
Specialized DNA Structures Act as Genomic Beacons for Integration by Evolutionarily Diverse Retroviruses
 , , ,
, , ,
Abstract
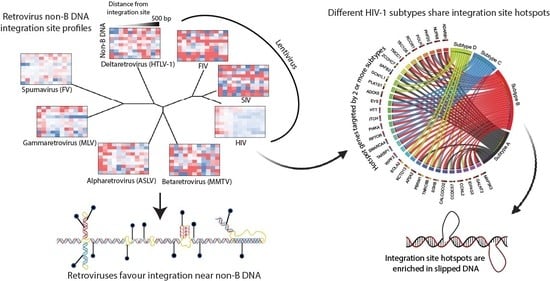
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
3.1. Integration Site Dataset Acquisition and Analyses
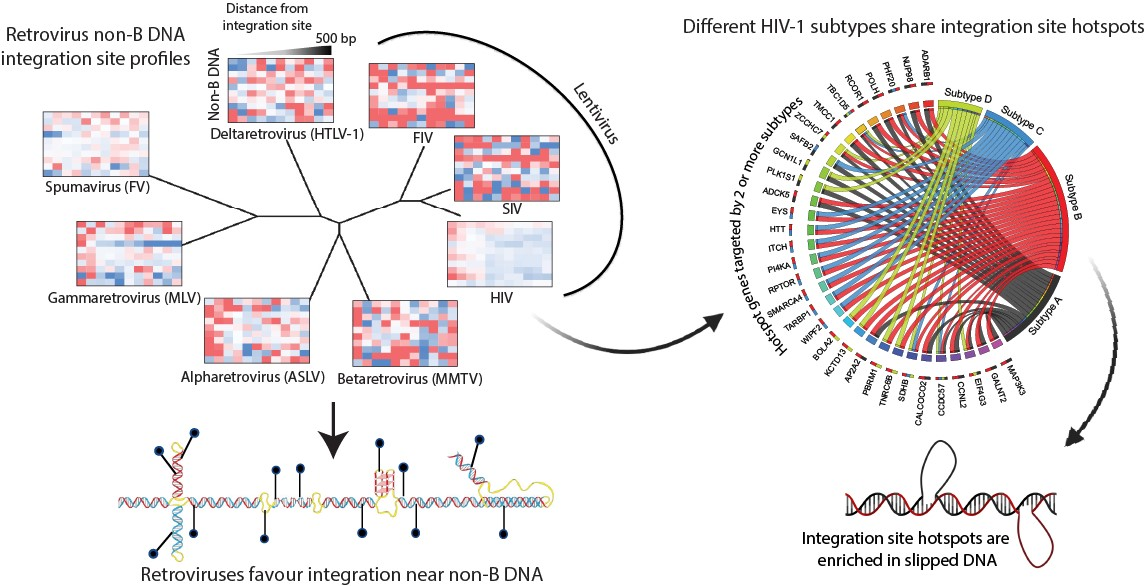
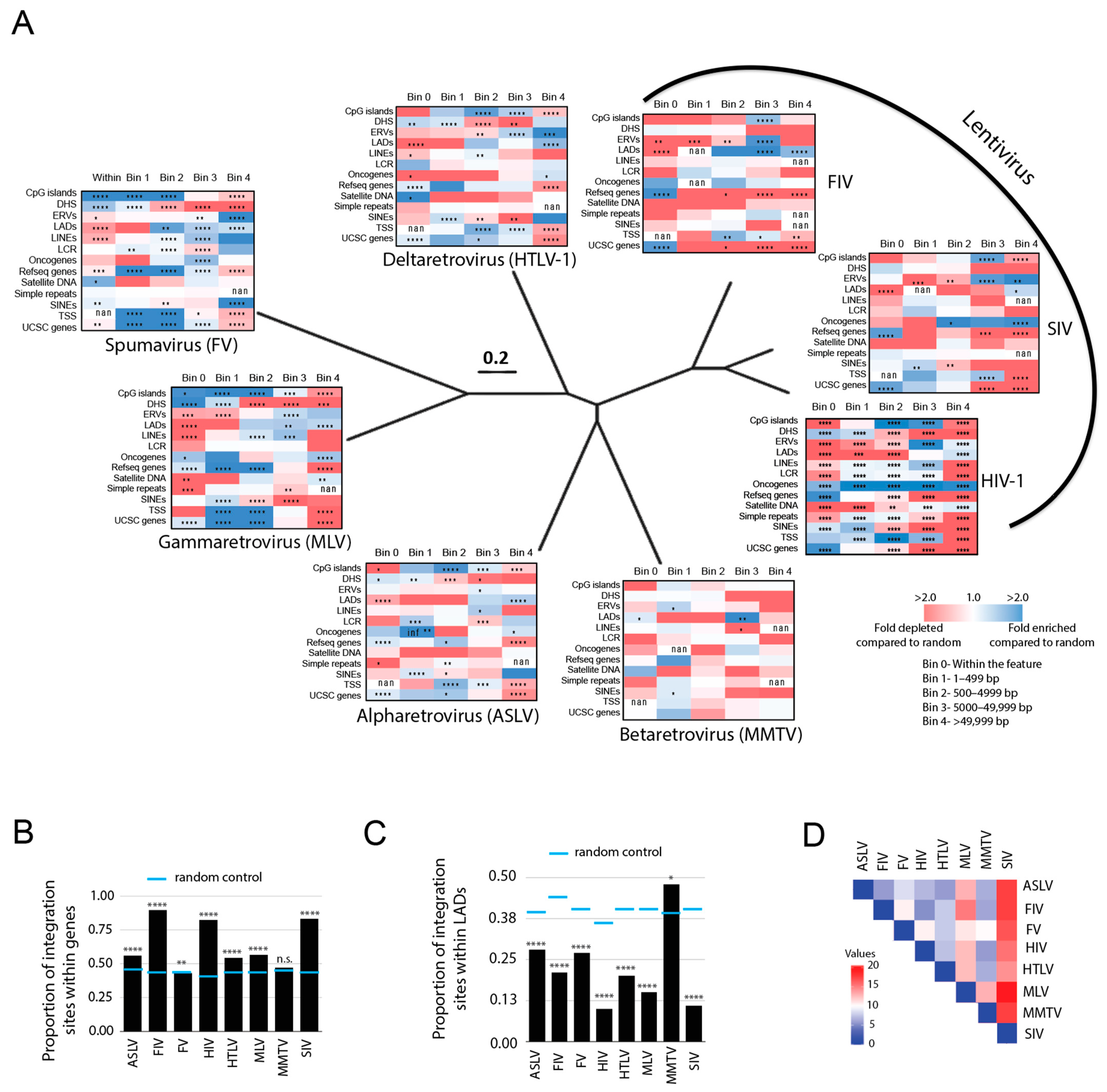
3.2. Evolutionarily Divergent Retroviruses Exhibit Distinct Integration Site Profiles
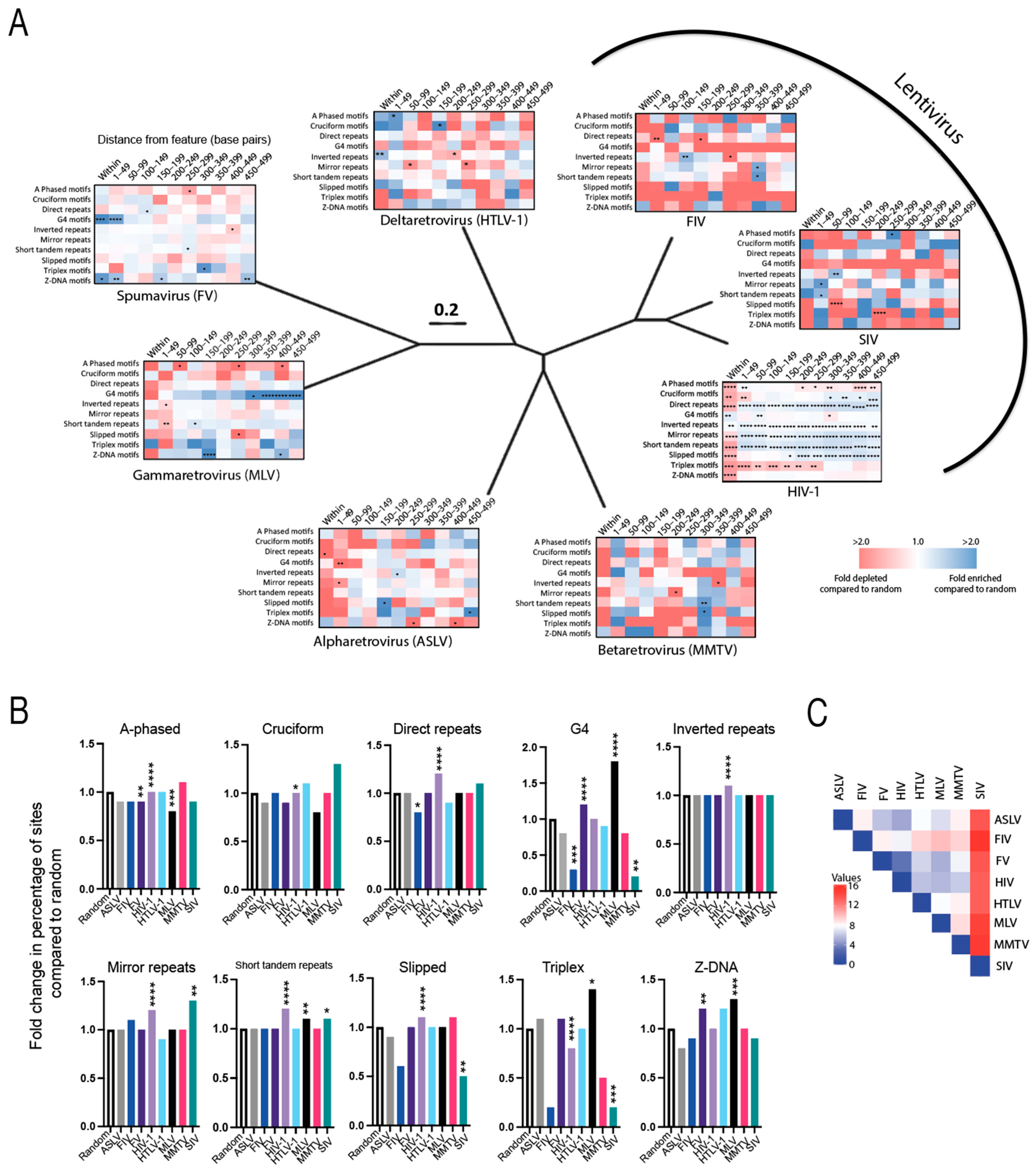
3.3. Evolutionarily Divergent Retroviruses Target Non-B DNA for Integration
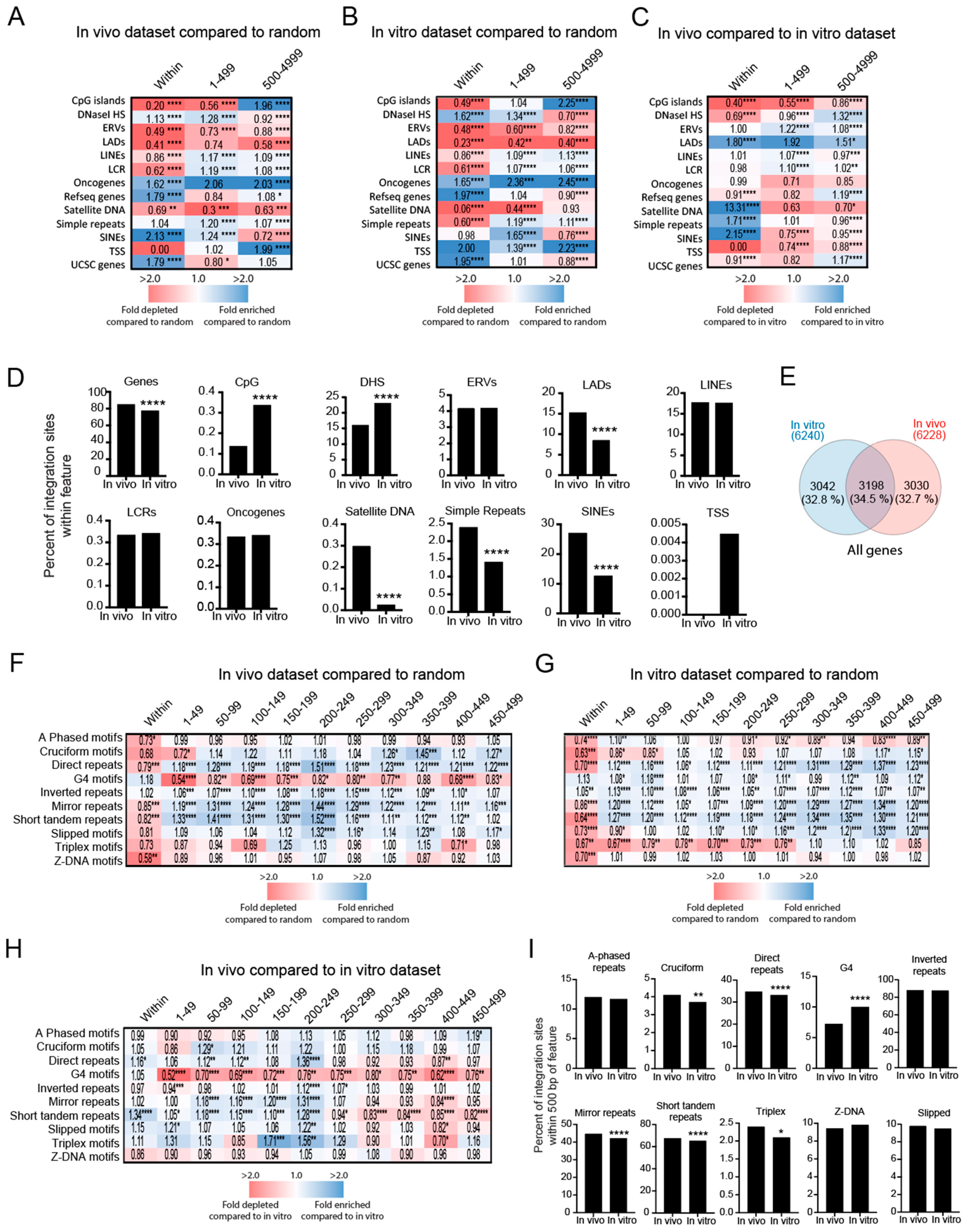
3.4. HIV-1 Integration Site Profiles Differ between In Vitro- and In Vivo-Derived Datasets
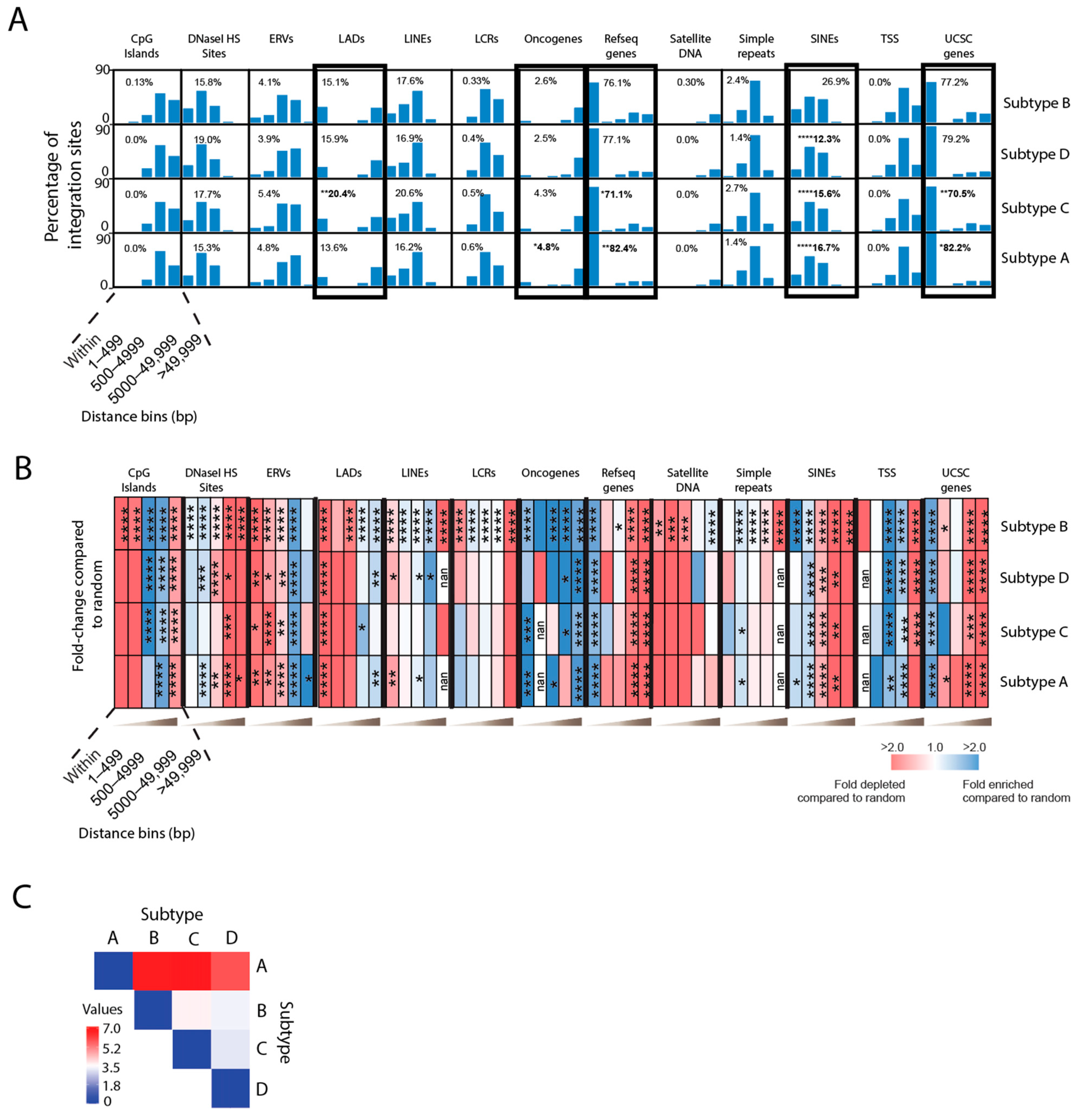
3.5. Integration Site Profiles Differ in Individuals Infected with HIV-1 Subtype A, B, C or D
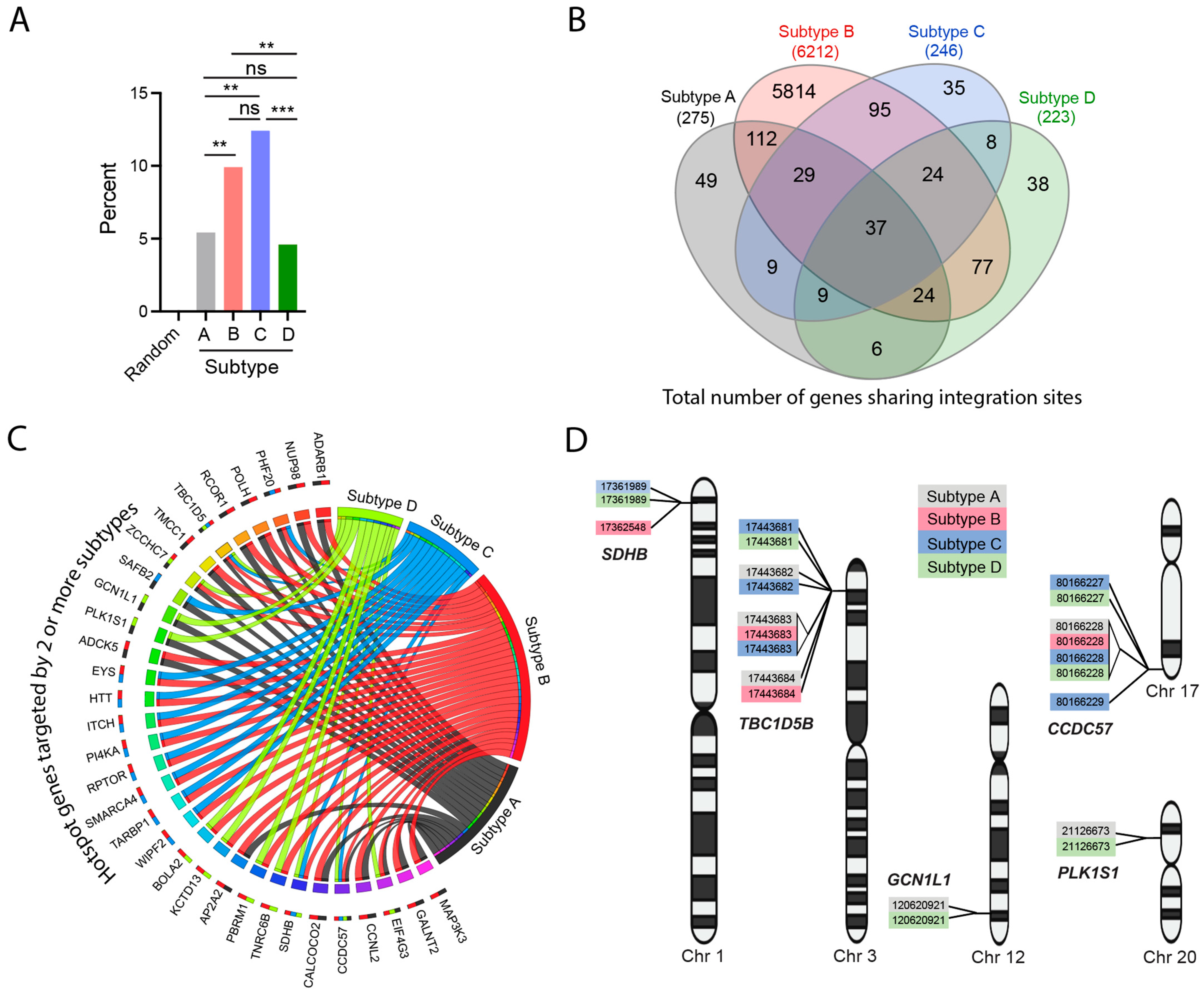
3.6. Integration Site Hotspots Are Shared between HIV-1 Subtypes
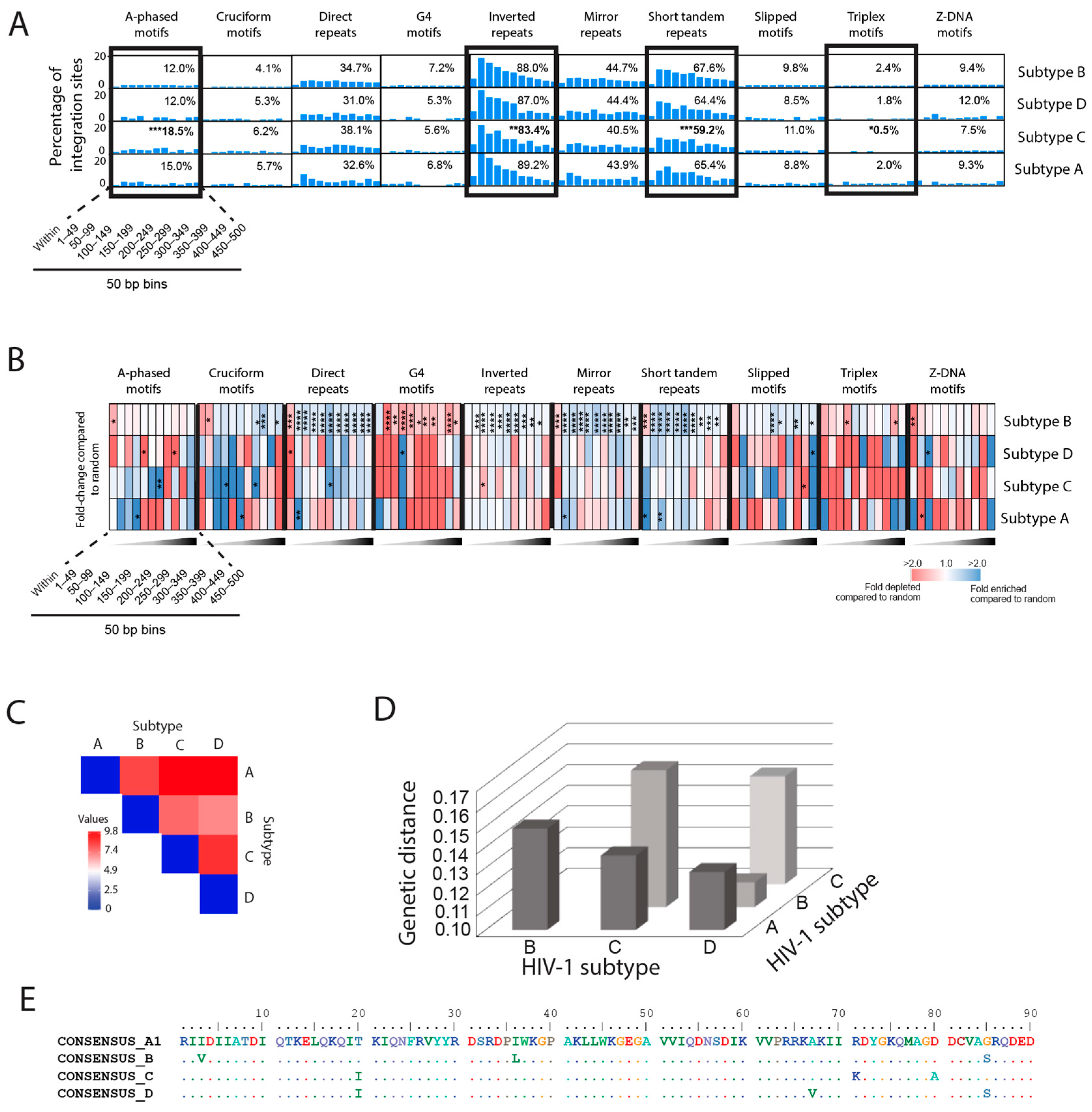
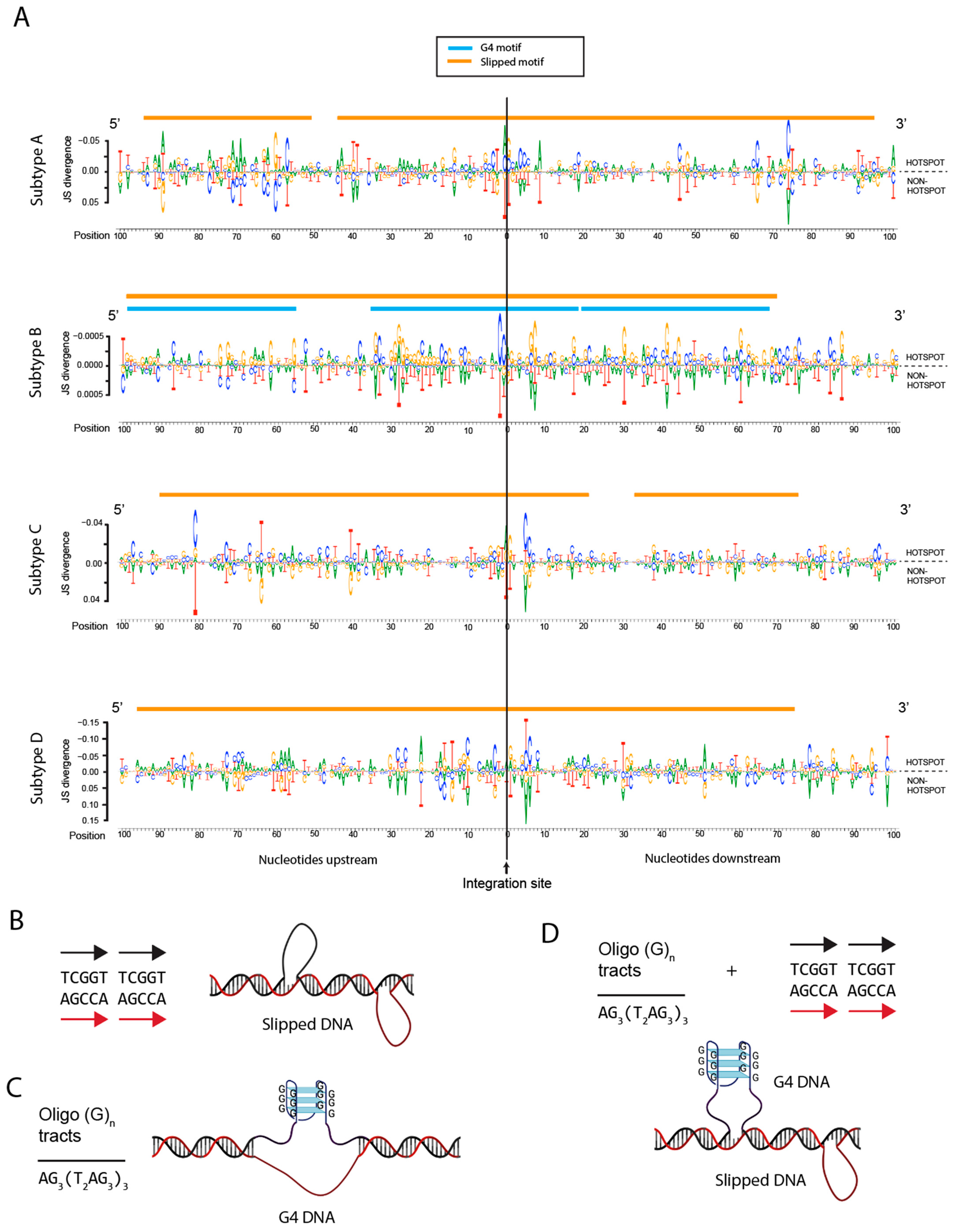
3.7. Integration Site Hotspots Are Located in Slipped DNA Motifs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daniel, R.; Greger, J.G.; Katz, R.A.; Taganov, K.D.; Wu, X.; Kappes, J.C.; Skalka, A.M. Evidence That Stable Retroviral Transduction and Cell Survival Following DNA Integration Depend on Components of the Nonhomologous End Joining Repair Pathway. J. Virol. 2004, 78, 8573–8581. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M. Transcription Start Regions in the Human Genome Are Favored Targets for MLV Integration. Science 2003, 300, 1749–1751. [Google Scholar] [CrossRef] [PubMed]
- Felice, B.; Cattoglio, C.; Cittaro, D.; Testa, A.; Miccio, A.; Ferrari, G.; Luzi, L.; Recchia, A.; Mavilio, F. Transcription Factor Binding Sites Are Genetic Determinants of Retroviral Integration in the Human Genome. PLoS ONE 2009, 4, e4571. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Miller, D.G.; Jacobs, M.A.; Allen, J.M.; Kiem, H.-P.; Kaul, R.; Russell, D.W. Foamy Virus Vector Integration Sites in Normal Human Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 1498–1503. [Google Scholar] [CrossRef]
- Barr, S.D.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F.D. Integration Targeting by Avian Sarcoma-Leukosis Virus and Human Immunodeficiency Virus in the Chicken Genome. J. Virol. 2005, 79, 12035–12044. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.; Beitzel, B.; Schroder, A.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA Integration: ASLV, HIV, and MLV Show Distinct Target Site Preferences. PLoS Biol. 2004, 2, E234. [Google Scholar] [CrossRef]
- Narezkina, A.; Taganov, K.D.; Litwin, S.; Stoyanova, R.; Hayashi, J.; Seeger, C.; Skalka, A.M.; Katz, R.A. Genome-Wide Analyses of Avain Sarcoma Virus Integration Sites. J. Virol. 2004, 78, 11656–11663. [Google Scholar] [CrossRef]
- Brady, T.; Lee, Y.N.; Ronen, K.; Malani, N.; Berry, C.C.; Bieniasz, P.D.; Bushman, F.D. Integration Target Site Selection by a Resurrected Human Endogenous Retrovirus. Genes Dev. 2009, 23, 633–642. [Google Scholar] [CrossRef]
