

Genomic Analysis of G2P[4] Group A Rotaviruses in Zambia Reveals Positive Selection in Amino Acid Site 7 of Viral Protein 3

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Stool Specimen and Strain Description
2.3. Data Collection from GenBank
2.4. Double-Stranded RNA Extraction
2.5. cDNA Synthesis
2.6. DNA Library Preparations and Whole-Genome Sequencing
2.7. Genome Assembly
2.8. Whole-Genome Genotyping
2.9. Phylogenetic Analysis
2.10. Inference of Selective Pressures
3. Results
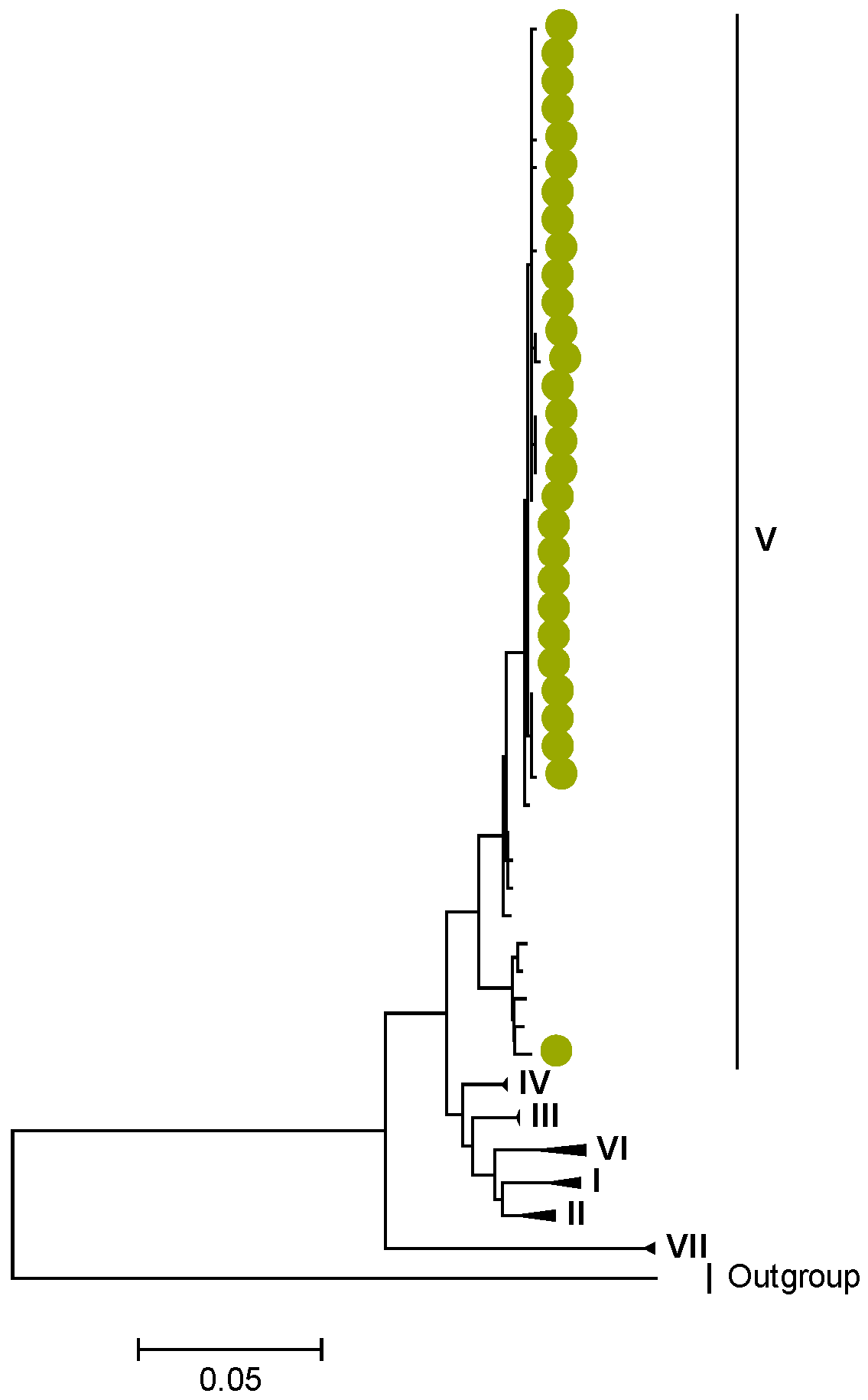
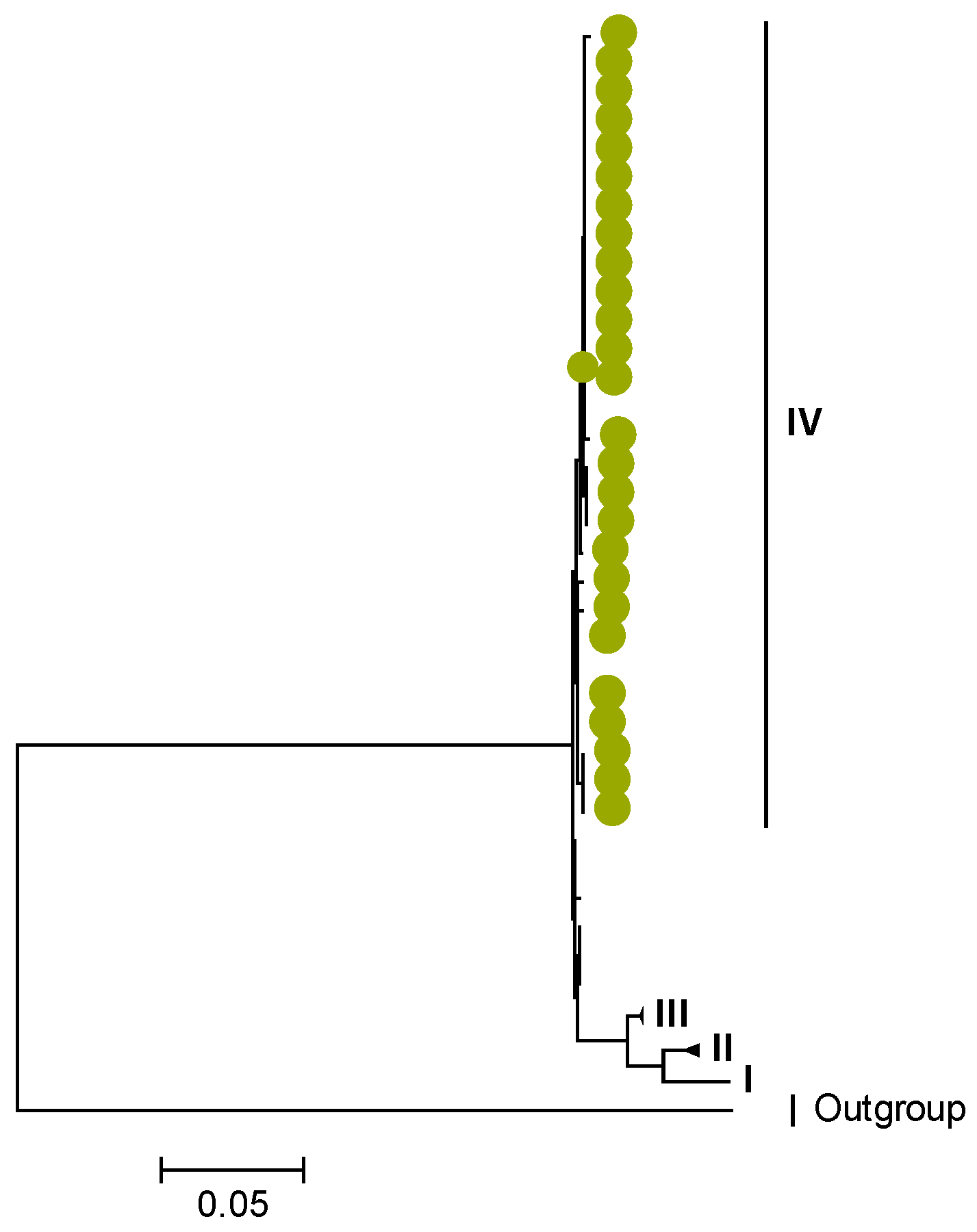
3.1. Whole Genotype Analysis
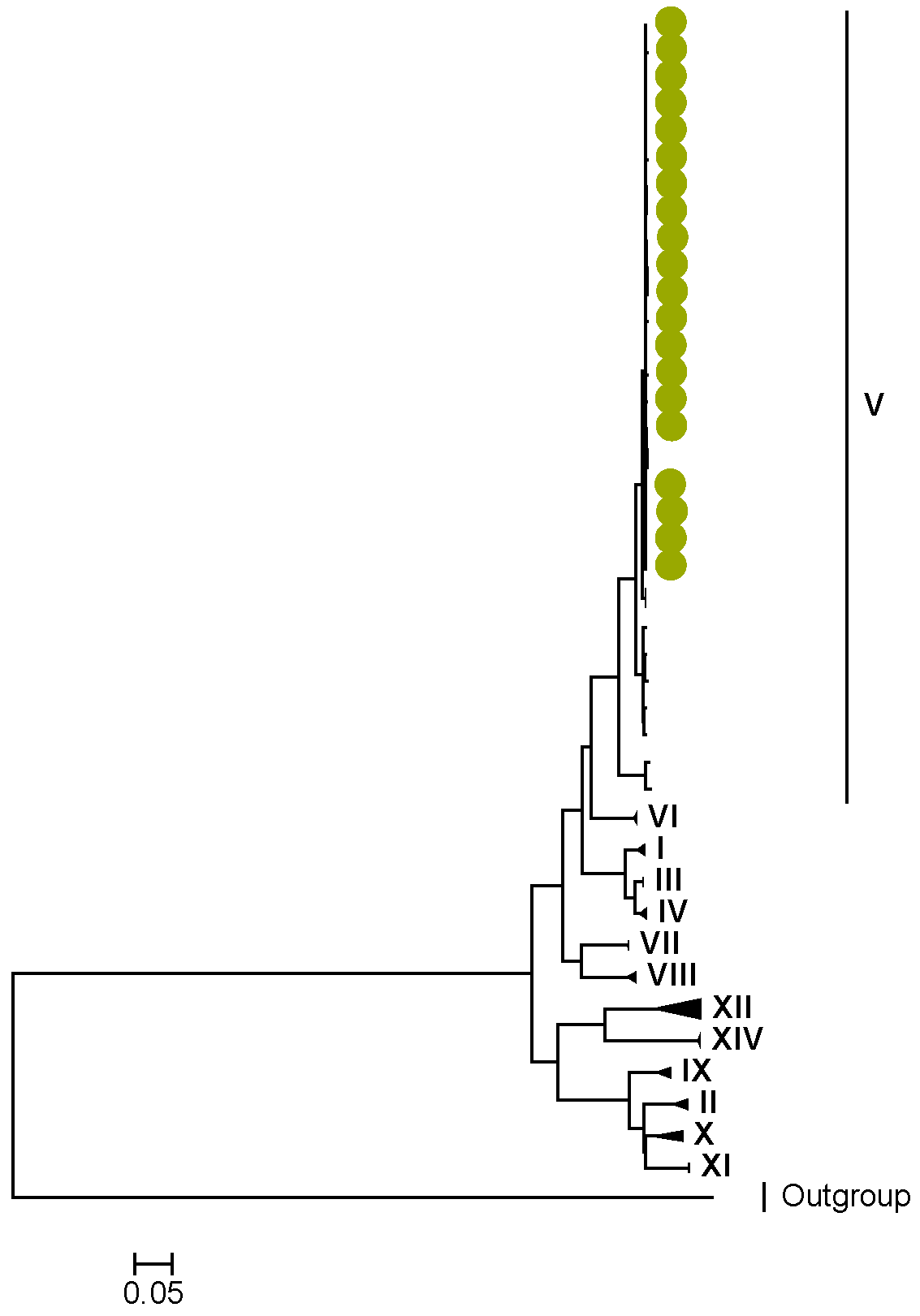
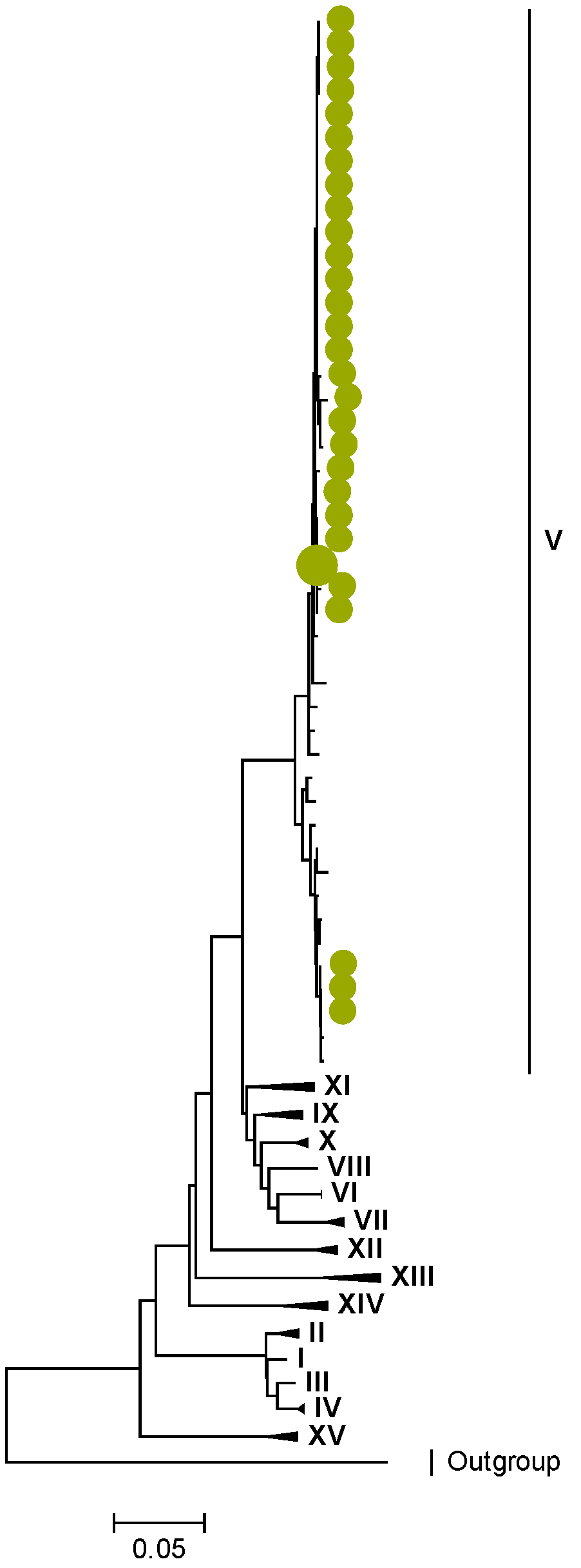
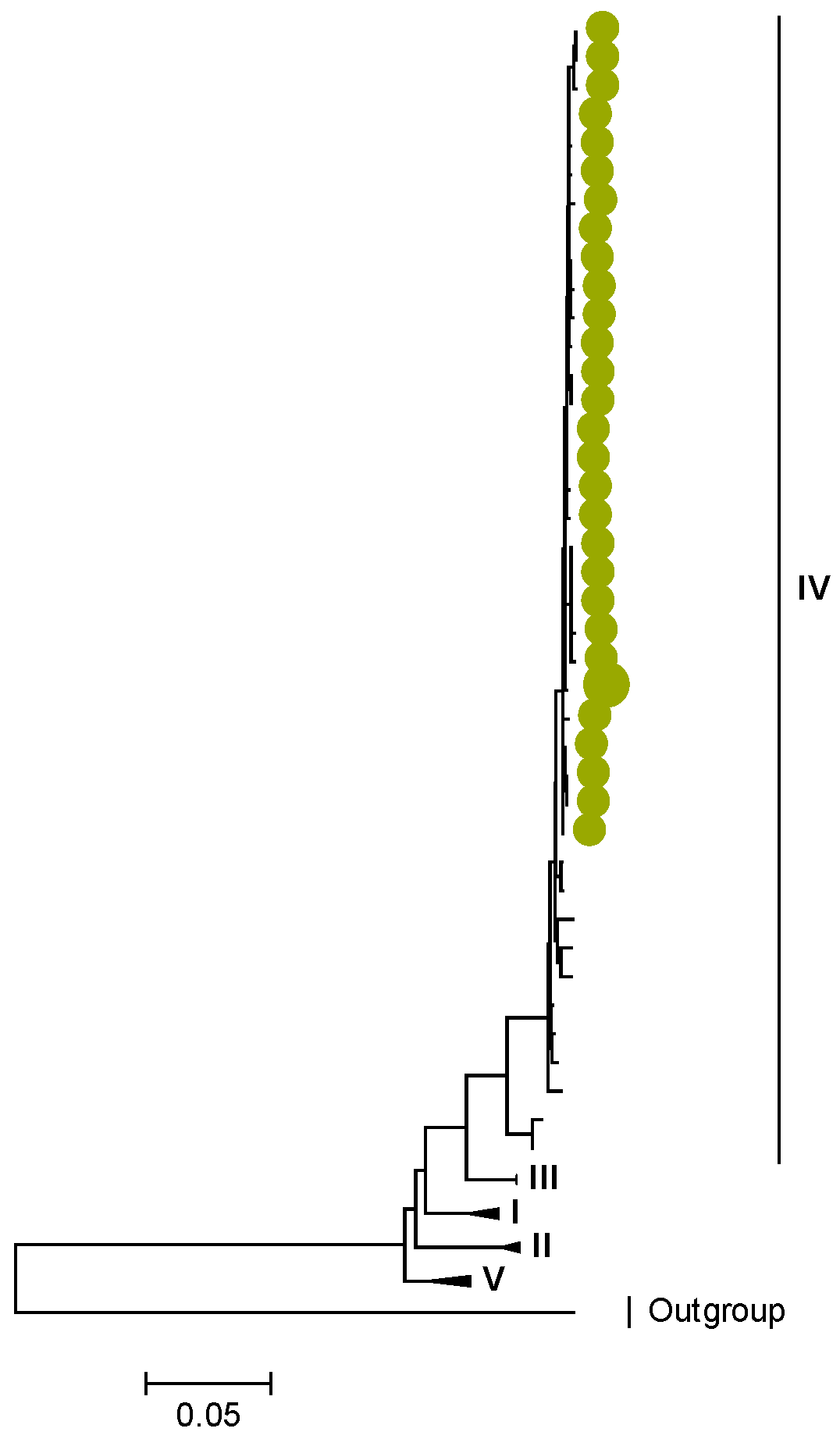
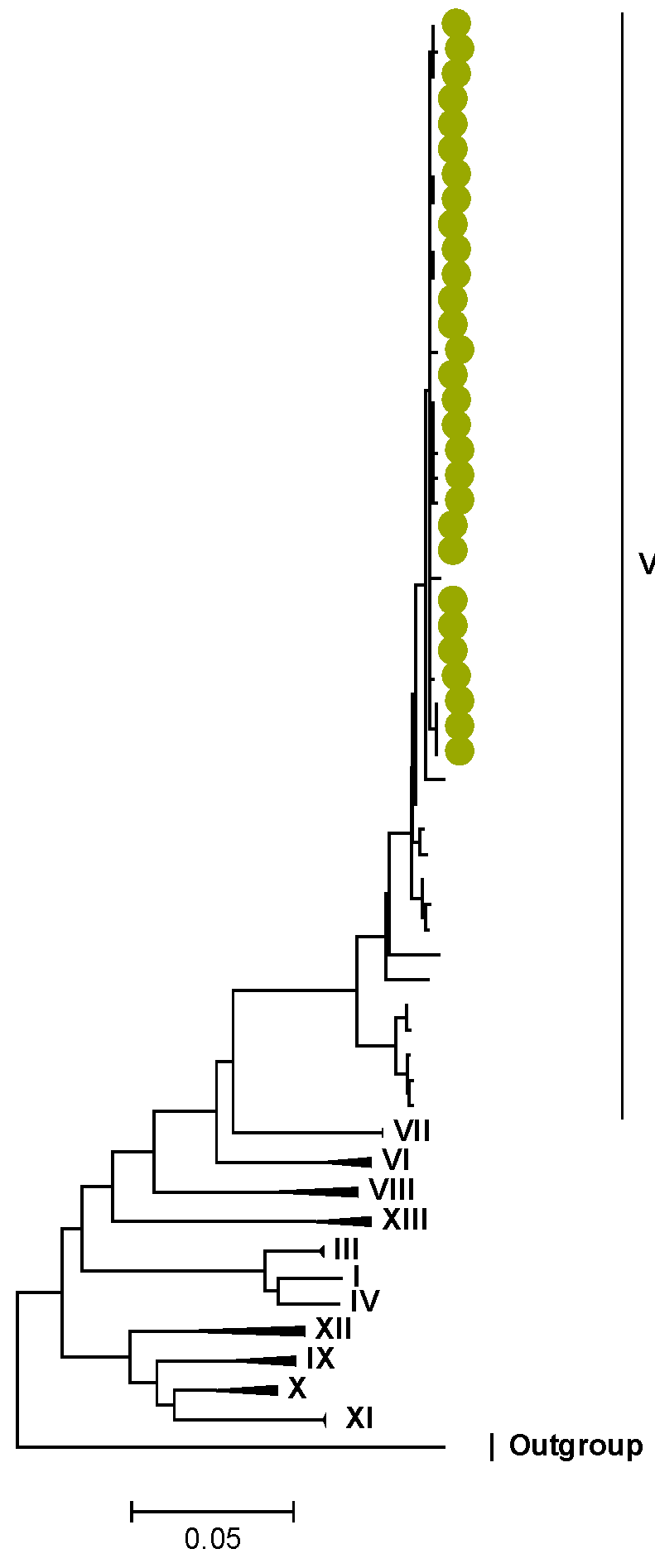
3.2. Phylogenetic and Sequence Analysis
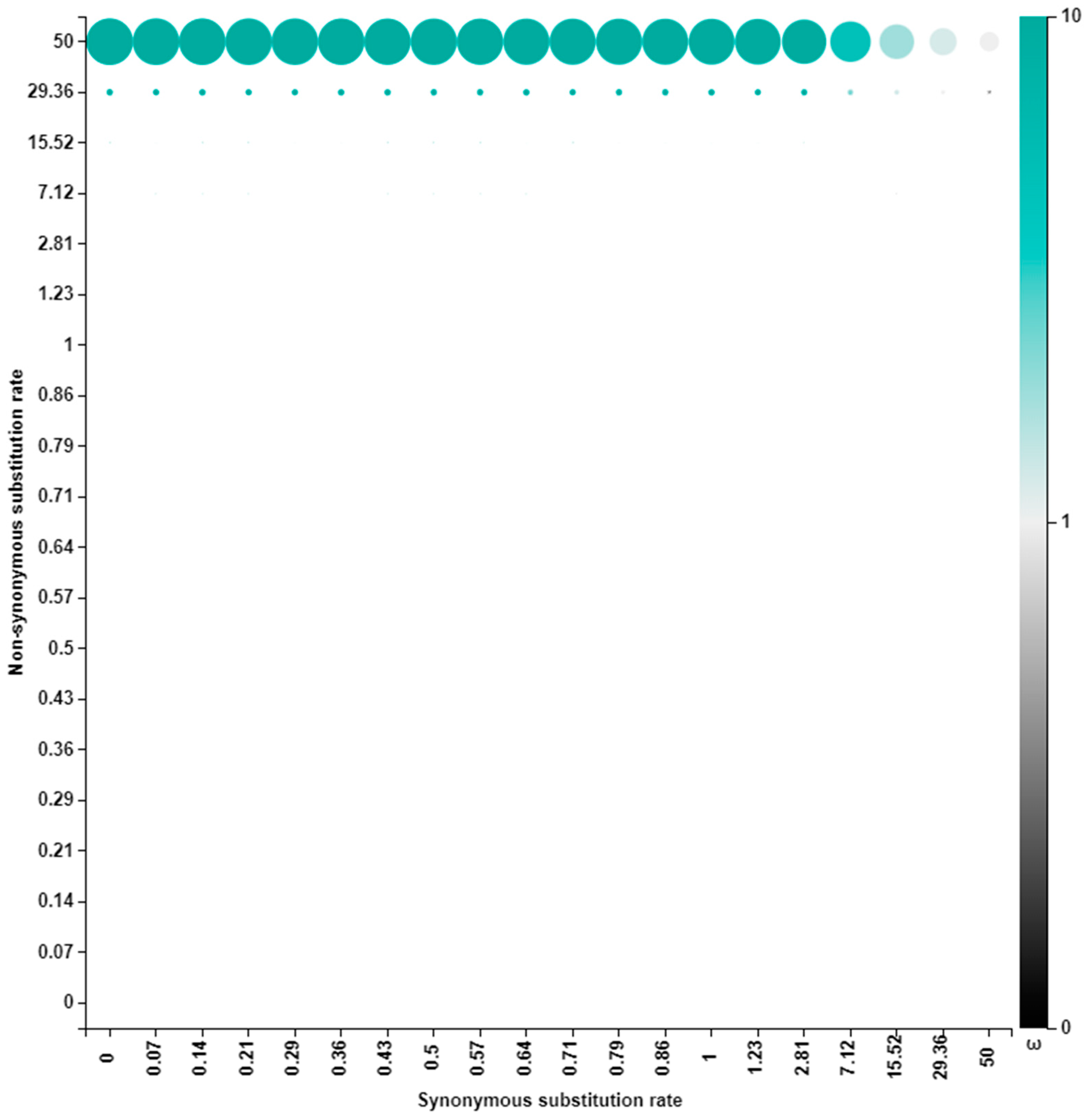
3.3. Selection Pressure Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Troeger, C.; Khalil, I.A.; Rao, P.C.; Cao, S.; Blacker, B.F.; Ahmed, T.; Armah, G.; Bines, J.E.; Brewer, T.G.; Colombara, D.V.; et al. Rotavirus vaccination and the global burden of rotavirus diarrhea among children younger than 5years. JAMA Pediatr. 2018, 172, 958–965. [Google Scholar] [CrossRef]
- Parashar, U.D.; Gibson, C.J.; Bresee, J.S.; Glass, R.I. Rotavirus and severe childhood diarrhea. Emerg. Infect. Dis. 2006, 12, 304–306. [Google Scholar] [CrossRef]
- Mpabalwani, E.M.; Simwaka, C.J.; Mwenda, J.M.; Mubanga, C.P.; Monze, M.; Matapo, B.; Parashar, U.D.; Tate, J.E. Impact of rotavirus vaccination on diarrhoeal hospitalisations in children aged <5 Years in Lusaka, Zambia. Clin. Infect. Dis. 2016, 62, S183–S187. [Google Scholar] [CrossRef]
- Chilengi, R.; Rudd, C.; Bolton, C.; Guffey, B.; Masumbu, P.K.; Stringer, J. Successes, challenges and lessons learned in accelerating introduction of rotavirus immunisation in Zambia. World J. Vaccines 2015, 5, 43–53. [Google Scholar] [CrossRef]
