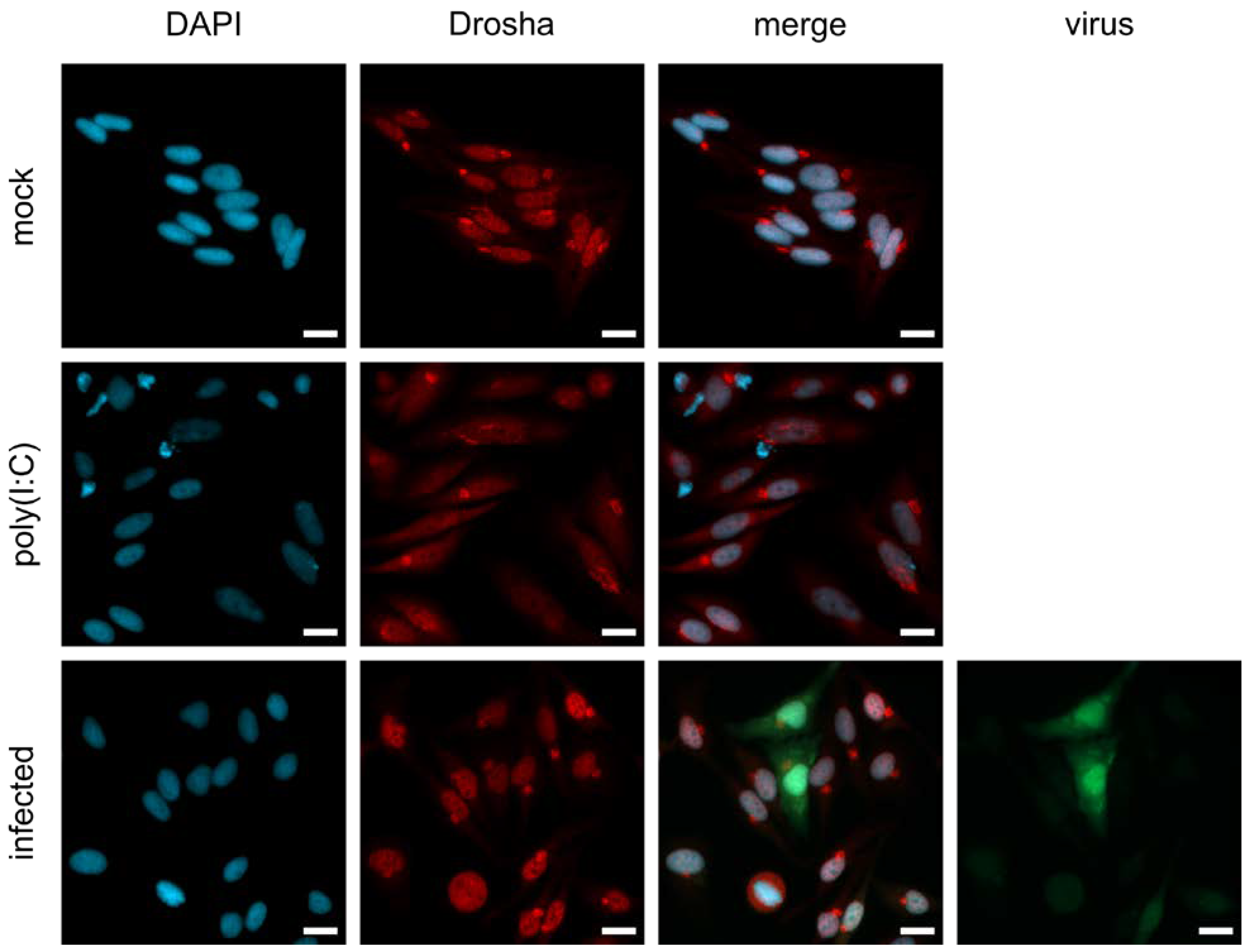
3.4. Nuclear Localization of Drosha in MeV-Infected Cells Might Be Responsible for the Limited Processing of MeV-Derived miRNA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kelly, E.; Russell, S.J. History of oncolytic viruses: Genesis to genetic engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Macedo, N.; Miller, D.M.; Haq, R.; Kaufman, H.L. Clinical landscape of oncolytic virus research in 2020. J. Immunother. Cancer 2020, 8, e001486. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leber, M.F.; Neault, S.; Jirovec, E.; Barkley, R.; Said, A.; Bell, J.C.; Ungerechts, G. Engineering and combining oncolytic measles virus for cancer therapy. Cytokine Growth Factor Rev. 2020, 56, 39–48. [Google Scholar] [CrossRef]
- Cattaneo, R.; Miest, T.; Shashkova, E.V.; Barry, M.A. Reprogrammed viruses as cancer therapeutics: Targeted, armed and shielded. Nat. Rev. Microbiol. 2008, 6, 529–540. [Google Scholar] [CrossRef]
- Ungerechts, G.; Springfeld, C.; Frenzke, M.E.; Lampe, J.; Johnston, P.B.; Parker, W.B.; Sorscher, E.J.; Cattaneo, R. Lymphoma chemovirotherapy: CD20-targeted and convertase-armed measles virus can synergize with fludarabine. Cancer Res. 2007, 67, 10939–10947. [Google Scholar] [CrossRef] [Green Version]
- Zaoui, K.; Bossow, S.; Grossardt, C.; Leber, M.F.; Springfeld, C.; Plinkert, P.K.; Kalle, C.; Ungerechts, G. Chemovirotherapy for head and neck squamous cell carcinoma with EGFR-targeted and CD/UPRT-armed oncolytic measles virus. Cancer Gene Ther. 2012, 19, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Grossardt, C.; Engeland, C.E.; Bossow, S.; Halama, N.; Zaoui, K.; Leber, M.F.; Springfeld, C.; Jaeger, D.; von Kalle, C.; Ungerechts, G. Granulocyte-macrophage colony-stimulating factor-armed oncolytic measles virus is an effective therapeutic cancer vaccine. Hum. Gene Ther. 2013, 24, 644–654. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.M.; Leber, M.F.; Bossow, S.; Engeland, C.E.; Dessila, J.; Grossardt, C.; Zaoui, K.; Bell, J.C.; Jager, D.; von Kalle, C.; et al. MicroRNA-sensitive oncolytic measles virus for chemovirotherapy of pancreatic cancer. Mol. Ther. Oncolytics 2021, 21, 340–355. [Google Scholar] [CrossRef]
- O’Cathail, S.M.; Pokrovska, T.D.; Maughan, T.S.; Fisher, K.D.; Seymour, L.W.; Hawkins, M.A. Combining Oncolytic Adenovirus with Radiation-A Paradigm for the Future of Radiosensitization. Front. Oncol. 2017, 7, 153. [Google Scholar] [CrossRef]