- Schroder, A.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F.D. HIV-1 Integration in the Human Genome Favors Active Genes and Local Hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Crise, B.; Li, Y.; Yuan, C.; Morcock, D.R.; Whitby, D.; Munroe, D.J.; Arthur, L.O.; Wu, X. Simian Immunodeficiency Virus Integration Preference Is Similar to That of Human Immunodeficiency Virus Type 1. J. Virol. 2005, 79, 12199–12204. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Moressi, C.J.; Scheetz, T.E.; Xie, L.; Tran, D.T.; Casavant, T.L.; Ak, P.; Benham, C.J.; Davidson, B.L.; McCray, P.B. Integration Site Choice of a Feline Immunodeficiency Virus Vector. J. Virol. 2006, 80, 8820–8823. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.; Lewinski, M.; Ciuffi, A.; Barr, S.; Leipzig, J.; Hannenhalli, S.; Hoffmann, C. Genome-Wide Analysis of Retroviral DNA Integration. Nat. Rev. Microbiol. 2005, 3, 848–858. [Google Scholar] [CrossRef]
- Taylor, B.; Sobieszczyk, M.; McCutchan, F.; Hammer, S.M. The Challenge of HIV-1 Subtipe Diversity. N. Engl. J. Med. 2008, 358, 1590–1602. [Google Scholar] [CrossRef] [PubMed]
- Pruss, D.; Bushman, F.D.; Wolffe, A.P. Human Immunodeficiency Virus Integrase Directs Integration to Sites of Severe DNA Distortion within the Nucleosome Core. Proc. Natl. Acad. Sci. USA 1994, 91, 5913–5917. [Google Scholar] [CrossRef] [PubMed]
- Pruss, D.; Reeves, R.; Bushman, F.D.; Wolffe, A.P. The Influence of DNA and Nucleosome Structure on Integration Events Directed by HIV Integrase. J. Biol. Chem. 1994, 269, 25031–25041. [Google Scholar] [CrossRef] [PubMed]
- Bor, Y.C.; Bushman, F.D.; Orgel, L.E. In Vitro Integration of Human Immunodeficiency Virus Type 1 CDNA into Targets Containing Protein-Induced Bends. Proc. Natl. Acad. Sci. USA 1995, 92, 10334–10338. [Google Scholar] [CrossRef]
- Muller, H.-P.; Varmus, H.E. DNA Bending Creates Favored Sites for Retroviral Integration: An Explanation for Preferred Insertion Sites in Nucleosomes. EMBO J. 1994, 13, 4704–4714. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Cherepanov, P. The Lentiviral Integrase Binding Protein LEDGF/P75 and HIV-1 Replication. PLoS Pathog. 2008, 4, e1000046. [Google Scholar] [CrossRef]
- Poeschla, E.M. Integrase, LEDGF/P75 and HIV Replication. Cell. Mol. Life Sci. 2008, 65, 1403–1424. [Google Scholar] [CrossRef] [PubMed]
- Llano, M.; Saenz, D.T.; Meehan, A.; Wongthida, P.; Peretz, M.; Walker, W.H.; Teo, W.; Poeschla, E.M. An Essential Role for LEDGF/P75 in HIV Integration. Science 2006, 314, 461–464. [Google Scholar] [CrossRef]
- Larue, R.C.; Plumb, M.R.; Crowe, B.L.; Shkriabai, N.; Sharma, A.; DiFiore, J.; Malani, N.; Aiyer, S.S.; Roth, M.J.; Bushman, F.D.; et al. Bimodal High-Affinity Association of Brd4 with Murine Leukemia Virus Integrase and Mononucleosomes. Nucleic Acids Res. 2014, 42, 4868–4881. [Google Scholar] [CrossRef]
- Sharma, A.; Larue, R.C.; Plumb, M.R.; Malani, N.; Male, F.; Slaughter, A.; Kessl, J.J.; Shkriabai, N.; Coward, E.; Aiyer, S.S.; et al. BET Proteins Promote Efficient Murine Leukemia Virus Integration at Transcription Start Sites. Proc. Natl. Acad. Sci. USA 2013, 110, 12036–12041. [Google Scholar] [CrossRef]