- Shah, M.P.; Mwenda, J.M.; Tate, J.E.; Steele, A.D.; Parashar, U.D. Estimated reductions in hospitalizations and deaths from childhood diarrhea following implementation of rotavirus vaccines in Africa. Expert Rev. Vaccines 2017, 16, 987–995. [Google Scholar] [CrossRef]
- Zeller, M.; Heylen, E.; Tamim, S.; McAllen, J.K.; Kirkness, E.F.; Akopov, A.; De Coster, S.; Van Ranst, M.; Matthijnssens, J. Comparative analysis of the Rotarix™ vaccine strain and G1P[8] rotaviruses detected before and after vaccine introduction in Belgium. PeerJ 2017, 5, e2733. [Google Scholar] [CrossRef]
- Estes, M.K.; Greenberg, H.B. Rotaviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer Heath/Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2013; pp. 1347–1401. [Google Scholar]
- Matthijnssens, J.; Ciarlet, M.; Rahman, M.; Attoui, H.; Bányai, K.; Estes, M.K.; Gentsch, J.R.; Iturriza-Gómara, M.; Kirkwood, C.D.; Martella, V.; et al. Recommendations for the classification of group A rotaviruses using all 11 genomic RNA segments. Arch. Virol. 2008, 153, 1621–1629. [Google Scholar] [CrossRef]
- Dóró, R.; László, B.; Martella, V.; Leshem, E.; Gentsch, J.; Parashar, U.; Bányai, K. Review of global rotavirus strain prevalence data from six years post vaccine licensure surveillance: Is there evidence of strain selection from vaccine pressure? Infect. Genet. Evol. 2014, 28, 446–461. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Ciarlet, M.; McDonald, S.M.; Attoui, H.; Bányai, K.; Brister, J.R.; Buesa, J.; Esona, M.D.; Estes, M.K.; Gentsch, J.R.; et al. Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG). Arch. Virol. 2011, 156, 1397–1413. [Google Scholar] [CrossRef]
- Mwangi, P.N.; Mogotsi, M.T.; Rasebotsa, S.P.; Seheri, M.L.; Mphahlele, M.J.; Ndze, V.N.; Dennis, F.E.; Jere, K.C.; Nyaga, M.M. Uncovering the first atypical ds-1-like G1P[8] rotavirus strains that circulated during pre-rotavirus vaccine introduction era in South Africa. Pathogens 2020, 9, 391. [Google Scholar] [CrossRef]