- Engeland, C.E.; Grossardt, C.; Veinalde, R.; Bossow, S.; Lutz, D.; Kaufmann, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W.; et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. 2014, 22, 1949–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langlois, R.A.; Shapiro, J.S.; Pham, A.M.; tenOever, B.R. In vivo delivery of cytoplasmic RNA virus-derived miRNAs. Mol. Ther. 2012, 20, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Rouha, H.; Thurner, C.; Mandl, C.W. Functional microRNA generated from a cytoplasmic RNA virus. Nucleic Acids Res. 2010, 38, 8328–8337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, J.S.; Varble, A.; Pham, A.M.; Tenoever, B.R. Noncanonical cytoplasmic processing of viral microRNAs. RNA 2010, 16, 2068–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varble, A.; Chua, M.A.; Perez, J.T.; Manicassamy, B.; Garcia-Sastre, A.; tenOever, B.R. Engineered RNA viral synthesis of microRNAs. Proc. Natl. Acad. Sci. USA 2010, 107, 11519–11524. [Google Scholar] [CrossRef] [Green Version]
- Engeland, C.E.; Ungerechts, G. Measles Virus as an Oncolytic Immunotherapy. Cancers 2021, 13, 544. [Google Scholar] [CrossRef]
- Leber, M.F.; Baertsch, M.A.; Anker, S.C.; Henkel, L.; Singh, H.M.; Bossow, S.; Engeland, C.E.; Barkley, R.; Hoyler, B.; Albert, J.; et al. Enhanced Control of Oncolytic Measles Virus Using MicroRNA Target Sites. Mol. Ther. Oncolyt. 2018, 9, 30–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, W.; Chen, Q.; Ma, L.; Liu, J.; Yang, Z.; Shen, J.; Cui, Y.; Bian, X.W.; Qian, C. Oncolytic adenovirus co-expressing miRNA-34a and IL-24 induces superior antitumor activity in experimental tumor model. J. Mol. Med. 2013, 91, 715–725. [Google Scholar] [CrossRef]
- Ruiz, A.J.; Russell, S.J. MicroRNAs and oncolytic viruses. Curr. Opin. Virol. 2015, 13, 40–48. [Google Scholar] [CrossRef]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef]
- Gottwein, E.; Cullen, B.R. Viral and cellular microRNAs as determinants of viral pathogenesis and immunity. Cell Host Microbe 2008, 3, 375–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, B.R. Five questions about viruses and microRNAs. PLoS Pathog. 2010, 6, e1000787. [Google Scholar] [CrossRef] [Green Version]
- tenOever, B.R. RNA viruses and the host microRNA machinery. Nat. Rev. Microbiol. 2013, 11, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.S.; Langlois, R.A.; Pham, A.M.; Tenoever, B.R. Evidence for a cytoplasmic microprocessor of pri-miRNAs. RNA 2012, 18, 1338–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Calain, P.; Roux, L. The rule of six, a basic feature for efficient replication of Sendai virus defective interfering RNA. J. Virol. 1993, 67, 4822–4830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noland, C.L.; Doudna, J.A. Multiple sensors ensure guide strand selection in human RNAi pathways. RNA 2013, 19, 639–648. [Google Scholar] [CrossRef] [Green Version]
- RNAfold. MFE Secondary Structure miR-122. Available online: http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi?PAGE=3&ID=_Z6hlBsKyP (accessed on 21 October 2022).