- Gupta, S.S.; Maetzig, T.; Maertens, G.N.; Sharif, A.; Rothe, M.; Weidner-Glunde, M.; Galla, M.; Schambach, A.; Cherepanov, P.; Schulz, T.F. Bromo- and Extraterminal Domain Chromatin Regulators Serve as Cofactors for Murine Leukemia Virus Integration. J. Virol. 2013, 87, 12721–12736. [Google Scholar] [CrossRef]
- Aiyer, S.; Swapna, G.V.T.; Malani, N.; Aramini, J.M.; Schneider, W.M.; Plumb, M.R.; Ghanem, M.; Larue, R.C.; Sharma, A.; Studamire, B.; et al. Altering Murine Leukemia Virus Integration through Disruption of the Integrase and BET Protein Family Interaction. Nucleic Acids Res. 2014, 42, 5917–5928. [Google Scholar] [CrossRef]
- Shun, M.-C.; Raghavendra, N.K.; Vandegraaff, N.; Daigle, J.E.; Hughes, S.; Kellam, P.; Cherepanov, P.; Engelman, A. LEDGF/P75 Functions Downstream from Preintegration Complex Formation to Effect Gene-Specific HIV-1 Integration. Genes Dev. 2007, 21, 1767–1778. [Google Scholar] [CrossRef]
- Marshall, H.M.; Ronen, K.; Berry, C.; Llano, M.; Sutherland, H.; Saenz, D.; Bickmore, W.; Poeschla, E.; Bushman, F.D. Role of PSIP1/LEDGF/P75 in Lentiviral Infectivity and Integration Targeting. PLoS ONE 2007, 2, e1340. [Google Scholar] [CrossRef]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A Role for LEDGF/P75 in Targeting HIV DNA Integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar] [CrossRef]
- Vandekerckhove, L.; Christ, F.; Van Maele, B.; De Rijck, J.; Gijsbers, R.; Van den Haute, C.; Witvrouw, M.; Debyser, Z. Transient and Stable Knockdown of the Integrase Cofactor LEDGF/P75 Reveals Its Role in the Replication Cycle of Human Immunodeficiency Virus. J. Virol. 2006, 80, 1886–1896. [Google Scholar] [CrossRef]
- Goff, S.P. Host Factors Exploited by Retroviruses. Nat. Rev. Microbiol. 2007, 5, 253–263. [Google Scholar] [CrossRef]
- Engelman, A. The Roles of Cellular Factors in Retroviral Integration. Curr. Top. Microbiol. Immunol. 2003, 281, 209–238. [Google Scholar]
- Bushman, F.D.; Malani, N.; Fernandes, J.; D’Orso, I.; Cagney, G.; Diamond, T.L.; Zhou, H.; Hazuda, D.J.; Espeseth, A.S.; König, R.; et al. Host Cell Factors in HIV Replication: Meta-Analysis of Genome-Wide Studies. PLoS Pathog. 2009, 5, e1000437. [Google Scholar] [CrossRef]
- Greene, W.C.; Peterlin, B.M. Charting HIV’s Remarkable Voyage through the Cell: Basic Science as a Passport to Future Therapy. Nat. Med. 2002, 8, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Craigie, R. The Road to Chromatin—Nuclear Entry of Retroviruses. Nat. Rev. Microbiol. 2007, 5, 187–196. [Google Scholar] [CrossRef]
- Achuthan, V.; Perreira, J.M.; Sowd, G.A.; Puray-Chavez, M.; McDougall, W.M.; Paulucci-Holthauzen, A.; Wu, X.; Fadel, H.J.; Poeschla, E.M.; Multani, A.S.; et al. Capsid-CPSF6 Interaction Licenses Nuclear HIV-1 Trafficking to Sites of Viral DNA Integration. Cell Host Microbe 2018, 24, 392–404.e8. [Google Scholar] [CrossRef]
- Chin, C.R.; Perreira, J.M.; Savidis, G.; Portmann, J.M.; Aker, A.M.; Feeley, E.M.; Smith, M.C.; Brass, A.L. Direct Visualization of HIV-1 Replication Intermediates Shows That Capsid and CPSF6 Modulate HIV-1 Intra-Nuclear Invasion and Integration. Cell Rep. 2015, 13, 1717–1731. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Muranyi, W.; Glass, B.; Laketa, V.; Yant, S.R.; Tsai, L.; Cihlar, T.; Müller, B.; Kräusslich, H.-G. Quantitative Microscopy of Functional HIV Post-Entry Complexes Reveals Association of Replication with the Viral Capsid. Elife 2014, 3, e04114. [Google Scholar] [CrossRef]