- Jere, K.C.; Chaguza, C.; Bar-Zeev, N.; Lowe, J.; Peno, C.; Kumwenda, B.; Nakagomi, O.; Tate, J.E.; Parashar, U.D.; Heyderman, R.S.; et al. Emergence of double-and triple-gene reassortant G1P[8] rotaviruses possessing a DS-1-like backbone after rotavirus vaccine introduction in Malawi. J. Virol. 2018, 92, e01246-17. [Google Scholar] [CrossRef]
- Mhango, C.; Banda, A.; Chinyama, E.; Mandolo, J.J.; Kumwenda, O.; Malamba-Banda, C.; Barnes, K.G.; Kumwenda, B.; Jambo, K.; Donato, C.M.; et al. Comparative whole genome analysis reveals re-emergence of typical human Wa-like and DS-1-like G3 rotaviruses after Rotarix vaccine introduction in Malawi. medRxiv 2022. [Google Scholar] [CrossRef]
- Mwanga, M.J.; Verani, J.R.; Omore, R.; Tate, J.E.; Parashar, U.D.; Murunga, N.; Gicheru, E.; Breiman, R.F.; Nokes, D.J.; Agoti, C.N. Multiple introductions and predominance of rotavirus group A genotype G3P[8] in coastal Kenya in 2018, 4 years after nationwide vaccine introduction. Pathogens 2020, 9, 981. [Google Scholar] [CrossRef]
- Fukuda, S.; Tacharoenmuang, R.; Guntapong, R.; Upachai, S.; Singchai, P.; Ide, T.; Hatazawa, R.; Sutthiwarakom, K.; Kongjorn, S.; Onvimala, N.; et al. Full genome characterization of novel DS-1-like G9P[8] rotavirus strains that have emerged in Thailand. PLoS ONE 2020, 15, e0231099. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Van Ranst, M. Genotype constellation and evolution of group A rotaviruses infecting humans. Curr. Opin. Virol. 2012, 2, 426–433. [Google Scholar] [CrossRef]
- Maringa, W.M.; Simwaka, J.; Mwangi, P.N.; Mpabalwani, E.M.; Mwenda, J.M.; Mphahlele, M.J.; Seheri, M.L.; Nyaga, M.M. Whole Genome Analysis of Human Rotaviruses Reveals Single Gene Reassortant Rotavirus Strains in Zambia. Viruses 2021, 13, 1872. [Google Scholar] [CrossRef]
- Wandera, E.A.; Hatazawa, R.; Tsutsui, N.; Kurokawa, N.; Kathiiko, C.; Mumo, M.; Waithira, E.; Wachira, M.; Mwaura, B.; Nyangao, J.; et al. Genomic characterization of an African G4P[6] human rotavirus strain identified in a diarrheic child in Kenya: Evidence for porcine-to-human interspecies transmission and reassortment. Infect. Genet. Evol. 2021, 96, 105133. [Google Scholar] [CrossRef]
