

Hyperinflammatory Response in COVID-19: A Systematic Review

 ,
, 
Abstract
:1. Introduction
2. Material and Methods
2.1. Study Design
2.2. Participants
2.3. Interventions and Comparators
2.4. Systematic Review Protocol
2.5. Search Strategy
2.6. Data Sources
2.7. Eligibility Criteria
2.8. Data Extraction
2.9. Methodological Quality and Risk of Bias Assessment
2.10. Data Analysis
3. Results
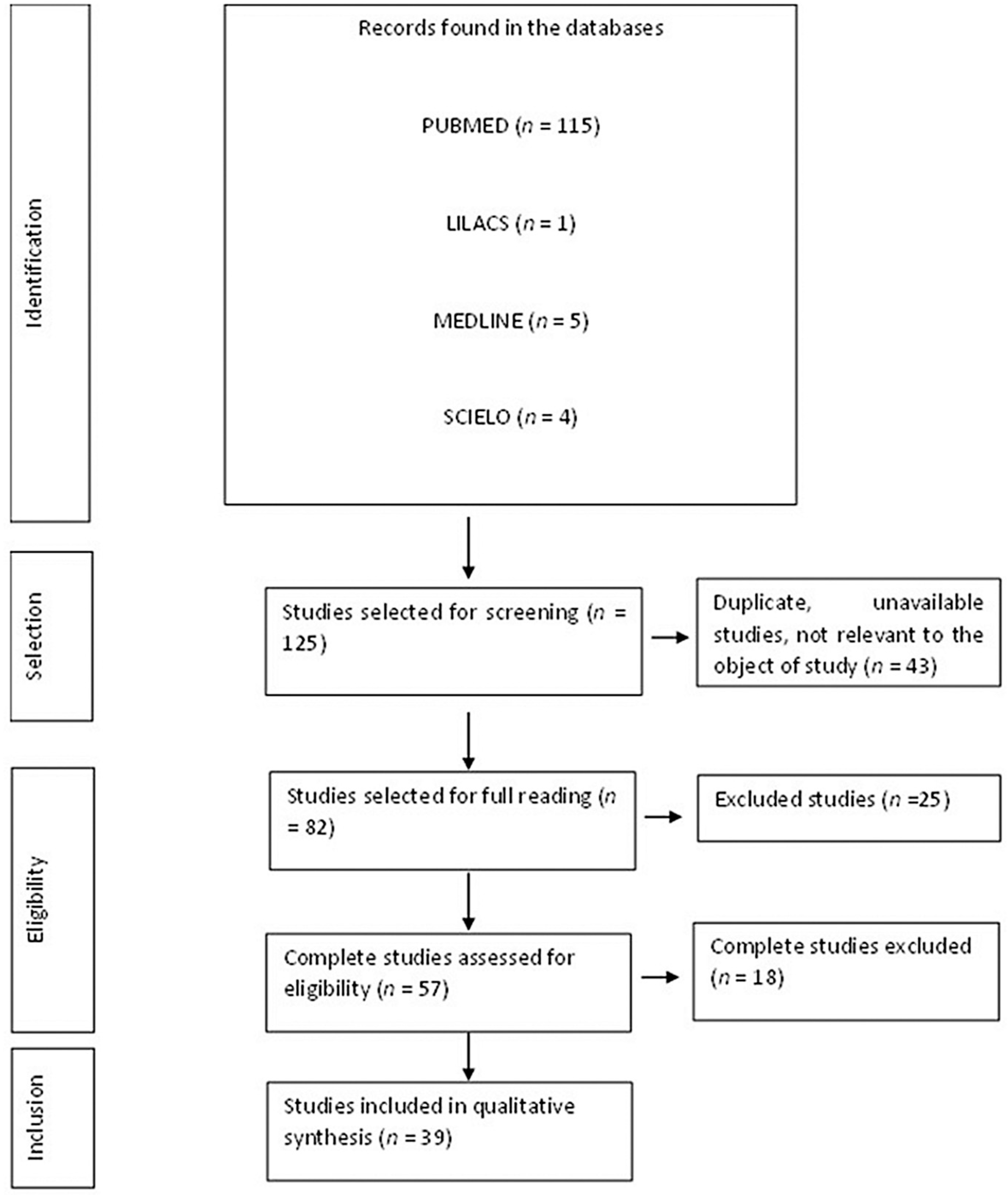
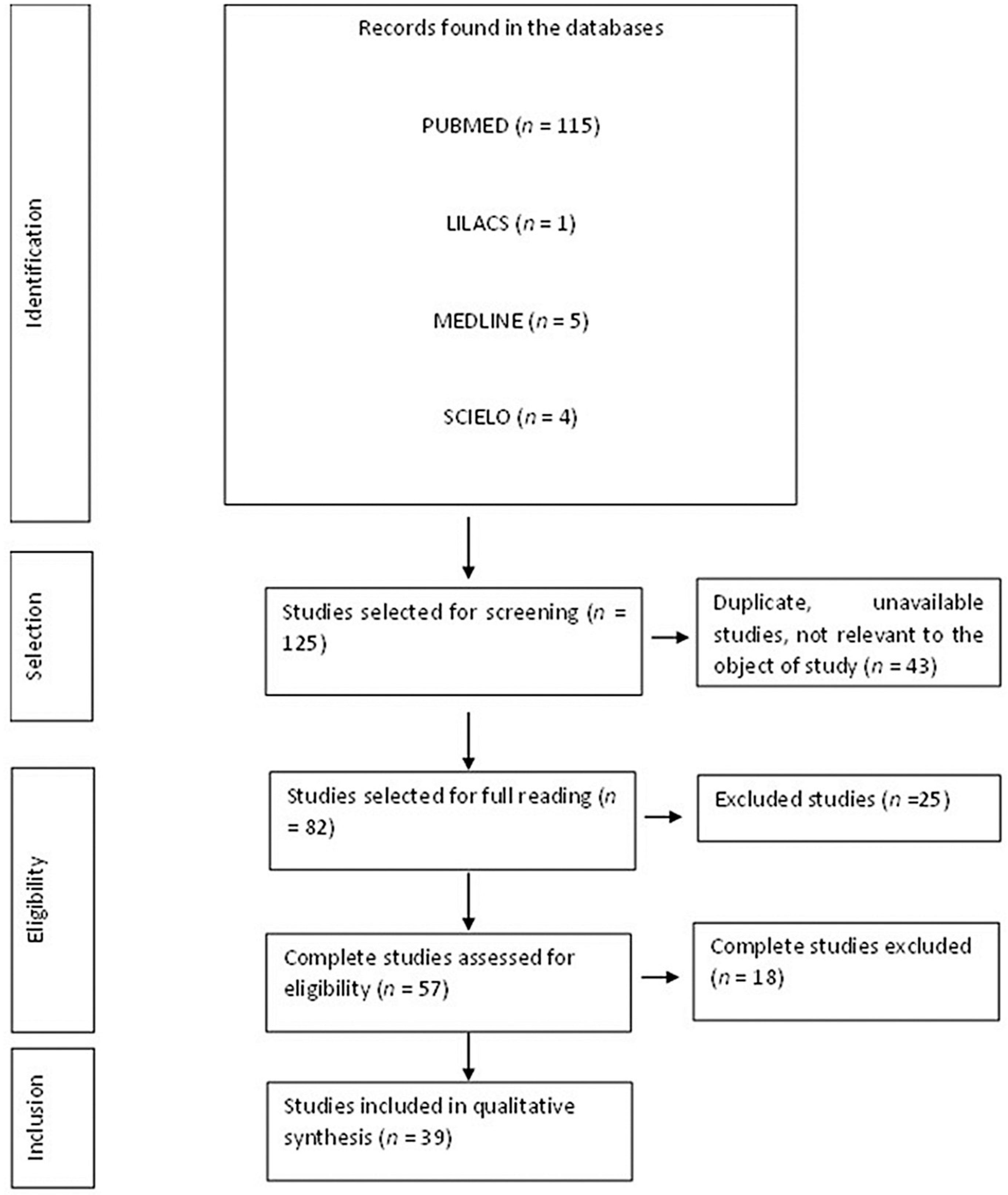
3.1. Flow Diagram of the Studies Retrieved for This Review
3.2. Study Selection and Characteristics
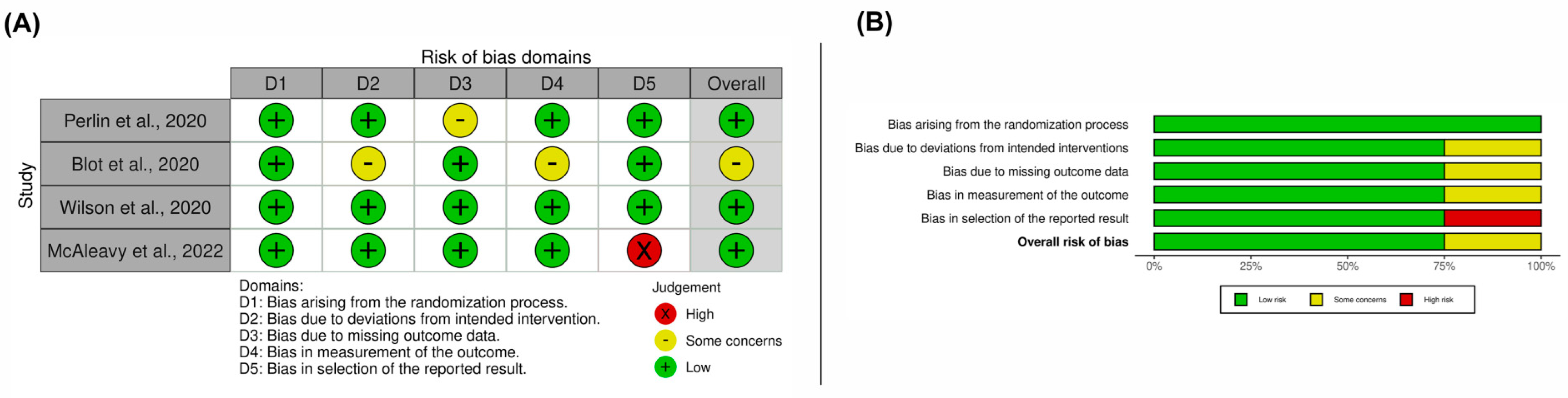
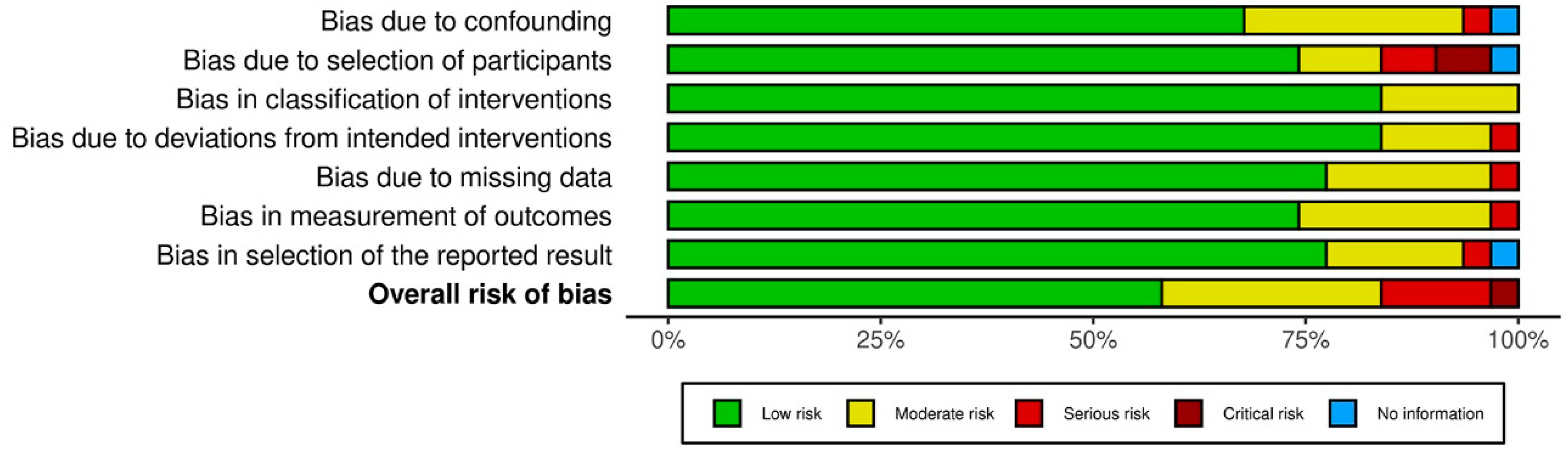
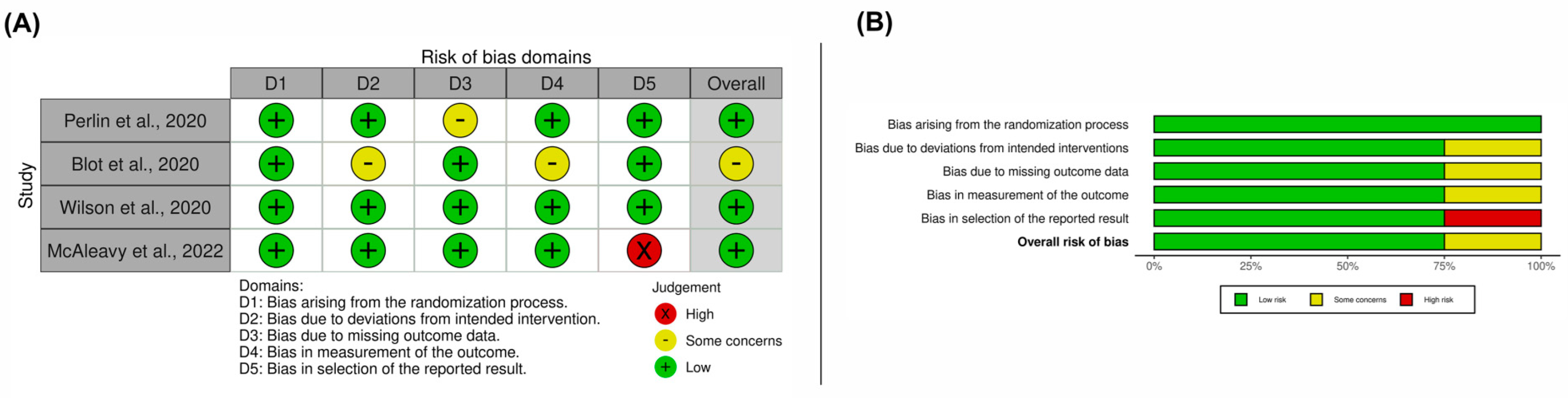
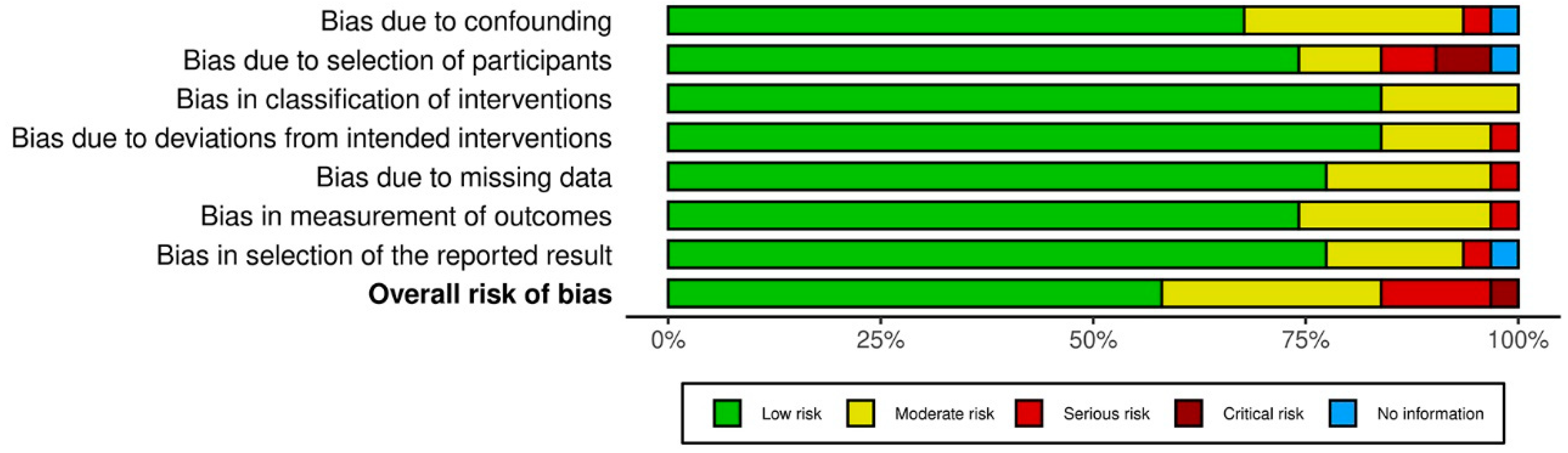
3.3. Results on Methodological Quality and Risk of Bias
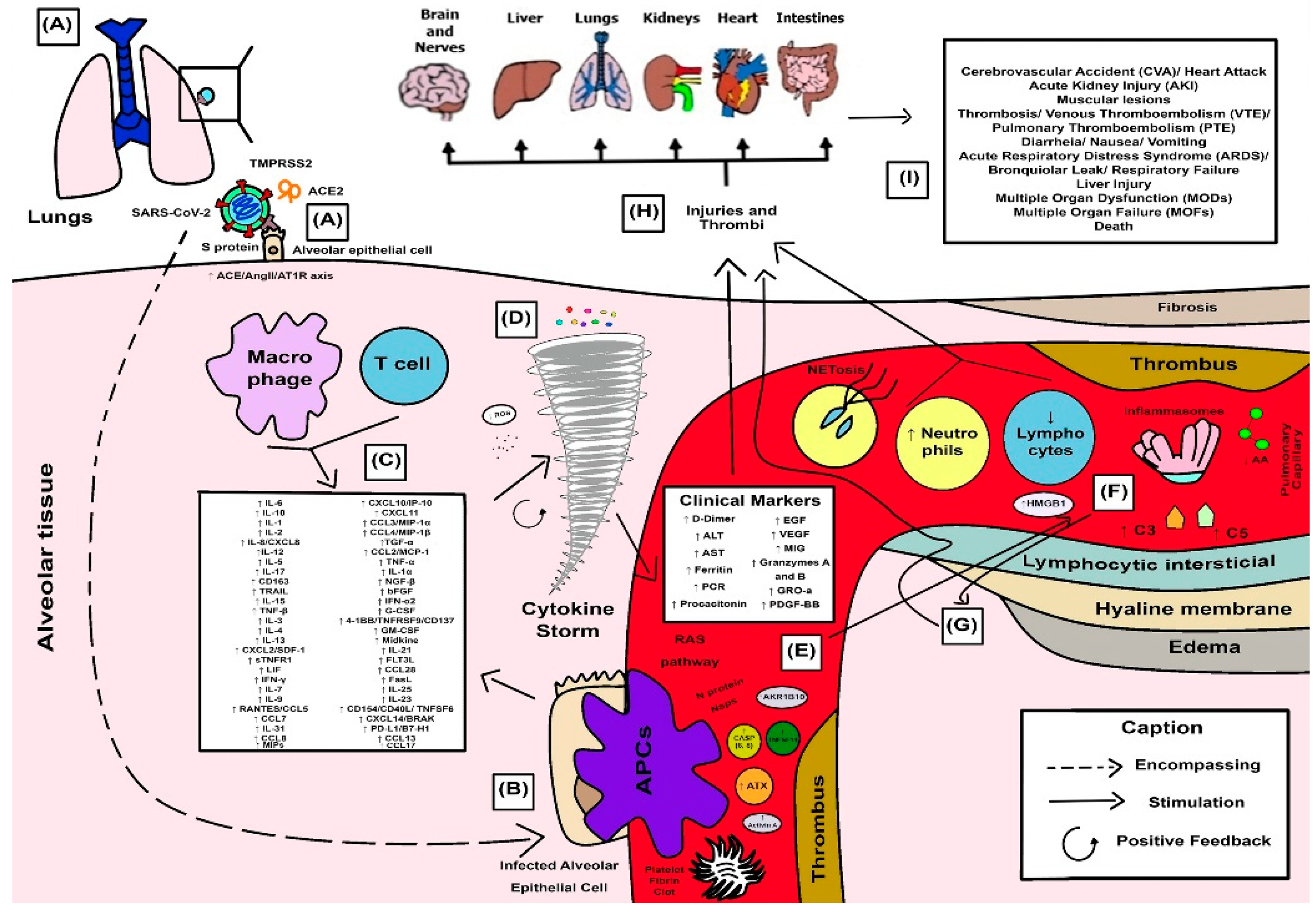
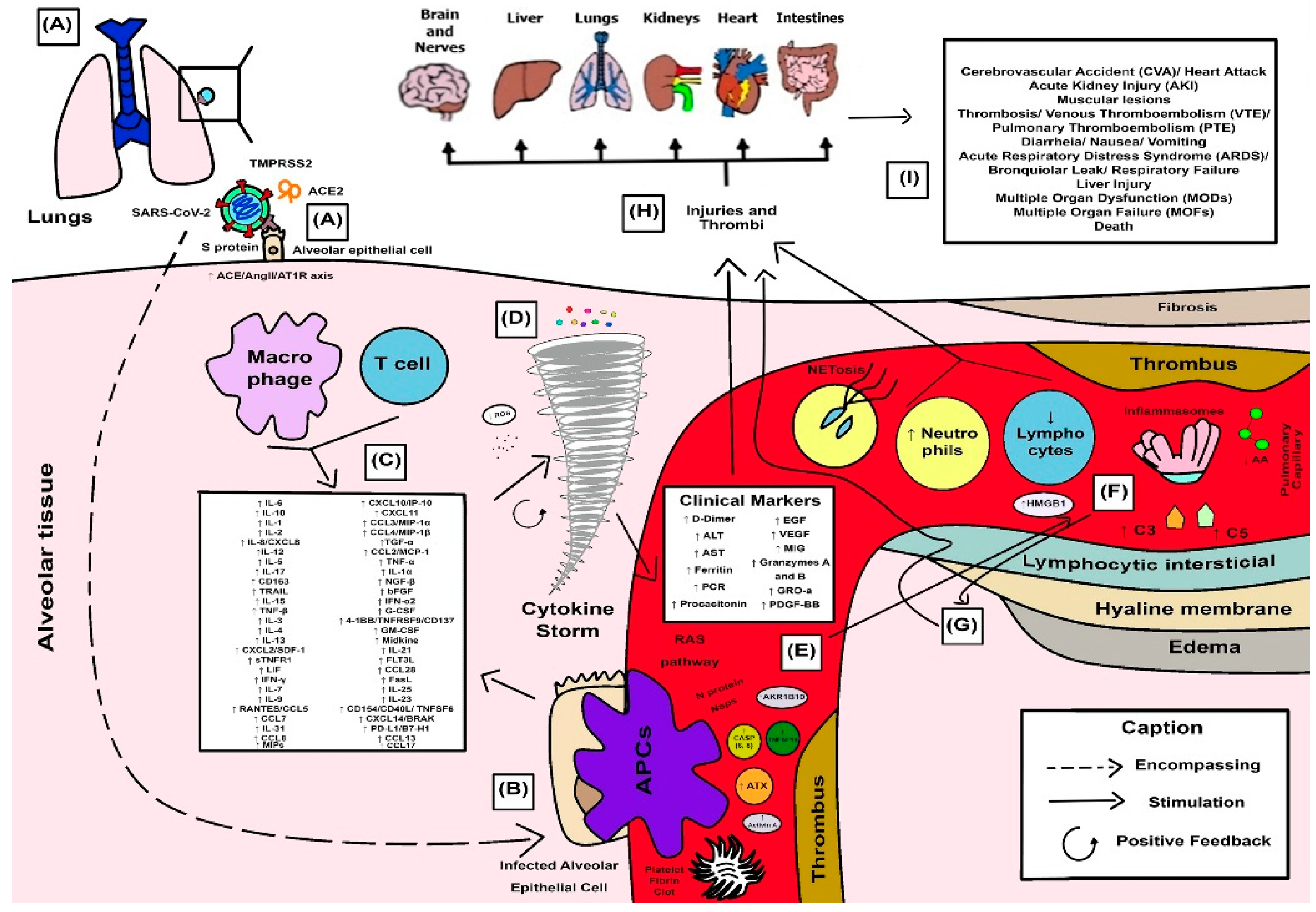
3.4. Synthesized Findings as a Didactic Illustrative Scheme for COVID-19 Response
4. Discussion
4.1. SARS-CoV-2 Spike Proteins and Their Combined Effects on Pro-Inflammatory Cellular Pathways
4.2. Cellular Protease-Mediated Pathogenic Device
4.3. Role of Leukocytes and the Excessive Exacerbation of Induced Innate Immune Response in a Hyperinflammatory Phenotype
4.4. Cytokine Storm and the Anger of Inflammation
4.5. Neutrophil Extracellular Traps (NETs) and Systemic Complications from COVID-19
4.6. Cytokines and Their Influence on Clinical and Laboratory Parameters Observed in Patients with Severe COVID-19
4.7. SARS-CoV-2 and Proteolytic Storm by Proteolytic Cascades (Coagulation, Fibrinolysis, Kinin, and Complement)
4.7.1. Coagulation and Fibrinolysis
4.7.2. Kinin Cascade
4.7.3. Complement Pathway
4.8. Co-Stimulatory Molecules Role in Tissue Inflammation
4.9. Final Considerations and Future Perspectives
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sethi, B.A.; Sethi, A.; Ali, S.; Aamir, H.S. Impact of Coronavirus disease (COVID-19) pandemic on health professionals. Pak. J. Med. Sci. 2020, 36, S6–S11. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.L.; Donaldson, E.F.; Baric, R.S. A decade after SARS: Strategies for controlling emerging coronaviruses. Nat. Rev. Microbiol. 2013, 11, 836–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabassum, A.; Iqbal, M.S.; Sultan, S.; Alhuthali, R.A.; Alshubaili, D.I.; Sayyam, R.S.; Abyad, L.M.; Qasem, A.H.; Arbaeen, A.F. Dysregulated Bradykinin: Mystery in the Pathogenesis of COVID-19. Mediat. Inflamm. 2022, 2022, e7423537. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Jia, H.P.; Look, D.C.; Shi, L.; Hickey, M.; Pewe, L.; Netland, J.; Farzan, M.; Wohlford-Lenane, C.; Perlman, S.; McCray, P.B., Jr. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 2005, 79, 14614–14621. [Google Scholar] [CrossRef] [Green Version]