- Dharan, A.; Talley, S.; Tripathi, A.; Mamede, J.I.; Majetschak, M.; Hope, T.J.; Campbell, E.M. KIF5B and Nup358 Cooperatively Mediate the Nuclear Import of HIV-1 during Infection. PLoS Pathog. 2016, 12, e1005700. [Google Scholar] [CrossRef] [PubMed]
- Buffone, C.; Martinez-Lopez, A.; Fricke, T.; Opp, S.; Severgnini, M.; Cifola, I.; Petiti, L.; Frabetti, S.; Skorupka, K.; Zadrozny, K.K.; et al. Nup153 Unlocks the Nuclear Pore Complex for HIV-1 Nuclear Translocation in Nondividing Cells. J. Virol. 2018, 92, e00648-18. [Google Scholar] [CrossRef]
- Ajoge, H.O.; Renner, T.M.; Bélanger, K.; Greig, M.; Dankar, S.; Kohio, H.P.; Coleman, M.D.; Ndashimye, E.; Arts, E.J.; Langlois, M.A.; et al. Antiretroviral APOBEC3 Cytidine Deaminases Alter HIV-1 Provirus Integration Site Profiles. Nat. Commun. 2023, 14, 16. [Google Scholar] [CrossRef]
- Bushman, F.D. Tethering Human Immunodeficiency Virus 1 Integrase to a DNA Site Directs Integration to Nearby Sequences. Proc. Natl. Acad. Sci. USA 1994, 91, 9233–9237. [Google Scholar] [CrossRef]
- Pryciak, P.M.; Varmus, H.E. Nucleosomes, DNA-Binding Proteins, and DNA Sequence Modulate Retroviral Integration Target Site Selection. Cell 1992, 69, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M.; Munroe, D.J. Weak Palindromic Consensus Sequences Are a Common Feature Found at the Integration Target Sites of Many Retroviruses. J. Virol. 2005, 79, 5211–5214. [Google Scholar] [CrossRef] [PubMed]
- McAllister, R.G.; Liu, J.; Woods, M.W.; Tom, S.K.; Rupar, C.A.; Barr, S.D. Lentivector Integration Sites in Ependymal Cells from a Model of Metachromatic Leukodystrophy: Non-B DNA as a New Factor Influencing Integration. Mol. Ther. Nucleic Acids 2014, 3, e187. [Google Scholar] [CrossRef]
- Choi, J.; Majima, T. Conformational Changes of Non-BDNA. Chem. Soc. Rev. 2011, 40, 5893–5909. [Google Scholar] [CrossRef]
- Bacolla, A.; Wells, R.D. Non-B DNA Conformations, Genomic Rearrangements, and Human Disease. J. Biol. Chem. 2004, 279, 47411–47414. [Google Scholar] [CrossRef]
- Ajoge, H.O.; Kohio, H.P.; Paparisto, E.; Coleman, M.D.; Wong, K.; Tom, S.K.; Bain, K.L.; Berry, C.C.; Arts, E.J.; Barr, S.D. G-Quadruplex DNA and Other Non-Canonical B-Form DNA Motifs Influence Productive and Latent HIV-1 Integration and Reactivation Potential. Viruses 2022, 14, 2494. [Google Scholar] [CrossRef]
- Venner, C.M.; Nankya, I.; Kyeyune, F.; Demers, K.; Kwok, C.; Chen, P.-L.; Rwambuya, S.; Munjoma, M.; Chipato, T.; Byamugisha, J.; et al. Infecting HIV-1 Subtype Predicts Disease Progression in Women of Sub-Saharan Africa. EBioMedicine 2016, 13, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.S.; Chen, P.-L.; Nankya, I.; Rinaldi, A.; Van Der Pol, B.; Ma, Y.-R.; Chipato, T.; Mugerwa, R.; Dunbar, M.; Arts, E.; et al. Hormonal Contraceptive Use and HIV Disease Progression Among Women in Uganda and Zimbabwe. JAIDS J. Acquir. Immune Defic. Syndr. 2011, 57, 157–164. [Google Scholar] [CrossRef]