- Maringa, W.M.; Mwangi, P.N.; Simwaka, J.; Mpabalwani, E.M.; Mwenda, J.M.; Peenze, I.; Esona, M.D.; Mphahlele, M.J.; Seheri, M.L.; Nyaga, M.M. Molecular characterisation of a rare reassortant porcine-like G5P[6] rotavirus strain detected in an unvaccinated child in Kasama, Zambia. Pathogens 2020, 9, 663. [Google Scholar] [CrossRef]
- Rasebotsa, S.; Mwangi, P.N.; Mogotsi, M.T.; Sabiu, S.; Magagula, N.B.; Rakau, K.; Uwimana, J.; Mutesa, L.; Muganga, N.; Murenzi, D.; et al. Whole genome and in-silico analyses of G1P[8] rotavirus strains from pre-and post-vaccination periods in Rwanda. Sci. Rep. 2020, 10, 1–22. [Google Scholar] [CrossRef]
- Nyaga, M.M.; Tan, Y.; Seheri, M.L.; Halpin, R.A.; Akopov, A.; Stucker, K.M.; Fedorova, N.B.; Shrivastava, S.; Steele, A.D.; Mwenda, J.M.; et al. Whole-genome sequencing and analyses identify high genetic heterogeneity, diversity and endemicity of rotavirus genotype P[6] strains circulating in Africa. Infect. Genet. Evol. 2018, 63, 79–88. [Google Scholar] [CrossRef]
- Donker, N.C.; Kirkwood, C.D. Selection and evolutionary analysis in the nonstructural protein NSP2 of rotavirus A. Infect. Genet. Evol. 2012, 12, 1355–1361. [Google Scholar] [CrossRef]
- Donato, C.M.; Zhang, Z.A.; Donker, N.C.; Kirkwood, C.D. Characterization of G2P[4] rotavirus strains associated with increased detection in Australian states using the RotaTeq® vaccine during the 2010-2011 surveillance period. Infect. Genet. Evol. 2014, 28, 398–412. [Google Scholar] [CrossRef]
- Gurgel, R.Q.; Cuevas, L.E.; Vieira, S.C.F.; Barros, V.C.F.; Fontes, P.B. Predominance of rotavirus P[4]G2 in a vaccinated population, Brazil. Emerg. Infect. Dis. 2007, 13, 1571–1573. [Google Scholar] [CrossRef]
- Mokomane, M.; Esona, M.D.; Bowen, M.D.; Tate, J.E.; Steenhoff, A.P.; Lechiile, K.; Gaseitsiwe, S.; Seheri, L.M.; Magagula, N.B.; Weldegebriel, G.; et al. Diversity of Rotavirus Strains Circulating in Botswana before and after introduction of the Monovalent Rotavirus Vaccine. Vaccine 2019, 37, 6324–6328. [Google Scholar] [CrossRef]