- Li, S.R.; Tang, Z.J.; Li, Z.H.; Liu, X. Searching therapeutic strategy of new coronavirus pneumonia from angiotensin-converting enzyme 2: The target of COVID-19 and SARS-CoV. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1021–1026. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Mahmudpour, M.; Roozbeh, J.; Keshavarz, M.; Farrokhi, S.; Nabipour, I. COVID-19 cytokine storm: The anger of inflammation. Cytokine 2020, 133, 155151. [Google Scholar] [CrossRef] [PubMed]
- Pacurari, M.; Kafoury, R.; Tchounwou, P.B.; Ndebele, K. The Renin-Angiotensin-aldosterone system in vascular inflammation and remodeling. Int. J. Inflamm. 2014, 2014, 689360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantazi, I.; Al-Qahtani, A.A.; Alhamlan, F.S.; Alothaid, H.; Matou-Nasri, S.; Sourvinos, G.; Vergadi, E.; Tsatsanis, C. SARS-CoV-2/ACE2 Interaction Suppresses IRAK-M Expression and Promotes Pro-Inflammatory Cytokine Production in Macrophages. Front. Immunol. 2021, 12, 683800. [Google Scholar] [CrossRef] [PubMed]
- Haybar, H.; Maniati, M.; Saki, N.; Zayeri, Z.D. COVID-19: Imbalance of multiple systems during infection and importance of therapeutic choice and dosing of cardiac and anti-coagulant therapies. Mol. Biol. Rep. 2021, 48, 2917–2928. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, S.; Liu, J.; Zhang, Z.; Wan, X.; Huang, B.; Chen, Y.; Zhang, Y. COVID-19: Immunopathogenesis and Immunotherapeutics. Signal Transduct. Target. Ther. 2020, 5, 128. [Google Scholar] [CrossRef] [PubMed]
- Molina-Molina, M. Secuelas y consecuencias de la COVID-19. Med. Respir. 2020, 13, 71–77. [Google Scholar]
- Coperchini, F.; Chiovato, L.; Rotondi, M. Interleukin-6, CXCL10 and Infiltrating Macrophages in COVID-19-Related Cytokine Storm: Not One for All But All for One! Front. Immunol. 2021, 12, 668507. [Google Scholar] [CrossRef]
- Nouri, Y.; Weinkove, R.; Perret, R. T-cell intrinsic Toll-like receptor signaling: Implications for cancer immunotherapy and CAR T-cells. J. Immunother. Cancer 2021, 9, e003065. [Google Scholar] [CrossRef]
- Cheon, S.Y.; Koo, B.N. Inflammatory Response in COVID-19 Patients Resulting from the Interaction of the Inflammasome and SARS-CoV-2. Int. J. Mol. Sci. 2021, 22, 7914. [Google Scholar] [CrossRef]
- Chen, S.H.; Scott, X.O.; Marcelo, Y.F.; Almeida, V.W.; Blackwelder, P.L.; Yavagal, D.R.; Peterson, E.C.; Starke, R.M.; Dietrich, W.D.; Keane, R.W.; et al. Netosis and inflammasomes in large vessel occlusion thrombi. Front. Pharmacol. 2021, 11, 607287. [Google Scholar] [CrossRef]