- Morrison, C.S.; Richardson, B.A.; Mmiro, F.; Chipato, T.; Celentano, D.D.; Luoto, J.; Mugerwa, R.; Padian, N.; Rugpao, S.; Brown, J.M.; et al. Hormonal Contraception and the Risk of HIV Acquisition. AIDS 2007, 21, 85–95. [Google Scholar] [CrossRef]
- Lemonovich, T.L.; Watkins, R.R.; Morrison, C.S.; Kwok, C.; Chipato, T.; Musoke, R.; Arts, E.J.; Nankya, I.; Salata, R.A. Differences in Clinical Manifestations of Acute and Early HIV-1 Infection between HIV-1 Subtypes in African Women. J. Int. Assoc. Provid. AIDS Care (JIAPAC) 2015, 14, 415–422. [Google Scholar] [CrossRef]
- Ciuffi, A.; Barr, S.D. Identification of HIV Integration Sites in Infected Host Genomic DNA. Methods 2011, 53, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Cer, R.Z.; Donohue, D.E.; Mudunuri, U.S.; Temiz, N.A.; Loss, M.A.; Starner, N.J.; Halusa, G.N.; Volfovsky, N.; Yi, M.; Luke, B.T.; et al. Non-B DB v2.0: A Database of Predicted Non-B DNA-Forming Motifs and Its Associated Tools. Nucleic Acids Res. 2013, 41, D94–D100. [Google Scholar] [CrossRef] [PubMed]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain Organization of Human Chromosomes Revealed by Mapping of Nuclear Lamina Interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef] [PubMed]
- Barr, S.D.; Ciuffi, A.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F.D. HIV Integration Site Selection: Targeting in Macrophages and the Effects of Different Routes of Viral Entry. Mol. Ther. 2006, 14, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11.12.1–11.12.34. [Google Scholar] [CrossRef]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-Enabled Heat Mapping for All. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- Derse, D.; Crise, B.; Li, Y.; Princler, G.; Lum, N.; Stewart, C.; McGrath, C.F.; Hughes, S.H.; Munroe, D.J.; Wu, X. Human T-Cell Leukemia Virus Type 1 Integration Target Sites in the Human Genome: Comparison with Those of Other Retroviruses. J. Virol. 2007, 81, 6731–6741. [Google Scholar] [CrossRef] [PubMed]
- Faschinger, A.; Rouault, F.; Sollner, J.; Lukas, A.; Salmons, B.; Gunzburg, W.H.; Indik, S. Mouse Mammary Tumor Virus Integration Site Selection in Human and Mouse Genomes. J. Virol. 2008, 82, 1360–1367. [Google Scholar] [CrossRef]
- Marini, B.; Kertesz-Farkas, A.; Ali, H.; Lucic, B.; Lisek, K.; Manganaro, L.; Pongor, S.; Luzzati, R.; Recchia, A.; Mavilio, F.; et al. Nuclear Architecture Dictates HIV-1 Integration Site Selection. Nature 2015, 521, 227–231. [Google Scholar] [CrossRef]
- Battivelli, E.; Dahabieh, M.S.; Abdel-Mohsen, M.; Svensson, J.P.; Tojal Da Silva, I.; Cohn, L.B.; Gramatica, A.; Deeks, S.; Greene, W.C.; Pillai, S.K.; et al. Distinct Chromatin Functional States Correlate with HIV Latency Reactivation in Infected Primary CD4+ T Cells. Elife 2018, 7, e34655. [Google Scholar] [CrossRef]
- Poletti, V.; Mavilio, F. Interactions between Retroviruses and the Host Cell Genome. Methods Clin. Dev. 2018, 8, 31–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattoglio, C.; Facchini, G.; Sartori, D.; Antonelli, A.; Miccio, A.; Cassani, B.; Schmidt, M.; von Kalle, C.; Howe, S.; Thrasher, A.J.; et al. Hot Spots of Retroviral Integration in Human CD34+ Hematopoietic Cells. Blood 2007, 110, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Nettling, M.; Treutler, H.; Grau, J.; Keilwagen, J.; Posch, S.; Grosse, I. DiffLogo: A Comparative Visualization of Sequence Motifs. BMC Bioinform. 2015, 16, 387. [Google Scholar] [CrossRef]