- Khandoker, N.; Thongprachum, A.; Takanashi, S.; Okitsu, S.; Nishimura, S. Molecular epidemiology of rotavirus gastroenteritis in Japan during 2014-2015: Characterization of re-emerging G2P[4] after rotavirus vaccine introduction. J. Med. Virol. 2018, 90, 1040–1046. [Google Scholar] [CrossRef]
- Wandera, E.A.; Mohammad, S.; Bundi, M.; Komoto, S.; Nyangao, J.; Kathiiko, C.; Odoyo, E.; Miring’u, G.; Taniguchi, K.; Ichinose, Y. Impact of rotavirus vaccination on rotavirus and all-cause gastroenteritis in peri-urban Kenyan children. Vaccine 2017, 35, 5217–5223. [Google Scholar] [CrossRef]
- Mhango, C.; Mandolo, J.J.; Wachepa, R.; Kanjerwa, O.; Malamba-Banda, C.; Matambo, P.B.; Barnes, K.G.; Chaguza, C.; Shawa, I.T.; Nyaga, M.M.; et al. Rotavirus Genotypes in Hospitalized Children with Acute Gastroenteritis Before and After Rotavirus Vaccine Introduction in Blantyre, Malawi, 1997–2019. J. Infect. Dis. 2020, 225, 2127–2136. [Google Scholar] [CrossRef]
- Al-Ayed, M.S.Z.; Asaad, A.M.; Qureshi, M.A.; Hawan, A.A. Epidemiology of group A rotavirus infection after the introduction of monovalent vaccine in the National Immunization Program of Saudi Arabia. J. Med. Virol. 2017, 89, 429–434. [Google Scholar] [CrossRef]
- Page, N.A.; Steele, A.D. Antigenic and genetic characterization of serotype G2 human rotavirus strains from South Africa from 1984 to 1998. J. Med. Virol. 2004, 72, 320–327. [Google Scholar] [CrossRef]
- Vizzi, E.; Piñeros, O.A.; Oropeza, M.D.; Naranjo, L.; Suárez, J.A. Human rotavirus strains circulating in Venezuela after vaccine introduction: Predominance of G29[P4] and reemergence of G1P[8]. Virol. J. 2017, 14, 58. [Google Scholar] [CrossRef] [Green Version]
- Matthijnssens, J.; Zeller, M.; Heylen, E.; De Coster, S.; Vercauteren, J.; Braeckman, T.; Van Herck, K.; Meyer, N.; Pirçon, J.Y.; Soriano-Gabarro, M.; et al. Higher proportion of G2P[4] rotaviruses in vaccinated hospitalized cases compared with unvaccinated hospitalized cases, despite high vaccine effectiveness against heterotypic G2P[4] rotaviruses. Clin. Microbiol. Infect. 2014, 20, O702–O710. [Google Scholar] [CrossRef]