- Planès, R.; Pinilla, M.; Santoni, K.; Hessel, A.; Passemar, C.; Lay, K.; Paillette, P.; Valadão, A.-L.C.; Robinson, K.S.; Bastard, P.; et al. Human NLRP1 is a sensor of pathogenic coronavirus 3CL proteases in lung epithelial cells. Mol. Cell. 2022, 82, 2385–2400.e9. [Google Scholar] [CrossRef]
- Colarusso, C.; Terlizzi, M.; Maglio, A.; Molino, A.; Candia, C.; Vitale, C.; Hansbro, P.M.; Vatrella, A.; Pinto, A.; Sorrentino, R. Activation of the AIM2 Receptor in Circulating Cells of Post-COVID-19 Patients With Signs of Lung Fibrosis Is Associated With the Release of IL-1α, IFN-α and TGF-β. Front. Immunol. 2022, 13, 934264. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Xie, X.; Feng, X.-L.; Xu, L.; Han, J.-B.; Yu, D.; Zou, Q.-C.; Liu, Q.; Li, X.; Ma, G.; et al. Specific inhibition of the NLRP3 inflammasome suppresses immune overactivation and alleviates COVID-19 like pathology in mice. EBioMedicine 2021, 75, 103803. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.S.; de Sá, K.S.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Gonçalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2020, 218, e20201707. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.Y.R.; Perlman, S. Proviral role of caspase-6 in coronavirus infections. Cell Res. 2022, 33, 7–8. [Google Scholar] [CrossRef]
- Cordeiro, L.P.; Crespo, I.R.G.; de Falcão, Q.M.L.B.; de Leite, Y.S.; Magalhães, I.E.; Silva, V.L. CONSIDERAÇÕES IMUNOLÓGICAS SOBRE A PATOGENIA DA INFECÇÃO PELO SARS-COV-2. Rev. Científica FMC 2020, 15, 69–86. [Google Scholar] [CrossRef]
- Görlinger, K.; Dirkmann, D.; Gandhi, A.; Simioni, P. COVID-19-Associated Coagulopathy and Inflammatory Response: What Do We Know Already and What Are the Knowledge Gaps? Anesth. Analg. 2020, 131, 1324–1333. [Google Scholar] [CrossRef]
- Basehore, M.J.; Howard, T.D.; Lange, L.A.; Moore, W.C.; Hawkins, G.A.; Marshik, P.L.; Harkins, M.S.; Meyers, D.A.; Bleecker, E.R. A comprehensive evaluation of IL4 variants in ethnically diverse populations: Association of total serum IgE levels and asthma in white subjects. J. Allergy Clin. Immunol. 2004, 114, 80–87. [Google Scholar] [CrossRef]
- Ruh, A.C.; Fernandes, D.; Artoni, R.F.; Favero, G.M. Inflamação: Entre a regeneração e a cicatrização. Publ. UEPG Ci Biol. Saúde. 2013, 19, 11–19. [Google Scholar] [CrossRef]
- Aarabi, S.; Longaker, M.T.; Gurtner, G.C. Hypertrophic scar formation following burns and trauma: New approaches to treatment. PLoS Med. 2007, 4, e234. [Google Scholar] [CrossRef]
- do Antonio, M.V.N.; Imperador, C.H.L.; Junior, C.R.E.; Chin, C.M.; Bosquesi, P.L. Tempestade de citocinas na COVID-19. ULAKES J. Med. 2020, 1. Available online: http://revistas.unilago.edu.br/index.php/ulakes/article/view/255 (accessed on 30 December 2021).