- Kirk, P.D.W.; Huvet, M.; Melamed, A.; Maertens, G.N.; Bangham, C.R.M. Retroviruses Integrate into a Shared, Non-Palindromic DNA Motif. Nat. Microbiol. 2016, 2, 16212. [Google Scholar] [CrossRef] [PubMed]
- Sinden, R.R.; Pytlos-Sinden, M.J.; Potaman, V.N. Slipped Strand DNA Structures. Front. Biosci. 2007, 12, 4788–4799. [Google Scholar] [CrossRef]
- Wahls, W.P.; Wallace, L.J.; Moore, P.D. The Z-DNA Motif d(TG)30 Promotes Reception of Information during Gene Conversion Events While Stimulating Homologous Recombination in Human Cells in Culture. Mol. Cell Biol. 1990, 10, 785–793. [Google Scholar] [PubMed]
- Chin, J.Y.; Schleifman, E.B.; Glazer, P.M. Repair and Recombination Induced by Triple Helix DNA. Front. Biosci. 2007, 12, 4288. [Google Scholar] [CrossRef]
- Brázda, V.; Laister, R.C.; Jagelská, E.B.; Arrowsmith, C. Cruciform Structures Are a Common DNA Feature Important for Regulating Biological Processes. BMC Mol. Biol. 2011, 12, 33. [Google Scholar] [CrossRef]
- Holtz, C.M.; Sadler, H.A.; Mansky, L.M. APOBEC3G Cytosine Deamination Hotspots Are Defined by Both Sequence Context and Single-Stranded DNA Secondary Structure. Nucleic Acids Res. 2013, 41, 6139–6148. [Google Scholar] [CrossRef]
- Ziegler, S.J.; Liu, C.; Landau, M.; Buzovetsky, O.; Desimmie, B.A.; Zhao, Q.; Sasaki, T.; Burdick, R.C.; Pathak, V.K.; Anderson, K.S.; et al. Insights into DNA Substrate Selection by APOBEC3G from Structural, Biochemical, and Functional Studies. PLoS ONE 2018, 13, e0195048. [Google Scholar] [CrossRef]
- Mazumder, A.N.; Neamati, J.O.; Ojwang, S.; Sunder, S.; Rando, R.F.; Pommier, Y. Inhibition of Human Immunodeficiency Virus Type 1 Integrase by Guanosine Quartet Structures. Biochemistry 1996, 35, 13762–13771. [Google Scholar] [CrossRef]
- Einkauf, K.B.; Osborn, M.R.; Gao, C.; Sun, W.; Sun, X.; Lian, X.; Parsons, E.M.; Gladkov, G.T.; Seiger, K.W.; Blackmer, J.E.; et al. Parallel analysis of transcription, integration, and sequence of single HIV-1 proviruses. Cell 2022, 185, 266–282.e15. [Google Scholar] [CrossRef] [PubMed]
- Einkauf, K.B.; Lee, G.Q.; Gao, C.; Sharaf, R.; Sun, X.; Hua, S.; Chen, S.M.; Jiang, C.; Lian, X.; Chowdhury, F.Z.; et al. Intact HIV-1 proviruses accumulate at distinct chromosomal positions during prolonged antiretroviral therapy. J. Clin. Investig. 2019, 129, 988–998. [Google Scholar] [CrossRef]
- Lian, X.; Gao, C.; Sun, X.; Jiang, C.; Einkauf, K.B.; Seiger, K.W.; Chevalier, J.M.; Yuki, Y.; Martin, M.; Hoh, R.; et al. Signatures of immune selection in intact and defective proviruses distinguish HIV-1 elite controllers. Sci. Transl. Med. 2021, 13, eabl4097. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Seiger, K.W.; Parsons, E.M.; Gao, C.; Sun, W.; Gladkov, G.T.; Roseto, I.C.; Einkauf, K.B.; Osborn, M.R.; Chevalier, J.M.; et al. Progressive transformation of the HIV-1 reservoir cell profile over two decades of antiviral therapy. Cell Host Microbe 2023, 31, 83–96.e5. [Google Scholar] [CrossRef]