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neubock, R.; Hofacker, I.L. The Vienna RNA websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef] [Green Version]
- Leber, M.F.; Bossow, S.; Leonard, V.H.; Zaoui, K.; Grossardt, C.; Frenzke, M.; Miest, T.; Sawall, S.; Cattaneo, R.; von Kalle, C.; et al. MicroRNA-sensitive oncolytic measles viruses for cancer-specific vector tropism. Mol. Ther. 2011, 19, 1097–1106. [Google Scholar] [CrossRef]
- Takeda, M.; Nakatsu, Y.; Ohno, S.; Seki, F.; Tahara, M.; Hashiguchi, T.; Yanagi, Y. Generation of measles virus with a segmented RNA genome. J. Virol. 2006, 80, 4242–4248. [Google Scholar] [CrossRef]
- Shapiro, J.S.; Schmid, S.; Aguado, L.C.; Sabin, L.R.; Yasunaga, A.; Shim, J.V.; Sachs, D.; Cherry, S.; tenOever, B.R. Drosha as an interferon-independent antiviral factor. Proc. Natl. Acad. Sci. USA 2014, 111, 7108–7113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, S.; Grund, S.E.; Diederichs, S. Alternative splicing affects the subcellular localization of Drosha. Nucleic Acids Res. 2016, 44, 5330–5343. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.D.; Gentner, B.; Cantore, A.; Colleoni, S.; Amendola, M.; Zingale, A.; Baccarini, A.; Lazzari, G.; Galli, C.; Naldini, L. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat. Biotechnol. 2007, 25, 1457–1467. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Pfaller, C.K.; Radeke, M.J.; Cattaneo, R.; Samuel, C.E. Measles virus C protein impairs production of defective copyback double-stranded viral RNA and activation of protein kinase R. J. Virol. 2014, 88, 456–468. [Google Scholar] [CrossRef] [Green Version]
- Ostertag, D.; Hoblitzell-Ostertag, T.M.; Perrault, J. Overproduction of double-stranded RNA in vesicular stomatitis virus-infected cells activates a constitutive cell-type-specific antiviral response. J. Virol. 2007, 81, 503–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maillard, P.V.; Ciaudo, C.; Marchais, A.; Li, Y.; Jay, F.; Ding, S.W.; Voinnet, O. Antiviral RNA interference in mammalian cells. Science 2013, 342, 235–238. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Li, M.; Tucker, L.; Ramratnam, B. Glycogen synthase kinase 3 beta (GSK3beta) phosphorylates the RNAase III enzyme Drosha at S300 and S302. PLoS ONE 2011, 6, e20391. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, Y.; Tucker, L.; Ramratnam, B. Phosphorylation of the RNase III enzyme Drosha at Serine300 or Serine302 is required for its nuclear localization. Nucleic Acids Res. 2010, 38, 6610–6619. [Google Scholar] [CrossRef]
- Cifuentes, D.; Xue, H.; Taylor, D.W.; Patnode, H.; Mishima, Y.; Cheloufi, S.; Ma, E.; Mane, S.; Hannon, G.J.; Lawson, N.D.; et al. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science 2010, 328, 1694–1698. [Google Scholar] [CrossRef]
- Okamura, K.; Hagen, J.W.; Duan, H.; Tyler, D.M.; Lai, E.C. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell 2007, 130, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plumet, S.; Herschke, F.; Bourhis, J.M.; Valentin, H.; Longhi, S.; Gerlier, D. Cytosolic 5′-triphosphate ended viral leader transcript of measles virus as activator of the RIG I-mediated interferon response. PLoS ONE 2007, 2, e279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikegame, S.; Takeda, M.; Ohno, S.; Nakatsu, Y.; Nakanishi, Y.; Yanagi, Y. Both RIG-I and MDA5 RNA helicases contribute to the induction of alpha/beta interferon in measles virus-infected human cells. J. Virol. 2010, 84, 372–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchie, A. First microRNA mimic enters clinic. Nat. Biotechnol. 2013, 31, 577. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anker, S.C.; Szczeponik, M.G.; Dessila, J.; Dittus, K.; Engeland, C.E.; Jäger, D.; Ungerechts, G.; Leber, M.F. Oncolytic Measles Virus Encoding MicroRNA for Targeted RNA Interference. Viruses 2023, 15, 308. https://doi.org/10.3390/v15020308
Anker SC, Szczeponik MG, Dessila J, Dittus K, Engeland CE, Jäger D, Ungerechts G, Leber MF. Oncolytic Measles Virus Encoding MicroRNA for Targeted RNA Interference. Viruses. 2023; 15(2):308. https://doi.org/10.3390/v15020308
Chicago/Turabian StyleAnker, Sophie C., Marie G. Szczeponik, Jan Dessila, Katia Dittus, Christine E. Engeland, Dirk Jäger, Guy Ungerechts, and Mathias F. Leber. 2023. "Oncolytic Measles Virus Encoding MicroRNA for Targeted RNA Interference" Viruses 15, no. 2: 308. https://doi.org/10.3390/v15020308