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Li, M.; Luo, G.; Wu, X.; Su, B.; Zhao, L.; Zhang, S.; Chen, X.; Jia, M.; Zhu, J.; et al. The Inflammatory Factors Associated with Disease Severity to Predict COVID-19 Progression. J. Immunol. 2021, 206, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, M.I.; Abdelmoneim, A.H.; Mahmoud, E.M.; Makhawi, A.M. Cytokine Storm in COVID-19 Patients, Its Impact on Organs and Potential Treatment by QTY Code-Designed Detergent-Free Chemokine Receptors. Mediat. Inflamm. 2020, 2020, 8198963. [Google Scholar] [CrossRef]
- Robba, C.; Battaglini, D.; Pelosi, P.; Rocco, P.R.M. Multiple organ dysfunction in SARS-CoV-2: MODS-CoV-2. Expert Rev. Respir. Med. 2020, 14, 865–868. [Google Scholar] [CrossRef]
- Silva, M.J.A.; Rodrigues, Y.C.; Lima, K.V.B.; Lima, L.N.G.C. Innate immunity to SARS-CoV-2 infection: A review. Epidemiol. Infect. 2022, 150, e142. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Rother, E.T. Revisão sistemática X revisão narrativa. Acta Paul. De Enferm. 2007, 20, v–vi. [Google Scholar] [CrossRef] [Green Version]
- da Santos, C.M.C.; de Pimenta, C.A.M.; Nobre, M.R.C. The PICO strategy for the research question construction and evidence search. Rev. Latino-Am. Enfermagem. 2007, 15, 508–511. [Google Scholar] [CrossRef] [Green Version]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Aromataris, E.; Munn, Z. JBI Manual for Evidence Synthesis; Joanna Briggs Institute: North Adelaide, Australia, 2020. [Google Scholar]
- Munn, Z.; Aromataris, E.; Tufanaru, C.; Stern, C.; Porritt, K.; Farrow, J.; Lockwood, C.; Stephenson, M.; Moola, S.; Lizarondo, L.; et al. The development of software to support multiple systematic review types: The Joanna Briggs Institute System for the Unified Management, Assessment and Review of Information (JBI SUMARI). Int. J. Evid. -Based Healthc. 2019, 17, 36–43. [Google Scholar] [CrossRef]
- Sterne, J.A.C.; Savović, J.; Page, M.J.; Elbers, R.G.; Blencowe, N.S.; Boutron, I.; Cates, C.J.; Cheng, H.Y.; Corbett, M.S.; Eldridge, S.M.; et al. RoB 2: A revised tool for assessing risk of bias in randomised trials. BMJ 2019, 28, l4898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterne, J.A.C.; Hernán, M.A.; Reeves, B.C.; Savović, J.; Berkman, N.D.; Viswanathan, M.; Henry, D.; Altman, D.G.; Ansari, M.T.; Boutron, I.; et al. ROBINS-I: A tool for assessing risk of bias in non-randomised studies of interventions. BMJ 2016, 12, i4919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samiei, M.; Shirazi, S.; Azar, F.P.; Fathifar, Z.; Ghojazadeh, M.; Alipour, M. The effect of different mixing methods on the properties of calcium-enriched mixture cement: A systematic review of in vitro studies. Iran. Endod. J. 2019, 14, 240–246. [Google Scholar] [PubMed]
- Neelakantan, P.; Ahmed, H.M.A.; Wong, M.C.M.; Matinlinna, J.P.; Cheung, G.S.P. Effect of root canal irrigation protocols on the dislocation resistance of mineral trioxide aggregate-based materials: A systematic review of laboratory studies. Int. Endod. J. 2018, 51, 847–861. [Google Scholar] [CrossRef]
- Wang, J.; Yang, X.; Li, Y.; Huang, J.-A.; Jiang, J.; Su, N. Specific cytokines in the inflammatory cytokine storm of patients with COVID-19-associated acute respiratory distress syndrome and extrapulmonary multiple-organ dysfunction. Virol. J. 2021, 18, 117. [Google Scholar] [CrossRef]
- Popadic, V.; Klasnja, S.; Milic, N.; Rajovic, N.; Aleksic, A.; Milenkovic, M.; Crnokrak, B.; Balint, B.; Todorovic-Balint, M.; Mrda, D.; et al. Predictors of Mortality in Critically Ill COVID-19 Patients Demanding High Oxygen Flow: A Thin Line between Inflammation, Cytokine Storm, and Coagulopathy. Oxidative Med. Cell. Longev. 2021, 2021, 6648199. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, D.; Hou, J.; Li, H.; Cao, D.; Guo, M.; Ling, Y.; Gao, M.; Zhou, Y.; Wan, Y.; et al. An inter-correlated cytokine network identified at the center of cytokine storm predicted COVID-19 prognosis. Cytokine 2021, 138, 155365. [Google Scholar] [CrossRef]
- Keddie, S.; Ziff, O.; Chou, M.; Taylor, R.; Heslegrave, A.; Garr, E.; Lakdawala, N.; Church, A.; Ludwig, D.; Manson, J.; et al. Laboratory biomarkers associated with COVID-19 severity and management. Clin. Immunol. 2020, 221, 108614. [Google Scholar] [CrossRef]
- Bouadma, L.; Wiedemann, A.; Patrier, J.; Surénaud, M.; Wicky, P.H.; Foucat, E.; Diehl, J.L.; Hejblum, B.P.; Sinnah, F.; de Montmollin, E.; et al. Immune Alterations in a Patient with SARS-CoV-2-Related Acute Respiratory Distress Syndrome. J. Clin. Immunol. 2020, 40, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Perlin, D.S.; Zafir-Lavie, I.; Roadcap, L.; Raines, S.; Ware, C.F.; Neil, G.A. Levels of the TNF-Related Cytokine LIGHT Increase in Hospitalized COVID-19 Patients with Cytokine Release Syndrome and ARDS. mSphere 2020, 5, e00699-20. [Google Scholar] [CrossRef] [PubMed]
- McElvaney, O.J.; McEvoy, N.L.; McElvaney, O.F.; Carroll, T.P.; Murphy, M.P.; Dunlea, D.M.; Ni Choileain, O.; Clarke, J.; O’Connor, E.; Hogan, G.; et al. Characterization of the Inflammatory Response to Severe COVID-19 Illness. Am. J. Respir. Crit. Care Med. 2020, 202, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Mandel, M.; Harari, G.; Gurevich, M.; Achiron, A. Cytokine prediction of mortality in COVID19 patients. Cytokine 2020, 134, 155190. [Google Scholar] [CrossRef]
- Bagheri-Hosseinabadi, Z.; Ostad Ebrahimi, H.; Bahrehmand, F.; Taghipour, G.; Abbasifard, M. The relationship between serum levels of interleukin-2 and IL-8 with circulating microRNA-10b in patients with COVID-19. Iran J. Immunol. 2021, 18, 65–73. [Google Scholar]
- Li, X.; Liu, H.; Meng, Y.; Yin, H.; Gao, W.; Yang, X.; Xu, D.; Cai, X.; Guan, Y.; Lerman, L.O.; et al. Critical roles of cytokine storm and secondary bacterial infection in acute kidney injury development in COVID-19: A multi-center retrospective cohort study. J. Med. Virol. 2021, 93, 6641–6652. [Google Scholar] [CrossRef]
- Gómez-Escobar, L.G.; Hoffman, K.L.; Choi, J.J.; Borczuk, A.; Salvatore, S.; Alvarez-Mulett, S.L.; Galvan, M.D.; Zhao, Z.; Racine-Brzostek, S.E.; Yang, H.S.; et al. Cytokine signatures of end organ injury in COVID-19. Sci. Rep. 2021, 11, 12606. [Google Scholar] [CrossRef]
- Gürsoy, B.; Sürmeli, C.D.; Alkan, M.; Satıcı, C.; Altunok, E.S.; Kamat, S.; Demirok, B.; Demirkol, M.A.; Börü, A. Cytokine storm in severe COVID-19 pneumonia. J. Med. Virol. 2021, 93, 5474–5480. [Google Scholar] [CrossRef]
- Ouwendijk, W.J.D.; Raadsen, M.P.; van Kampen, J.J.; Verdijk, R.M.; von der Thusen, J.H.; Guo, L.; Hoek, R.A.S.; Akker, J.P.C.V.D.; Endeman, H.; Langerak, T.; et al. High Levels of Neutrophil Extracellular Traps Persist in the Lower Respiratory Tract of Critically Ill Patients With Coronavirus Disease 2019. J. Infect. Dis. 2021, 223, 1512–1521. [Google Scholar] [CrossRef]
- Blot, M.; Jacquier, M.; Glele, L.-S.A.; Beltramo, G.; Nguyen, M.; Bonniaud, P.; Prin, S.; Andreu, P.; Bouhemad, B.; Bour, J.-B.; et al. CXCL10 could drive longer duration of mechanical ventilation during COVID-19 ARDS. Crit. Care. 2020, 24, 632. [Google Scholar] [CrossRef]
- Lorenz, G.; Moog, P.; Bachmann, Q.; La Rosée, P.; Schneider, H.; Schlegl, M.; Spinner, C.; Heemann, U.; Schmid, R.M.; Algül, H.; et al. Cytokine release syndrome is not usually caused by secondary hemophagocytic lymphohistiocytosis in a cohort of 19 critically ill COVID-19 patients. Sci. Rep. 2020, 10, 18277. [Google Scholar] [CrossRef] [PubMed]
- Notz, Q.; Schmalzing, M.; Wedekink, F.; Schlesinger, T.; Gernert, M.; Herrmann, J.; Sorger, L.; Weismann, D.; Schmid, B.; Sitter, M.; et al. Pro- and Anti-Inflammatory Responses in Severe COVID-19-Induced Acute Respiratory Distress Syndrome—An Observational Pilot Study. Front. Immunol. 2020, 11, 581338. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Calfee, C.S.; Cherian, S.; Brealey, D.; Cutler, S.; King, C.; Killick, C.; Richards, O.; Cheema, Y.; Bailey, C.; et al. Prevalence of phenotypes of acute respiratory distress syndrome in critically ill patients with COVID-19: A prospective observational study. Lancet Respir. Med. 2020, 8, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.G.; Simpson, L.J.; Ferreira, A.-M.; Rustagi, A.; Roque, J.; Asuni, A.; Ranganath, T.; Grant, P.M.; Subramanian, A.; Rosenberg-Hasson, Y.; et al. Cytokine profile in plasma of severe COVID-19 does not differ from ARDS and sepsis. JCI Insight 2020, 5, e140289. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xie, J.; Zhao, L.; Fei, X.; Zhang, H.; Tan, Y.; Nie, X.; Zhou, L.; Liu, Z.; Ren, Y.; et al. Alveolar macrophage dysfunction and cytokine storm in the pathogenesis of two severe COVID-19 patients. EBioMedicine 2020, 57, 102833. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.A.; Morales-Nebreda, L.; Markov, N.S.; Swaminathan, S.; Querrey, M.; Guzman, E.R.; Abbott, D.A.; Donnelly, H.K.; Donayre, A.; Goldberg, I.A.; et al. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature 2021, 590, 635–641. [Google Scholar] [CrossRef]
- Gu, S.X.; Tyagi, T.; Jain, K.; Gu, V.W.; Lee, S.H.; Hwa, J.M.; Kwan, J.M.; Krause, D.S.; Lee, A.I.; Halene, S.; et al. Thrombocytopathy and endotheliopathy: Crucial contributors to COVID-19 thromboinflammation. Nat. Rev. Cardiol. 2021, 18, 194–209. [Google Scholar] [CrossRef]
- Borges, L.; Pithon-Curi, T.C.; Curi, R.; Hatanaka, E. COVID-19 and Neutrophils: The Relationship between Hyperinflammation and Neutrophil Extracellular Traps. Mediat. Inflamm. 2020, 2020, 8829674. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target. Ther. 2020, 5, 235. [Google Scholar] [CrossRef]
- Panigrahy, D.; Gilligan, M.M.; Huang, S.; Gartung, A.; Cortés-Puch, I.; Sime, P.J.; Phipps, R.P.; Serhan, C.N.; Hammock, B.D. Inflammation resolution: A dual-pronged approach to averting cytokine storms in COVID-19? Cancer Metastasis Rev. 2020, 39, 337–340. [Google Scholar] [CrossRef]
- Quartuccio, L.; Fabris, M.; Sonaglia, A.; Peghin, M.; Domenis, R.; Cifù, A.; Curcio, F.; Tascini, C. Interleukin 6, soluble interleukin 2 receptor alpha (CD25), monocyte colony-stimulating factor, and hepatocyte growth factor linked with systemic hyperinflammation, innate immunity hyperactivation, and organ damage in COVID-19 pneumonia. Cytokine 2021, 140, 155438. [Google Scholar] [CrossRef] [PubMed]
- Welcome, M.O.; Mastorakis, N.E. Neuropathophysiology of coronavirus disease 2019: Neuroinflammation and blood brain barrier disruption are critical pathophysiological processes that contribute to the clinical symptoms of SARS-CoV-2 infection. Inflammopharmacol 2021, 29, 939–963. [Google Scholar] [CrossRef] [PubMed]