- Jiang, C.; Lian, X.; Gao, C.; Sun, X.; Einkauf, K.B.; Chevalier, J.M.; Chen, S.M.Y.; Hua, S.; Rhee, B.; Chang, K.; et al. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature 2020, 585, 261–267. [Google Scholar] [CrossRef]
- Jiang, J.-C.; Upton, K.R. Human Transposons Are an Abundant Supply of Transcription Factor Binding Sites and Promoter Activities in Breast Cancer Cell Lines. Mob. DNA 2019, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Issa, J.-P.J. Enrichment for Histone H3 Lysine 9 Methylation at Alu Repeats in Human Cells. J. Biol. Chem. 2003, 278, 27658–27662. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct Evidence for a G-Quadruplex in a Promoter Region and Its Targeting with a Small Molecule to Repress c-MYC Transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef]
- Verma, A.; Yadav, V.K.; Basundra, R.; Kumar, A.; Chowdhury, S. Evidence of Genome-Wide G4 DNA-Mediated Gene Expression in Human Cancer Cells. Nucleic Acids Res. 2009, 37, 4194–4204. [Google Scholar] [CrossRef]
- Waga, S.; Mizuno, S.; Yoshida, M. Chromosomal Protein HMG1 Removes the Transcriptional Block Caused by the Cruciform in Supercoiled DNA. J. Biol. Chem. 1990, 265, 19424–19428. [Google Scholar] [CrossRef] [PubMed]
- Waga, S.; Mizuno, S.; Yoshida, M. Nonhistone Protein HMG1 Removes the Transcriptional Block Caused by Left-Handed Z-Form Segment in a Supercoiled DNA. Biochem. Biophys. Res. Commun. 1988, 153, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Magistri, M.; Napoli, S.; Carbone, G.M.; Catapano, C.V. Mechanisms of Triplex DNA-Mediated Inhibition of Transcription Initiation in Cells. Biochimie 2010, 92, 317–320. [Google Scholar] [CrossRef]
- Maher, L.J.; Dervan, P.B.; Wold, B. Analysis of Promoter-Specific Repression by Triple-Helical DNA Complexes in a Eukaryotic Cell-Free Transcription System. Biochemistry 1992, 31, 70–81. [Google Scholar] [CrossRef]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA Secondary Structures: Stability and Function of G-Quadruplex Structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Delic, J.; Onclercq, R.; Moisan-Coppey, M. Inhibition and Enhancement of Eukaryotic Gene Expression by Potential Non-B DNA Sequences. Biochem. Biophys. Res. Commun. 1991, 180, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Tornaletti, S.; Park-Snyder, S.; Hanawalt, P.C. G4-Forming Sequences in the Non-Transcribed DNA Strand Pose Blocks to T7 RNA Polymerase and Mammalian RNA Polymerase II. J. Biol. Chem. 2008, 283, 12756–12762. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskii, B.P.; De Silva, E.; Tornaletti, S.; Wang, G.; Vasquez, K.M.; Hanawalt, P.C. A Triplex-Forming Sequence from the Human c-MYC Promoter Interferes with DNA Transcription. J. Biol. Chem. 2007, 282, 32433–32441. [Google Scholar] [CrossRef]
- Löwer, R.; Löwer, J.; Kurth, R. The Viruses in All of Us: Characteristics and Biological Significance of Human Endogenous Retrovirus Sequences. Proc. Natl. Acad. Sci. USA 1996, 93, 5177–5184. [Google Scholar] [CrossRef]
- Han, G.-Z.; Worobey, M. An Endogenous Foamy-like Viral Element in the Coelacanth Genome. PLoS Pathog. 2012, 8, e1002790. [Google Scholar] [CrossRef]