- Giammanco, G.M.; Bonura, F.; Zeller, M.; Heylen, E.; Van Ranst, M.; Martella, V.; Banyai, K.; Matthijnssens, J.; De Grazia, S. Evolution of DS-1-like human G2P[4] rotaviruses assessed by complete genome analyses. J. Gen. Virol. 2014, 95, 91–109. [Google Scholar] [CrossRef]
- Doan, Y.H.; Nakagomi, T.; Agbemabiese, C.A.; Nakagomi, O. Changes in the distribution of lineage constellations of G2P[4] Rotavirus A strains detected in Japan over 32 years (1980–2011). Infect. Genet. Evol. 2015, 34, 423–433. [Google Scholar] [CrossRef]
- Mwangi, P.N.; Page, N.A.; Seheri, M.L.; Mphahlele, M.J.; Nadan, S.; Esona, M.D.; Kumwenda, B.; Kamng’ona, A.W.; Donato, C.M.; Steele, A.D.; et al. Evolutionary changes between pre-and post-vaccine South African group A G2P[4] rotavirus strains, 2003–2017. Microb. Genom. 2022, 8, 000809. [Google Scholar] [CrossRef]
- Simwaka, J.C.; Mpabalwani, E.M.; Seheri, M.; Peenze, I.; Monze, M.; Matapo, B.; Parashar, U.D.; Mufunda, J.; Mphahlele, J.M.; Tate, J.E.; et al. Diversity of rotavirus strains circulating in children under five years of age who presented with acute gastroenteritis before and after rotavirus vaccine introduction, University Teaching Hospital, Lusaka, Zambia, 2008–2015. Vaccine 2018, 36, 7243–7247. [Google Scholar] [CrossRef]
- Gentsch, J.R.; Glass, R.I.; Woods, P.; Gouvea, V.; Gorziglia, M.; Flores, J.; Das, B.K.; Bhan, M.K. Identification of group A rotavirus gene 4 types by polymerase chain reaction. J. Clin. Microbiol. 1992, 30, 1365–1373. [Google Scholar] [CrossRef]
- Gouvea, V.; Glass, R.I.; Woods, P.; Taniguchi, K.; Clark, H.F.; Forrester, B.; Fang, Z.Y. Polymerase chain reaction amplification and typing of rotavirus nucleic acid from stool specimens. J. Clin. Microbiol. 1990, 28, 276–282. [Google Scholar] [CrossRef]
- Andrews, S.F. A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 October 2022).