- Gando, S.; Wada, T. Thromboplasminflammation in COVID-19 Coagulopathy: Three Viewpoints for Diagnostic and Therapeutic Strategies. Front. Immunol. 2021, 12, 649122. [Google Scholar] [CrossRef] [PubMed]
- Hoxha, M. What about COVID-19 and arachidonic acid pathway? Eur. J. Clin. Pharmacol. 2020, 76, 1501–1504. [Google Scholar] [CrossRef]
- Wang, J.; Kaplan, N.; Wysocki, J.; Yang, W.; Lu, K.; Peng, H.; Batlle, D.; Lavker, R.M. The ACE2-deficient mouse: A model for a cytokine storm-driven inflammation. FASEB J. 2020, 34, 10505–10515. [Google Scholar] [CrossRef]
- Morrell, E.D.; Bhatraju, P.K.; Sathe, N.A.; Lawson, J.; Mabrey, L.; Holton, S.E.; Presnell, S.R.; Wiedeman, A.; Acosta-Vega, C.; Mitchem, M.A.; et al. Chemokines, soluble PD-L1, and immune cell hyporesponsiveness are distinct features of SARS-CoV-2 critical illness. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2022, 323, L14–L26. [Google Scholar] [CrossRef]
- Ventura-Santana, E.; Ninan, J.R.; Snyder, C.M.; Okeke, E.B. Neutrophil Extracellular Traps, Sepsis and COVID-19—A Tripod Stand. Front Immunol. 2022, 13, 902206. [Google Scholar] [CrossRef]
- Chabert, C.; Vitte, A.L.; Iuso, D.; Chuffart, F.; Trocme, C.; Buisson, M.; Poignard, P.; Lardinois, B.; Debois, R.; Rousseaux, S.; et al. AKR1B10, One of the Triggers of Cytokine Storm in SARS-CoV2 Severe Acute Respiratory Syndrome. Int. J. Mol. Sci. 2022, 23, 1911. [Google Scholar] [CrossRef]
- Nishitsuji, H.; Iwahori, S.; Ohmori, M.; Shimotohno, K.; Murata, T. Ubiquitination of SARS-CoV-2 NSP6 and ORF7a Facilitates NF-κB Activation. mBio 2022, 13, e00971-22. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Alkazmi, L.; Habotta, O.A.; Batiha, G.E.-S. High-mobility group box 1 (HMGB1) in COVID-19: Extrapolation of dangerous liaisons. Inflammopharmacology 2022, 30, 811–820. [Google Scholar] [CrossRef]
- Combadière, B.; Adam, L.; Guillou, N.; Quentric, P.; Rosenbaum, P.; Dorgham, K.; Bonduelle, O.; Parizot, C.; Sauce, D.; Mayaux, J.; et al. LOX-1-Expressing Immature Neutrophils Identify Critically-Ill COVID-19 Patients at Risk of Thrombotic Complications. Front. Immunol. 2021, 12, 752612. [Google Scholar] [CrossRef] [PubMed]
- Nikitopoulou, I.; Fanidis, D.; Ntatsoulis, K.; Moulos, P.; Mpekoulis, G.; Evangelidou, M.; Vassiliou, A.G.; Dimakopoulou, V.; Jahaj, E.; Tsipilis, S.; et al. Increased Autotaxin Levels in Severe COVID-19, Correlating with IL-6 Levels, Endothelial Dysfunction Biomarkers, and Impaired Functions of Dendritic Cells. Int. J. Mol. Sci. 2021, 22, 10006. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Wang, A.; Fang, Y.; Shu, T.; Wu, D.; Wang, C.; Huang, M.; Min, J.; Jin, L.; Zhou, W.; et al. SARS-CoV-2 Membrane Glycoprotein M Triggers Apoptosis With the Assistance of Nucleocapsid Protein N in Cells. Front. Cell Infect. Microbiol. 2021, 11, 706252. [Google Scholar] [CrossRef] [PubMed]
- McAleavy, M.; Zhang, Q.; Ehmann, P.J.; Xu, J.; Wipperman, M.F.; Ajithdoss, D.; Pan, L.; Wakai, M.; Simonson, R.; Gadi, A.; et al. The Activin/FLRG Pathway Associates with Poor COVID-19 Outcomes in Hospitalized Patients. Mol. Cell Biol. 2022, 42, e00467–e00521. [Google Scholar] [CrossRef] [PubMed]
- Jamilloux, Y.; Henry, T.; Belot, A.; Viel, S.; Fauter, M.; El Jammal, T.; Walzer, T.; François, B.; Sève, P. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun. Rev. 2020, 19, 102567. [Google Scholar] [CrossRef]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.; Savani, R.C.; Zaki, H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. bioRxiv 2021, 10, e68563. [Google Scholar]
- El-Shimy, I.A.; Mohamed, M.M.A.; Hasan, S.S.; Hadi, M.A. Targeting host cell proteases as a potential treatment strategy to limit the spread of SARS-CoV-2 in the respiratory tract. Pharmacol. Res. Perspect. 2021, 9, e00698. [Google Scholar] [CrossRef]
- He, R.; Lu, Z.; Zhang, L.; Fan, T.; Xiong, R.; Shen, X.; Feng, H.; Meng, H.; Lin, W.; Jiang, W.; et al. The clinical course and its correlated immune status in COVID-19 pneumonia. J. Clin. Virol. 2020, 127, 104361. [Google Scholar] [CrossRef]
- Lu, G.; Wang, J. Dynamic changes in routine blood parameters of a severe COVID-19 case. Clin. Chim. Acta 2020, 508, 98–102. [Google Scholar] [CrossRef]
- Zhu, B.; Feng, X.; Jiang, C.; Mi, S.; Yang, L.; Zhao, Z.; Zhang, Y.; Zhang, L. Correlation between white blood cell count at admission and mortality in COVID-19 patients: A retrospective study. BMC Infect. Dis. 2021, 21, 574. [Google Scholar] [CrossRef]
- Swain, S.L.; McKinstry, K.K.; Strutt, T.M. Expanding roles for CD4+ T cells in immunity to viruses. Nat. Rev. Immunol. 2012, 12, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Hirawat, R.; Saifi, M.A.; Godugu, C. Targeting inflammatory cytokine storm to fight against COVID-19 associated severe complications. Life Sci. 2021, 267, 118923. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Fang, Y.-Y.; Deng, Y.; Liu, W.; Wang, M.-F.; Ma, J.-P.; Xiao, W.; Wang, Y.-N.; Zhong, M.-H.; Li, C.-H.; et al. Clinical characteristics of novel coronavirus cases in tertiary hospitals in Hubei Province. Chin. Med. J. 2020, 133, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Miyamae, T.; Kuroiwa, Y.; Nishioka, K. Novel Coronavirus Disease 2019 (COVID-19) and Cytokine Storms for More Effective Treatments from an Inflammatory Pathophysiology. J. Clin. Med. 2021, 10, 801. [Google Scholar] [CrossRef]
- Petiz, L.L.; Glaser, T.; Scharfstein, J.; Ratajczak, M.Z.; Ulrich, H. P2Y14 Receptor as a Target for Neutrophilia Attenuation in Severe COVID-19 Cases: From Hematopoietic Stem Cell Recruitment and Chemotaxis to Thrombo-inflammation. Stem Cell Rev. Rep. 2021, 17, 241–252. [Google Scholar] [CrossRef]
- Alfaidi, M.; Wilson, H.; Daigneault, M.; Burnett, A.; Ridger, V.; Chamberlain, J.; Francis, S. Neutrophil Elastase Promotes Interleukin-1β Secretion from Human Coronary Endothelium. J. Biol. Chem. 2015, 290, 24067–24078. [Google Scholar] [CrossRef] [Green Version]
- Renu, K.; Prasanna, P.L.; Valsala Gopalakrishnan, A. Coronaviruses pathogenesis, comorbidities and multi-organ damage—A review. Life Sci. 2020, 255, 117839. [Google Scholar] [CrossRef]
- Cheng, Y.; Luo, R.; Wang, K.; Zhang, M.; Wang, Z.; Dong, L.; Li, J.; Yao, Y.; Ge, S.; Xu, G. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020, 97, 829–838. [Google Scholar] [CrossRef]
- Ji, P.; Zhu, J.; Zhong, Z.; Li, H.; Pang, J.; Li, B.; Zhang, J. Association of elevated inflammatory markers and severe COVID-19: A meta-analysis. Medicine 2020, 99, e23315. [Google Scholar] [CrossRef]
- Lazzaroni, M.G.; Piantoni, S.; Masneri, S.; Garrafa, E.; Martini, G.; Tincani, A.; Andreoli, L.; Franceschini, F. Coagulation dysfunction in COVID-19: The interplay between inflammation, viral infection and the coagulation system. Blood Rev. 2021, 46, 100745. [Google Scholar] [CrossRef]
- Meizoso, J.P.; Moore, H.B.; Moore, E.E. Fibrinolysis Shutdown in COVID-19: Clinical Manifestations, Molecular Mechanisms, and Therapeutic Implications. J. Am. Coll. Surg. 2021, 232, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Ng, M.H.; Li, C.K. Thrombocytopenia in patients with severe acute respiratory syndrome (review). Hematology 2005, 10, 101–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizuka, T.; Cheng, J.; Singh, H.; Vitto, M.D.; Manthati, V.L.; Falck, J.R.; Laniado-Schwartzman, M. 20-Hydroxyeicosatetraenoic acid stimulates nuclear factor-kappaB activation and the production of inflammatory cytokines in human endothelial cells. J. Pharmacol. Exp. Ther. 2008, 324, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Ripon, A.R.; Bhowmik, D.R.; Amin, M.T.; Hossain, M.S. Role of arachidonic cascade in COVID-19 infection: A review. Prostaglandins Other Lipid Mediat. 2021, 154, 106539. [Google Scholar] [CrossRef]
- Martens, C.P.; Van Mol, P.; Wauters, J.; Wauters, E.; Gangnus, T.; Noppen, B.; Callewaert, H.; Feyen, J.H.; Liesenborghs, L.; Heylen, E.; et al. Dysregulation of the kallikrein-kinin system in bronchoalveolar lavage fluid of patients with severe COVID-19. eBioMedicine 2022, 1, 83. Available online: https://www.thelancet.com/journals/ebiom/article/PIIS2352-3964(22)00377-2/fulltext (accessed on 8 October 2022). [CrossRef]
- Wagner, K.; Vito, S.; Inceoglu, B.; Hammock, B.D. The role of long chain fatty acids and their epoxide metabolites in nociceptive signaling. Prostaglandins Other Lipid Mediat. 2014, 113–115, 2–12. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, K. Soluble epoxide hydrolase: A new therapeutic target for depression. Expert Opin. Ther. Targets 2016, 20, 1149–1151. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Xie, X.; Tu, Z.; Fu, J.; Xu, D.; Zhou, Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct. Target. Ther. 2021, 6, 255. [Google Scholar] [CrossRef]
- Haljasmägi, L.; Salumets, A.; Rumm, A.P.; Jürgenson, M.; Krassohhina, E.; Remm, A.; Sein, H.; Kareinen, L.; Vapalahti, O.; Sironen, T.; et al. Longitudinal proteomic profiling reveals increased early inflammation and sustained apoptosis proteins in severe COVID-19. Sci. Rep. 2020, 10, 20533. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Balasubramaniam, V.R.; Othman, I.; Shaikh, M.F. Enlightening the role of high mobility group box 1 (HMGB1) in inflammation: Updates on receptor signalling. Eur. J. Pharmacol. 2019, 858, 172487. [Google Scholar] [CrossRef]
- Qu, L.; Chen, C.; Chen, Y.; Li, Y.; Tang, F.; Huang, H.; He, W.; Zhang, R.; Shen, L. High-Mobility Group Box 1 (HMGB1) and Autophagy in Acute Lung Injury (ALI): A Review. Med. Sci. Monit. 2019, 25, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Trovato, F.M.; Zia, R.; Napoli, S.; Wolfer, K.; Huang, X.; Morgan, P.E.; Husbyn, H.; Elgosbi, M.; Lucangeli, M.; Miquel, R. Dysregulation of the LPC-ATX-LPA axis in ACLF is associated with mortality and systemic inflammation via LPA-dependent monocyte activation. Hepatology 2021, 3, 22. [Google Scholar]
- Simonovich, V.A.; Pratx, L.D.B.; Scibona, P.; Beruto, M.V.; Vallone, M.G.; Vázquez, C.; Savoy, N.; Giunta, D.H.; Pérez, L.G.; Sánchez, M.D.L.; et al. A Randomized Trial of Convalescent Plasma in Covid-19 Severe Pneumonia. N. Engl. J. Med. 2021, 384, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Trendelenburg, A.U.; Meyer, A.; Jacobi, C.; Feige, J.N.; Glass, D.J. TAK-1/p38/nNFκB signaling inhibits myoblast differentiation by increasing levels of Activin A. Skelet. Muscle 2012, 2, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campanella, E.; Marino, A.; Ceccarelli, M.; Gussio, M.; Cosentino, F.; Moscatt, V.; Micali, C.; Nunnari, G.; Celesia, B.M.; Cacopardo, B. Pain crisis management in a patient with sickle cell disease during SARS-CoV-2 infection: A case report and literature review. World Acad. Sci. J. 2022, 4, 14. [Google Scholar] [CrossRef]