- Switzer, W.M.; Salemi, M.; Shanmugam, V.; Gao, F.; Cong, M.; Kuiken, C.; Bhullar, V.; Beer, B.E.; Vallet, D.; Gautier-Hion, A.; et al. Ancient Co-Speciation of Simian Foamy Viruses and Primates. Nature 2005, 434, 376–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, S.R. Mouse Mammary Tumor Virus Molecular Biology and Oncogenesis. Viruses 2010, 2, 2000–2012. [Google Scholar] [CrossRef] [PubMed]
- Cohn, L.B.; Silva, I.T.; Oliveira, T.Y.; Rosales, R.A.; Parrish, E.H.; Learn, G.H.; Hahn, B.H.; Czartoski, J.L.; McElrath, M.J.; Lehmann, C.; et al. HIV-1 Integration Landscape during Latent and Active Infection. Cell 2015, 160, 420–432. [Google Scholar] [CrossRef]
- Kuniholm, J.; Coote, C.; Henderson, A.J. Defective HIV-1 genomes and their potential impact on HIV pathogenesis. Retrovirology 2022, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Winans, S.; Yu, H.J.; de los Santos, K.; Wang, G.Z.; KewalRamani, V.N.; Goff, S.P. A Point Mutation in HIV-1 Integrase Redirects Proviral Integration into Centromeric Repeats. Nat. Commun. 2022, 13, 1474. [Google Scholar] [CrossRef]
- Myers, R.E.; Pillay, D. Analysis of Natural Sequence Variation and Covariation in Human Immunodeficiency Virus Type 1 Integrase. J. Virol. 2008, 82, 9228–9235. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Liu, T.F.; Kiuchi, M.; Zioni, R.; Gifford, R.J.; Holmes, S.P.; Shafer, R.W. Natural Variation of HIV-1 Group M Integrase: Implications for a New Class of Antiretroviral Inhibitors. Retrovirology 2008, 5, 74. [Google Scholar] [CrossRef] [PubMed]
- Demeulemeester, J.; Vets, S.; Schrijvers, R.; Madlala, P.; De Maeyer, M.; De Rijck, J.; Ndung’u, T.; Debyser, Z.; Gijsbers, R.; De Maeyer, M.; et al. HIV-1 Integrase Variants Retarget Viral Integration and Are Associated with Disease Progression in a Chronic Infection Cohort. Cell Host Microbe 2014, 16, 651–662. [Google Scholar] [CrossRef]
- Binka, M.; Ooms, M.; Steward, M.; Simon, V. The Activity Spectrum of Vif from Multiple HIV-1 Subtypes against APOBEC3G, APOBEC3F, and APOBEC3H. J. Virol. 2012, 86, 49–59. [Google Scholar] [CrossRef] [Green Version]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kohio, H.P.; Ajoge, H.O.; Coleman, M.D.; Ndashimye, E.; Gibson, R.M.; Arts, E.J.; Barr, S.D. Specialized DNA Structures Act as Genomic Beacons for Integration by Evolutionarily Diverse Retroviruses. Viruses 2023, 15, 465. https://doi.org/10.3390/v15020465
Kohio HP, Ajoge HO, Coleman MD, Ndashimye E, Gibson RM, Arts EJ, Barr SD. Specialized DNA Structures Act as Genomic Beacons for Integration by Evolutionarily Diverse Retroviruses. Viruses. 2023; 15(2):465. https://doi.org/10.3390/v15020465
Chicago/Turabian StyleKohio, Hinissan P., Hannah O. Ajoge, Macon D. Coleman, Emmanuel Ndashimye, Richard M. Gibson, Eric J. Arts, and Stephen D. Barr. 2023. "Specialized DNA Structures Act as Genomic Beacons for Integration by Evolutionarily Diverse Retroviruses" Viruses 15, no. 2: 465. https://doi.org/10.3390/v15020465
APA StyleKohio, H. P., Ajoge, H. O., Coleman, M. D., Ndashimye, E., Gibson, R. M., Arts, E. J., & Barr, S. D. (2023). Specialized DNA Structures Act as Genomic Beacons for Integration by Evolutionarily Diverse Retroviruses. Viruses, 15(2), 465. https://doi.org/10.3390/v15020465