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef]
- Hatcher, E.L.; Zhdanov, S.A.; Bao, Y.; Blinkova, O.; Nawrocki, E.P.; Ostapchuck, Y.; Schäffer, A.A.; Brister, J.R. Virus Variation Resource–improved response to emergent viral outbreaks. Nucleic Acids Res. 2017, 45, D482–D490. [Google Scholar] [CrossRef]
- Sayers, E.W.; Agarwala, R.; Bolton, E.E.; Brister, J.R.; Canese, K.; Clark, K.; Connor, R.; Fiorini, N.; Funk, K.; Hefferon, T.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2019, 47, D23. [Google Scholar] [CrossRef]
- Doan, Y.H.; Nakagomi, T.; Cunliffe, N.A.; Pandey, B.D.; Sherchand, J.B. The occurrence of amino acid substitutions D96N and S242N in VP7 of emergent G2P[4] rotaviruses in Nepal in 2004–2005: A global and evolutionary perspective. Arch. Virol. 2011, 156, 1960–1978. [Google Scholar] [CrossRef]
- Agbemabiese, C.A.; Nakagomi, T.; Damanka, S.A.; Dennis, F.E.; Lartey, B.L. Sub-genotype phylogeny of the non-G, non-P genes of genotype 2 Rotavirus A strains. PLoS ONE 2019, 14, e0217422. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Agbemabiese, C.A.; Nakagomi, T.; Doan, Y.H.; Do, L.P.; Damanka, S. Genomic constellation and evolution of Ghanaian G2P[4] rotavirus strains from a global perspective. Infect. Genet. Evol. 2016, 45, 122–131. [Google Scholar] [CrossRef]
- Dennis, A.F.; McDonald, S.M.; Payne, D.C.; Mijatovic-Rustempasic, S.; Esona, M.D.; Edwards, K.M.; Chappell, J.D.; Patton, J.T. Molecular epidemiology of contemporary G2P[4] human rotaviruses cocirculating in a single US community: Footprints of a globally transitioning genotype. J. Virol. 2014, 88, 3789–3801. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Foley, B.; Giorgi, E.E.; Bhattacharya, T.; Parker, M.D.; et al. Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. BioRxiv 2020. [Google Scholar]
- Franco, M.A.; Tin, C.; Greenberg, H.B. CD8+ T cells can mediate almost complete short-term and partial long-term immunity to rotavirus in mice. J. Virol. 1997, 71, 4165–4170. [Google Scholar] [CrossRef]
- Aledo, J.C. Methionine in proteins: The Cinderella of the proteinogenic amino acids. Protein Sci. 2019, 28, 1785–1796. [Google Scholar] [CrossRef]
- Manta, B.; Gladyshev, V.N. Regulated methionine oxidation by monooxygenases. Free. Radic. Biol. Med. 2017, 109, 141–155. [Google Scholar] [CrossRef]
- Keating, D.H.; Cronan, J.E. An isoleucine to valine substitution in Escherichia coli acyl carrier protein results in a functional protein of decreased molecular radius at elevated pH. J. Biol. Chem. 1996, 271, 15905–15910. [Google Scholar] [CrossRef]
- Fuentes-Pananá, E.M.; López, S.; Gorziglia, M.; Arias, C.F. Mapping the hemagglutination domain of rotaviruses. J. Virol. 1995, 69, 2629–2632. [Google Scholar] [CrossRef] [PubMed]
- Betts, M.J.; Russell, R.B. Amino acid properties and consequences of substitutions. Bioinform. Genet. 2003, 317, 289. [Google Scholar]
- Dowling, W.; Denisova, E.; LaMonica, R.; Mackow, E.R. Selective membrane permeabilization by the rotavirus VP5* protein is abrogated by mutations in an internal hydrophobic domain. J. Virol. 2000, 74, 6368–6376. [Google Scholar] [CrossRef] [PubMed]
- Creixell, P.; Schoof, E.M.; Tan, C.S.H.; Linding, R. Mutational properties of amino acid residues: Implications for evolvability of phosphorylatable residues. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2584–2593. [Google Scholar] [CrossRef] [PubMed]
- Ogden, K.M.; Snyder, M.J.; Dennis, A.F.; Patton, J.T. Predicted structure and domain organization of rotavirus capping enzyme and innate immune antagonist VP3. J. Virol. 2014, 88, 9072–9085. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Singh, A.; Kumar, P.; Uversky, V.N.; Rao, C.D.; Giri, R. Understanding the penetrance of intrinsic protein disorder in rotavirus proteome. Int. J. Biol. Macromol. 2020, 144, 892–908. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.M.; Verma, N.C.; Rao, C.; Uversky, V.N.; Nandi, C.K. Intrinsically disordered proteins of viruses: Involvement in the mechanism of cell regulation and pathogenesis. Prog. Mol. Biol. Transl. Sci. 2020, 174, 1–78. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lineages and Sub-Lineages | Representative G2 Sequences | AA and Position | |||
|---|---|---|---|---|---|
| 44 | 96 | 178 | 287 | ||
| G2-Lineage I | RVA/Human-tc/USA/DS-1/1976/G2P[4] | I | D | S | I |
| RVA/Human-wt/TWN/TW6/1981/G2P[4] | I | D | S | I | |
| G2-Lineage II | RVA/Human-tc/KEN/D205/1989/G2P[4] | I | D | S | I |
| RVA/Human-wt/ITA/PAI11/1996/G2P[4] | I | D | S | I | |