- Marino, A.; Campanella, E.; Ceccarelli, M.; Larocca, L.; Bonomo, C.; Micali, C.; Munafò, A.; Celesia, B.M.; Nunnari, G.; Cacopardo, B. Sarilumab administration in patients with severe COVID-19: A report of four cases and a literature review. World Acad. Sci. J. 2022, 4, 24. [Google Scholar] [CrossRef]
- Marino, A.; Munafò, A.; Augello, E.; Bellanca, C.M.; Bonomo, C.; Ceccarelli, M.; Musso, N.; Cantarella, G.; Cacopardo, B.; Bernardini, R. Sarilumab Administration in COVID-19 Patients: Literature Review and Considerations. Infect. Dis. Rep. 2022, 14, 360–371. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors and Year of Publication | Database | Methodology | Objective | Methodological Quality (JBI Score) | Results |
|---|---|---|---|---|---|
| Wang et al., 2021 [48]. | PUBMED | Cohort study | To analyze the correlations of inflammatory cytokine levels with clinical and laboratory variables and explore the relationships of different cytokines with severe acute respiratory distress syndrome (ARDS) and extrapulmonary multiple organ dysfunction (MOD). | (9/11) | Cytokine storms contributed to ARDS and extrapulmonary MOD. Interleukins 6 (IL-6), IL-8/CXCL8, IL-10, and tumor necrosis factor α (TNF-α) had higher serum levels in the ARDS group than the controls, and these levels continuously increased after hospital (ICU) admission. The levels of these cytokines were correlated with coagulation and disseminated intravascular coagulation (DIC) parameters. IL-6 and TNF-α levels correlated with creatinine and urea nitrogen levels and were also higher in patients with ARDS and acute kidney injury (AKI). Elevated levels of cytokines were related to low O2 levels (PaO2)/inspired O2 fraction (FiO2). Elevated IL-6, IL-8/CXCL8, and TNF-α levels showed positive correlations with the Acute Physiology and Chronic Health Evaluation-II (APACHE-II) score. Nonsurvivors had higher levels of IL-6 and IL-10 on admission to the ICU and increased levels over time. The ARDS group had significantly higher levels of white blood cells, neutrophils, and incidence of lymphopenia. |
| Popadic et al., 2021 [49]. | PUBMED | Cohort study | To evaluate the potential independent predictors of mortality in critically ill patients with COVID-19 and ARDS. | (11/11) | Patients with moderate to severe ARDS showed elevated serum albumin, D-dimer, and IL-6 levels upon admission to the ICU, accompanied by an elevated chest CT severity score as independent predictors of mortality. Patients who died had lower levels of lymphocytes on admission to the hospital and, on admission to the ICU, had higher levels of D-dimer and IL-6 and lower levels of lymphocytes and serum albumin. The cytokine storm was presented as one of the crucial pathophysiological mechanisms for multiple organ failure and death in patients with severe COVID-19 infection. |
| Liu et al., 2021 [50]. | PUBMED | Case-control | Analyze the peak and early (within 10 days of disease onset) concentrations of 12 cytokines in the plasma. | (10/11) | The cytokines IL-5, IL-2, IL-6, IL-10, IFN-γ, IL-8/CXCL8, IL-17, and IL-12p70 showed elevated concentrations among the 12 mediators. IL-5, IL-8/CXCL8, and IL-6 increased significantly among the deceased patients but did not differ significantly between those with mild/moderate and severe symptoms. Serum levels of IFN-α and IL-2 correlated significantly with the duration of the disease course. Additional analyzes showed that IL-6 and IL-8/CXCL8 were negatively correlated with the relative ratios (%) of CD3+ T cells—the number and percentage of CD3+ T cells in the peripheral blood, thus indirectly proving that IL-6 and IL-8/CXCL8 correlated with disease severity. These data imply that IL-6, IL-8/CXCL8, and IL-5 are central players in the COVID-19-related cytokine storm. |
| Keddie et al., 2020 [51]. | PUBMED | Cohort study | Compare clinical features of the disease and routine laboratory tests with specialized cytokine biomarkers associated with COVID-19 disease and its complications. | (11/11) | IL-6, C-reactive protein (CRP), IL-10, lactate dehydrogenase (LDH), and TNF-α are indicative of distinct aspects of COVID-19 severity, such as the need for oxygen, the presence of ARDS, and the need for intensive care support, including dialysis and ventilation. Elevated levels of these biomarkers are associated with the greater severity of COVID-19. IL-1β was the only biomarker that differed significantly by sex, with higher levels in men, and was related to higher mortality. D-dimer, ferritin, and lymphocytes were inversely correlated with all cytokine levels and indicative of severe COVID-19. |
| Bouadma et al., 2020 [52]. | PUBMED | Case Report | Provide a complete description of a fatal case of COVID-19 in Europe, including the chronological immune profile of the patient. | (8/8) | On day fourteen of the disease (D14), a storm of pro-inflammatory factors and Th1/Th2 cells was detected; some tended to decrease with follow-up, these being interferon-gamma (IFN-γ), MIP-1α/CCL3, MIP-1β/CCL4, transforming growth factor alpha (TGF-α), monocyte chemoattractant protein-1 (MCP-1/CCL2), TNF-α, interleukin-1 alpha (IL-1α), Nerve Growth Factor-beta (NGF-β), basic FGF (FGFb), IFN-α2, IL-5, G-CSF, as well as a burst of Th1 cytokines (IL-2, IL-3, IL-12), Th2 cytokines (IL-4, IL-5, IL-6), and an immunomodulator (IL-1RA). In the immune profile, there was a significant increase in the level of other markers: 4-1BB/TNFRSF9/CD137, GM-CSF, Neutrites growth-promoting factor 2 (Midkine), IL-21, Flt-3 ligand, chemokine ligand 28 (CCL28, also known as mucosae-associated epithelial chemokine-MEC, CCK1 and SCYA28), Fas/TNFSF6 ligand, IL-17E/IL-25, IL-23, CD40/CD154/TNFSF5 ligand, CXCL14/BRAK, IL-31, Granzyme A, and PD-L1/B7-H1 associated with T-cell activation, depletion, and apoptosis. IL-1RA decreased dramatically from day 15 to day 22 of the disease (D15 to D22), in contrast, to persistently high levels of IL-1. In D20, a significant increase in the level of biomarkers of cellular cytotoxicity, neutrophil chemotaxis, and endothelial activation was observed (MIG, VEGF, IL-7, granzyme B, CXCL1/GRO-a, PDGF-BB, RANTES/CCL5, IL-8/CXCL8, IL-9, EGF), and after that, a strong and rapid increase in blood neutrophil counts were observed from D21 to D23. After the dose of IFN-β 1a on D23, there was a dramatic increase in the level of several cytokines, reflecting T cell activation, monocytes, and inflammation (IL-2, IP-10/CXCL10, TNF-related apoptosis-inducing ligand [TRAIL], IL-17, IL-12 (p70), CD163, IL-12 (p40), IL-15, TNF-β, CXCL2/SDF-1a, LIF, IL-1β), as well as an anti-inflammatory profile (IL-3, IL-4, IL-13, IL-1RA) and a permeable bowel (I-FABP). On D24, death occurred. |
| Perlin et al., 2020 [53]. | PUBMED | Clinical Trial | To observe serum levels of tumor necrosis factor-TNF superfamily 14 (TNFSF14/LIGHT) in hospitalized patients with COVID-19 and compare this with healthy control patients matching by age and sex. | (13/13) | The bioavailable TNFSF14/LIGHT unbound to the decoy inhibitor receptor-3 (DcR3/TNFRSF6B) was detected. Hospitalized patients diagnosed with COVID-19, including patients with and without ventilator support, had significantly higher free LIGHT levels than healthy controls matched for age and sex. Higher levels of IL-6 were detected in ventilated patients. IL-6 levels that were measured in hospitalized patients older than 60 years who died were higher than those in patients who recovered. |
| McElvaney et al., 2020 [54]. | PUBMED | Case-control Study | Define the cytokine profile of COVID-19 and identify evidence of immunometabolic alterations in patients with severe disease. | (10/10) | IL-1β, IL-6, IL-8/CXCL8, and sTNFR1, were all increased in patients with COVID-19. Critically ill patients demonstrated higher levels of IL-1β, IL-6, and sTNFR1 but lower IL-10 than patients with severe community-acquired pneumonia requiring ICU support. COVID-19 neutrophils exhibited altered immunometabolism, with increased cytosolic PKM2 (pyruvate kinase M2), phosphorylated PKM2, HIF-1α (hypoxia-inducible factor 1α), and lactate. The production and sialylation of alpha-1 antitrypsin (AAT) increased in COVID-19 according to the proportion of serum IL-6 elevation, with the suppression of the anti-inflammatory response in severe disease. In critically ill patients with COVID-19, increases in IL-6: AAT predicted a prolonged stay in the ICU and mortality, while improvements in IL-6: AAT was associated with clinical resolution. COVID-ICU patients had elevated WBC counts and higher levels of circulating neutrophils, C-reactive protein (CRP), fibrinogen, and lactate compared to stable COVID-19 patients. |
| Mandel et al., 2020 [55]. | PUBMED | Cohort study | Assess alveolar inflammatory status in patients with moderate to severe COVID-19. | (11/11) | The burden of the pro-inflammatory cytokines IL-6 and IL-8/CXCL8 in the broncho-alveolar environment was associated with a negative clinical course in patients. |
| Bagheri-Hosseinabadi et al., 2021 [56]. | PUBMED | Cross-sectional study | To determine the association of microRNA (miRNA)-10b and serum levels of IL-2 and IL-8/CXCL8 in patients with COVID-19. | (7/8) | The microRNA (miRNA)-10b expression was significantly and negatively regulated in the peripheral blood of COVID-19 patients compared to healthy controls. The levels of IL-2 and IL-8/CXCL8 were significantly increased in the serum samples compared to healthy subjects. The expression level of miR-10b was significantly correlated with serum IL-2 and IL-8/CXCL8 levels, as well as with the age of the patients, and is therefore considered a cytokine storm induction factor. |
| Li et al., 2021 [57]. | MEDLINE | Cohort study | To investigate the incidence of AKI in hospitalized patients with COVID-19 from three medical centers within and beyond Wuhan, and to analyze the influencing factors of AKI in patients with COVID-19. | (10/11) | The median baseline levels of IL-6 were significantly higher in patients with severe disease compared to moderate COVID-19. Cytokine storm syndrome was observed only in those with severe COVID-19. Furthermore, COVID-19 patients with acute kidney injury (AKI) had significantly higher levels of IL-6 and a cytokine storm rate compared to the group without AKI. The cytokine storm exhibited a strong correlation and collinearity with WBC counts, lymphocyte counts, IL-6 levels, D-dimer, and positive contact history. |
| Gómez-Escobar et al., 2021 [58]. | MEDLINE | Cohort study | To evaluate the differences in inflammatory cytokines in patients with COVID-19 compared to contemporaneously hospitalized controls and, to analyze the relationship between these cytokines and the development of acute respiratory distress syndrome (ARDS), acute kidney injury (AKI), and mortality. | (11/11) | COVID-19 patients had lower absolute lymphocyte and platelet counts but higher hemoglobin levels compared to the controls. In serum chemistry, the patients had lower levels of albumin, alanine aminotransferase, aspartate aminotransferase, and lactate. There was significant overexpression of interferon gamma-induced protein 10 (IP-10/CXCL10), TNF-α, IFN-α2, IFN-γ, IL-1RA, MCP-3, M-CSF, IL-7, CCL2/MCP-1, MIP-1β/CCL4, IL-15, IL-12 (p40), PDGF AA, IL-6, FLT 3L, and IL-10 in COVID-19 patients. The C-reactive protein (CRP) was positively correlated with IL-6 expression. Serum creatinine levels were positively correlated with IL-12 fractalkine/CX3CL1 and Monokine induced by IFN-γ (MIG). In addition, there were significant correlations between ferritin levels and the expression of MIG, TNF-α, and IL-10. The most relevant cytokines that were significantly associated with acute respiratory distress syndrome (ARDS) were MCP-3, TNF-α, fractalkine/CX3CL1, M-CSF, and CCL2/MCP-1. The most relevant cytokines that were significantly associated with mortality in patients were IFN-β, IL-13, TNF-β, TGF-α, and IL-18/IGIF. |
| Gürsoy et al., 2021 [59]. | MEDLINE | Cohort study | To identify laboratory criteria that predict worsening disease and intensification of the ICU, as well as cytokine storm development. | (10/11) | Patients with COVID-19 pneumonia after the development of acute respiratory distress syndrome (ARDS) and admission to the ICU had elevated levels of LDH, highly sensitive troponin (hs-troponin), procalcitonin, triglycerides, CRP, and developed lymphopenia. |
| Ouwendijk et al., 2021 [60]. | MEDLINE | Cohort study | Investigate the presence of NETs and the correlates of pathogeny in blood and LRT samples from critically ill patients with COVID-19 | (9/11) | Plasma levels of extracellular neutrophil traps (NETs) were correlated with CRP and IL-6 levels in patients who required prolonged admission to the ICU but not in those released <14 days after admission to the ICU or those with a fatal illness. Longitudinal changes in NET levels and CRP or IL-6 were similar in most patients who required prolonged admission to the ICU, suggesting that inflammation and NET production are coregulated. Blood NET levels increased in critically ill patients, especially shortly after admission to the ICU, and were related to sputum viral RNA load and blood levels of neutrophil recruitment chemokines and inflammatory markers. |
| Blot et al., 2020 [61]. | MEDLINE | Clinical trial | Compare the cytokine response patterns, in the alveolar and systemic compartments, between COVID-19-related ARDS and non-COVID-19-related ARDS. | (13/13) | The 30-day mortality rate was higher in the COVID-19 group compared to the non-COVID-19 group. Patients with COVID-19 ARDS had significantly higher rates of CC chemokine ligand 5 (CCL5/RANTES) and non-significant increased levels of CXCL2/SDF-1, CXCL10/IP-10, CD40 ligand (CD40L/CD154), IL-10, and GM-CSF compared to those with non-COVID-19 ARDS. There were also significantly lower concentrations of plasma IL-2, the TNF-related apoptosis-inducing ligand (TRAIL), and G-CSF. Serum CXCL10/IP-10 concentration was independently associated with a higher number of ventilator-free days after adjustment for COVID-19 etiology, submission to noninvasive ventilation (NIV) prior to intubation, exposure to multiple antibiotics, plasma concentrations of CXCL2/SDF-1, CCL5/RANTES, and CD40 ligand/CD154. Higher cytokine concentrations in the epithelial lining fluid (ELF) of CXCL1/GRO-a, CXCL10/IP-10, granzyme B, TRAIL, and EGF were recorded in patients with COVID-19. Significantly lower ELF concentrations of IL-2, G-CSF, and IL-17A and a trend toward lower concentrations of CCL3/MIP-1 α were also identified. |
| Lorenz et al., 2020 [62]. | MEDLINE | Cohort study | Verify whether cytokine release syndrome is caused by secondary hemophagocytic lymphohistiocytosis in critically ill patients with COVID-19. | (10/11) | On admission to the ICU, most patients who had a hyperinflammatory immune response had a fever, increased C-reactive protein (CRP), elevated IL-6, serum ferritin, and soluble interleukin-2 receptor (sIL-2R), with relatively low procalcitonin. Serum IL-6 levels did not separate, and unfavorable clinical courses and D-dimers tended to increase in both groups. All patients had hyperfibrinogenemia. At the cellular level, varying absolute numbers of leukocytes, relative neutrophilia, lymphopenia, and a reduced percentage of monocytes were observed at ICU admission. Quite a small number of circulating CD8+ T cell subsets were found that tended to lower values in the unfavorable group. Relative neutrophilia, lymphopenia, and increased neutrophil/lymphocyte ratio (NLR) followed the clinical course of patients. |
| Notz et al., 2020 [63]. | MEDLINE | Cohort study | Characterize immune responses in patients suffering from severe COVID-19-induced acute respiratory distress syndrome (ARDS). | (9/11) | Peripheral blood lymphocytes were below the reference range on admission, but white blood cell counts and their subgroups nearly tripled over the course of ICU treatment. Elevated levels of IL-6 were detected, along with high levels of the C-reactive protein (CRP). There was a significant inverse correlation between the lymphocyte count and IL-6 levels on admission. There was a dynamic increase in IL-10. CXCL10/IP-10 was significantly elevated at all time points compared to its reference range and healthy controls. IFN-γ, IL-1β, IL2, IL-7, IL-17A, and GM-CSF were below their respective reference ranges. GDF-15/MIC-1 showed considerably higher levels in all patients with COVID-19. The absolute numbers of CD3+ T, CD19+ B, and CD3− CD56/CD16+ NK cells increased over time. |
| Sinha et al., 2020 [64]. | MEDLINE | Cohort study | To identify phenotypes in COVID-19-related ARDS. | (10/11) | The findings suggest that the prevalence of hyperinflammatory phenotypes was low. There was a high mortality rate with the hypoinflammatory phenotype in COVID-19. The lymphocyte count was slightly lower in the hypoinflammatory phenotype. The D-dimer and CRP values were similar between the phenotypes. Inflammation biomarkers IL-6, soluble tumor necrosis factor receptor 1 (TNFR1), D-dimer, and bilirubin were all significantly higher in nonsurvivors than in survivors in the COVID-19 cohort. Soluble levels of IL-6 and TNFR1 were similar or lower in patients with COVID-19-associated ARDS (from this cohort) than in patients with ARDS due to other causes (HARP-2). The rate of mortality for both phenotypes was considerably higher in the COVID-19 cohort than in the historical data for ARDS associated with other causes. |
| Wilson et al., 2020 [65]. | MEDLINE | Clinical trial | To contribute to a broader understanding of the inflammatory response in moderate and severe COVID-19. | (13/13) | The levels of cytokines IL-1β, IL-8/CXCL8, IL-18/IGIF, and TNF-α did not differ significantly between the groups of patients with moderate COVID-19, severe COVID-19, ARDS, and sepsis. There was a trend towards higher levels of IL-1RA and IL-6 in patients with severe COVID-19 compared to those with moderate COVID-19, which is consistent with previous reports. There was a trend of higher IL-18/IGIF in the severe COVID-19 group compared to the sepsis group; however, this was not significant after correcting for multiple comparisons. |
| Wang et al., 2020 [66]. | MEDLINE | Quasi-experimental study | To make a significant contribution to understanding the mechanisms underlying the phenotype of severe cases in COVID-19 and the appropriate development of treatment strategies. | (9/9) | Alveolar macrophages significantly increased and filled part of the alveolar cavities with scattered neutrophils and lymphocytes. Various chemokines and inflammatory cytokines are secreted by alveolar macrophages, including IL-6, IL-10, and TNF-α. IL-6 and TNF-α were moderately expressed in macrophages, while the expression of IL-10 was strong. There was an expression of ACE2 by hyperplastic type II alveolar epithelial cells, alveolar macrophages, and macrophages in the cortical sinuses of the lymph nodes of the pulmonary hilum. |
| Grant et al., 2021 [67]. | MEDLINE | Cohort study | To investigate the pathobiology of SARS-CoV-2 by characterizing the immune response in the alveoli of patients infected with the virus. | (11/11) | At the first collection of bronchoalveolar lavage (BAL), patients with severe SARS-CoV-2 pneumonia had higher levels of CRP compared to patients with other types of pneumonia, while other biomarkers of inflammation were found at similar levels. Mortality did not differ among patients with SARS-CoV-2 pneumonia compared to the entire cohort. Comparison of alveolar macrophage transcriptomic profiles between patients with severe SARS-CoV-2 pneumonia, patients with pneumonia secondary to other pathogens, controls without pneumonia, and healthy volunteers showed that most patients with COVID-19 were clustered. This clustering was characterized by genes involved in the response to interferon and also included genes encoding the CC chemokine ligand (CCL) 7, CCL8, and CCL13 chemokines, which drive monocyte and T-cell recruitment. |
| Gu et al., 2021 [68]. | PUBMED | Review | Summarize new lines of evidence that point to platelet and endothelial dysfunction as essential components of COVID-19 pathology and describe the mechanisms that may be responsible for the contribution of cardiovascular risk factors to the most severe outcomes in COVID-19. | (11/11) | Elevated plasma levels of D-dimers, thrombocytopenia, elevated levels of inflammatory markers (such as C-reactive protein, erythrocyte sedimentation rate, ferritin, and various cytokines, including IL-1β, IL-6, and TNF-α) lead to a storm picture of cytokines. Elevations in the levels of various circulating markers of endothelial injury were identified, such as von Willebrand factor (vWF), plasminogen activator inhibitor-1 (PAI1), soluble thrombomodulin, angiopoietin 2 and follistatin, which have a role in coagulopathy. |
| Borges et al., 2020 [69]. | PUBMED | Review | Summarize the role of neutrophils in the symptoms of COVID-19, considering the interaction between hyperinflammation (overproduction of NETs and cytokines) and the clearance function of neutrophils to eliminate viral infection. | (10/11) | An extensive NET formation can lead to a cascade of inflammatory reactions that destroy the surrounding tissues, favor microthrombosis and result in permanent damage to the pulmonary, cardiovascular, and renal systems. |