| G2-Lineage III | RVA/Human-wt/JPN/TMCII/1980/G2P[4] | I | D | S | I |
| RVA/Human-wt/JPN/KUN/1980/G2P[4] | I | D | S | I | |
| G2-Lineage IV-Sub-lineage IVa_1 | RVA/Human-wt/BRA/1250 06RJ/2006/G2P[4] | M | N | N | V |
| RVA/Human-wt/THA/CU438-KK/09/2009/G2P[4] | M | N | N | V | |
| G2-Lineage IV-Sub-lineage IVa_2 | RVA/Human-wt/ZAF/SA4476DB/97/1997/G2P[4] | M | N | N | V |
| RVA/Human-wt/ZAF/SA4419SB/97/1997/G2P[4] | M | N | N | V | |
| G2-Lineage IV-Sub-lineage IVa_3 | RVA/Human-wt/NPL/04N618/2004/G2P[4] | M | N | N | V |
| RVA/Human-wt/NPL/04N618/2004/G2P[4] | M | N | N | V | |
| RVA/Human-wt/ZMB/UFS-NGS-MRC-DPRU1724/2012/G2P[4] | M | N | N | V | |
| RVA/Human-wt/ZMB/UFS-NGS-MRC-DPRU1729/2012/G2P[4] | M | N | N | V | |
| RVA/Human-wt/ZMB/UFS-NGS-MRC-DPRU4691/2014/G2P[4] | M | N | N | V | |
| RVA/Human-wt/ZMB/UFS-NGS-MRC-DPRU4699/2014/G2P[4] | M | N | N | V | |
| RVA/Human-wt/ZMB/UFS-NGS-MRC-DPRU9556/2015/G2P[4] | M | N | N | V | |
| RVA/Human-wt/ZMB/UFS-NGS-MRC-DPRU9559/2015/G2P[4] | M | N | N | V | |
| RVA/Human-wt/ZMB/UFS-NGS-MRC-DPRU16639/2016/G2P[4] | M | N | N | V | |
| RVA/Human-wt/ZMB/UFS-NGS-MRC-DPRU16646/2016/G2P[4] | M | N | N | V | |
| G2-Lineage V | RVA/Human-wt/AUS/CK20055/2010/G2P[4] | I | D | S | I |
| RVA/Human-wt/AUS/CK20037/2008/G2P[4] | I | D | S | I | |
| Amino Acid Substitution | Region | Amino Acid Property |
|---|---|---|
| I44M | Cytotoxic T lymphocyte | No change in charge or polarity |
| D96N | Neutralization epitope region 7-1a | Polar negatively charged to polar neutral residue |
| S178N | β-barrel domain | No change in charge or polarity |
| I287V | Rossmann-fold domain |
| Lineages | Representative P[4] Sequences | AA and Position | ||
|---|---|---|---|---|
| 120 | 598 | 630 | ||
| P[4]-Lineage I | RVA/Human-wt/AUS/CK20001/1977/G2P4 | I | S | M |
| RVA/Human-tc/USA/DS-1/1976/G2P4 | I | S | M | |
| P[4]-Lineage II | RVA/Human-wt/MWI/BID124/2012/G2P4 | I | S | M |
| RVA/Human-wt/ITA/PAI11/1996/G2P[4] | I | S | M | |
| P[4]-Lineage III | RVA/Human-tc/JPN/KUN/1980/G2P4 | I | S | M |
| RVA/Human-wt/JPN/TMCII/1980/G2P[4] | I | S | M | |
| P[4]-Lineage IV | RVA/Human-wt/UGA/MRC-DPRU3710/2009/G2P4 | V | L | I |
| RVA/Human-wt/MWI/BID19T/2012/G2P4 | V | L | I | |
| RVA/Human-wt/ZMB/UFS-NGS-MRC-DPRU1724/2012/G2P[4] | V | L | I | |
| RVA/Human-wt/ZMB/UFS-NGS-MRC-DPRU1729/2012/G2P[4] | V | L | I | |
| RVA/Human-wt/ZMB/UFS-NGS-MRC-DPRU4691/2014/G2P[4] | V | L | I | |
| RVA/Human-wt/ZMB/UFS-NGS-MRC-DPRU4699/2014/G2P[4] | V | L | I | |
| RVA/Human-wt/ZMB/UFS-NGS-MRC-DPRU9556/2015/G2P[4] | V | L | I | |
| RVA/Human-wt/ZMB/UFS-NGS-MRC-DPRU9559/2015/G2P[4] | V | L | I | |
| RVA/Human-wt/ZMB/UFS-NGS-MRC-DPRU16639/2016/G2P[4] | V | L | I | |
| RVA/Human-wt/ZMB/UFS-NGS-MRC-DPRU16646/2016/G2P[4] | V | L | I | |
| Amino Acid Substitution | Region | Amino Acid Property |
|---|---|---|
| I120V | Hemagglutination domain | No change in charge or polarity |
| S598L | VP5 * | Polar neutral to nonpolar neutrally charged residue |
| M630I | VP5 * | No change in charge or polarity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mwangi, P.N.; Potgieter, R.-L.; Simwaka, J.; Mpabalwani, E.M.; Mwenda, J.M.; Mogotsi, M.T.; Magagula, N.; Esona, M.D.; Steele, A.D.; Seheri, M.L.; et al. Genomic Analysis of G2P[4] Group A Rotaviruses in Zambia Reveals Positive Selection in Amino Acid Site 7 of Viral Protein 3. Viruses 2023, 15, 501. https://doi.org/10.3390/v15020501
Mwangi PN, Potgieter R-L, Simwaka J, Mpabalwani EM, Mwenda JM, Mogotsi MT, Magagula N, Esona MD, Steele AD, Seheri ML, et al. Genomic Analysis of G2P[4] Group A Rotaviruses in Zambia Reveals Positive Selection in Amino Acid Site 7 of Viral Protein 3. Viruses. 2023; 15(2):501. https://doi.org/10.3390/v15020501
Chicago/Turabian StyleMwangi, Peter N., Robyn-Lee Potgieter, Julia Simwaka, Evans M. Mpabalwani, Jason M. Mwenda, Milton T. Mogotsi, Nonkululeko Magagula, Mathew D. Esona, A. Duncan Steele, Mapaseka L. Seheri, and et al. 2023. "Genomic Analysis of G2P[4] Group A Rotaviruses in Zambia Reveals Positive Selection in Amino Acid Site 7 of Viral Protein 3" Viruses 15, no. 2: 501. https://doi.org/10.3390/v15020501
APA StyleMwangi, P. N., Potgieter, R.-L., Simwaka, J., Mpabalwani, E. M., Mwenda, J. M., Mogotsi, M. T., Magagula, N., Esona, M. D., Steele, A. D., Seheri, M. L., & Nyaga, M. M. (2023). Genomic Analysis of G2P[4] Group A Rotaviruses in Zambia Reveals Positive Selection in Amino Acid Site 7 of Viral Protein 3. Viruses, 15(2), 501. https://doi.org/10.3390/v15020501