| Li et al., 2020 [70]. | PUBMED | In vitro assay | Report the underlying mechanisms of the inflammatory responses triggered by the virus. | * | The activation of caspase-8 (CASP8) plays a leading role in SARS-CoV-2-induced apoptosis and inflammatory responses. SARS-CoV-2 induces an upregulated level of caspase-8 activation to process pro-IL-1β and, in the meantime, allows sufficient necroptosis activation to release IL-1β. The necroptosis inhibitor in the study did not fully block IL-1β secretion during SARS-CoV-2 infection, indicating that other pathways, such as pyroptosis, may also be involved in inflammatory responses. |
| Panigrahy et al., 2020 [71]. | PUBMED | Review | To present the use of pro-resolution mediators for COVID-19 as a potential complement to current antiviral strategies. | (11/11) | The “Eicosanoid storm” caused by cell (“fragment”) death, including prostaglandins and leukotrienes, in turn, triggers a strong inflammatory response. Endogenous autologous lipid mediators called eicosanoids to play a key role in the induction of inflammation and the production of pro-inflammatory cytokines. Both pro-resolution specialized lipid autacoid mediators (SMPs) and soluble epoxide hydrolase (sEH) inhibitors that promote regression can promote the regression of COVID-19 inflammation, thereby reducing acute respiratory distress syndrome (ARDS) and other complications related to virus-induced inflammation. |
| Quartuccio et al., 2021 [72]. | PUBMED | Cohort study | To determine which cytokines are associated with respiratory failure in patients hospitalized for COVID-19. | (11/11) | The PaO2/FiO2 (P/F) ratio was calculated in all patients at hospital admission and was strongly correlated with IL-6, M-CSF, interleukin-2 receptor alpha subunit (sIL-2Rα), and hepatocyte growth (HGF) showing a stronger association with P/F levels below 300. ROC curve analyses for IL-6, M-CSF, HGF, and sIL-2Rα showed that these four soluble factors were significantly high and correlated with lactate dehydrogenase (LDH), white blood cell count, neutrophil count, lymphocyte count, and CRP. All but sIL-2Rα correlated with the D-dimer, while only HGF correlated slightly with the cardiac marker creatine phosphokinase (CPK). |
| Welcome; Mastorakis, 2021 [73]. | PUBMED | Review | To review the literature on the possible neuropathogenic mechanisms of SARS-CoV-2-induced brain damage. | (9/11) | In SARS-CoV-2 brain nerve invasion, the downregulation of angiotensin-converting enzyme 2 (ACE2) and increased activity of transmembrane serine 2 protease (TMPRSS2) and cathepsin L can lead to the up-regulation of pro-inflammatory mediators. Inflammatory and reactive oxygen species (ROS) promote a neuroinflammatory response and destroy the blood–brain barrier. Furthermore, the dysregulation of hormone and neurotransmitter signals can constitute the basic mechanism involved in the neuropathic sequelae of SARS-CoV-2 infection. |
| Cattle; Wada, 2021 [74]. | PUBMED | Review | Investigate thromboplasmin inflammation in COVID-19-caused coagulopathy for diagnostic and therapeutic implications. | (11/11) | Histones and NETs trigger the release of inflammatory cytokines and initiate clotting by expressing the tissue factor in monocytes and endothelial cells and by activating factor (F) XII, which is amplified by reduced anticoagulant factors and impaired fibrinolysis. Platelets are also activated by histones and NETs, leading to the procoagulant phenotype through the expression of P-selectin. Thrombin generation is enhanced by prothrombinase comprising FVa and FXa, while histones act as prothrombinase surrogates to promote the cleavage of FXa from prothrombin to form active thrombin and initiate disseminated intravascular coagulation (DIC). COVID-19 coagulopathy is a disease of thromboplasmin inflammation with immunothrombosis associated with the condition of immunoinflammation consisting of AngII-induced coagulopathy, FXIIa hyperfibrinolysis, the kallikrein-kinin system (KKS), and DIC. |
| Hoxha, 2020 [75]. | PUBMED | Review | Define and summarize all findings on the possible association between the arachidonic acid (AA) pathway and the pathophysiology of COVID-19. | (11/11) | The data clarify that COX-2 and prostaglandins (PGs), particularly PGE 2, have pro-inflammatory effects on the pathophysiology of COVID-19. A deficiency of AA makes humans more susceptible to COVID-19. Targeting these pro-inflammatory mediators can help reduce the mortality and morbidity of COVID-19 patients. |
| Mustafa et al., 2020 [34]. | PUBMED | Review | To synthesize data on the cytokine storm in COVID-19 patients, its impact on the body organs, and potential treatment by chemokine receptors. | (9/11) | Excessive infiltration of inflammatory cells, such as monocytes and neutrophils, into lung tissue can cause lung injury. Another source of lung damage is the cytokine-induced apoptosis of lung epithelial cells. IFN-α and β and IFN-γ induce inflammatory cell infiltration through two main mechanisms that involve the Fas ligand (FasL) or the TRAIL-death receptor 5 (DR5) and cause apoptosis of the airways and airway alveolar epithelial cells. Increased levels of cytokines in SRC include IL-1β, IL-2, IL-7, IL-8/CXCL8, IL-9, IL-10, IL-17, G-CSF, GM-CSF, IFN-γ, TNF-α, IP-10/CXCL10, MCP-1/CCL2, MIP-1α/CCL3, and MIP-1β/CCL4 which are associated with increased severity of the disease along with the development of ARDS and cardiac injury in patients with underlying heart problems. |
| Wang et al., 2020 [76]. | PUBMED | In vivo assay | Test the hypothesis that ACE2-deficient mice are “prepared” for a corneal inflammatory response, which, once initiated, would persist. | * | ACE2, which is present in human tissues and the corneal limbus of mice, and a genetic deficiency of ACE2 results in a marked inflammatory response in corneal epithelial and stromal tissues. Furthermore, ACE2 deficiency results in the marked upregulation of AngII, the main peptide that is normally degraded by ACE2. In ACE2-deficient mice, once an inflammatory response is initiated, the inflammation persists and becomes permanent, remodeling the stromal microenvironment. This microenvironmental change results in a wide range of epithelial phenotypes. They revealed that interleukins (IL-1α, IL-1β), chemokines (CCL2/MCP-1, CXCL8/IL-8), and TNF-α are all significantly elevated, resulting in a cytokine storm-like phenotype. |
| Morrell et al., 2022 [77]. | PUBMED | Cohort study | Identify immunological signatures enriched specifically in critically ill patients with COVID-19 compared to patients without COVID-19. | (9/11) | Plasma levels of CXCL10/IP-10, soluble programmed death ligand 1 (sPD-L1), IFN-γ, CCL26/MIP-4α, C-reactive protein (CRP), and TNF-α were found to be markedly higher in SARS-CoV-2-positive patients compared to negative patients. CCL17 was associated with more severe respiratory failure in SARS-CoV-2-positive patients. CD4 + CXCR3 + cells were negatively correlated with plasma levels of CXCL10/IP-10 in SARS-CoV-2 negative individuals. On the contrary, these cells were positively correlated with plasma levels of CXCL10/IP-10 in SARS-CoV-2 positive patients. |
| Ventura-Santana et al., 2022 [78]. | PUBMED | Review | Present evidence indicating that severe COVID-19 has clinical presentations that are consistent with definitions of viral sepsis. | (11/11) | SARS-CoV-2 replicates in neutrophils and triggers NETosis, contributing to the pathology of COVID-19. Elevated levels of markers of NET formation, including cell-free DNA, myeloperoxidase (MPO)-DNA, citrullinated histone H3, and neutrophil elastase (NE), which are associated with disease severity, were observed in sera from COVID-19 patients. 19. NET formation associated with systemic inflammation and cytokine storms contribute to mortality in severe COVID-19. |
| Chabert et al., 2022 [79]. | PUBMED | Cohort study | Highlight the relationship between the production of de Aldo-Keto Reductase Family 1 Member B10 (AKR1B10) and severe forms of COVID-19. | (10/11) | Patients who developed a severe form of the disease (in the ICU) had higher concentrations of Aldo-Keto Reductase Family 1 Member B10 (AKR1B10) when compared to outpatients in the ICU. AKR1B10 concentration was found to be negatively correlated with the lymphocyte count and positively correlated with lactate dehydrogenase (LDH) levels. |
| Nishitsuji et al., 2022 [80]. | PUBMED | In vitro assay | To provide new molecular insights into the pathogenesis of SARS-CoV-2 and the host’s immune response to infection. | * | Nonstructural protein 6 (NSP6) and open reading frame 7a (ORF7a) lead to the activation of nuclear factor beta (NF-κB) through associations with protein kinase mitogen-activated kinase 7 (MAP3K7/TAK1). The K63-linked polyubiquitination of NSP6 and ORF7a by Tripartite Motif Containing 13 (TRIM13) and Ring Finger Protein 121 (RNF121), respectively, appears to be essential for the activation of NF-κB. |
| Al-kuraishy et al., 2022 [81]. | PUBMED | Review | To elucidate the potential role of the high mobility group box 1 protein (HMGB1) in the pathogenesis of SARS-CoV-2 infection. | (10/11) | Data clarify that high serum levels of the high-mobility group box 1 protein (HMGB1) could be observed in patients with COVID-19 associated with the severity of the disease, the development of the cytokine storm, acute lung injury (ALI), lung syndrome, and Acute respiratory distress (ARDS). Critically ill ICU patients had high circulating HMGB1. It has also been identified as a critical mediator of thrombosis through platelet activations, the stimulation of inflammatory reactions, neutrophil recruitment, and NET formation. |
| Combadiere et al., 2021 [82]. | MEDLINE | Cohort study | To investigate the subsets and functions of neutrophils in the blood and bronchoalveolar lavage (BAL) of patients with COVID-19 based on the clinical characteristics of the patient. | (10/11) | Approximately 80% of patients in the ICU developed strong myelemia with CD10-CD64 + immature neutrophils (ImNs). Cellular profiling revealed three distinct subsets of neutrophils expressing oxidized low-density lipoprotein receptor-1 (LOX-1), interleukin-3 alpha receptor (CD123), or programmed death ligand 1 (PD-L1), which were up-regulated in ICU patients compared to non-ICU patients. The proportion of ImNs expressing LOX-1 or CD123 was positively correlated with clinical severity, cytokine storm (IL-1β, IL-6, IL-8/CXCL8, TNF-α), and ARDS. |
| Nikitopoulou et al., 2021 [83]. | MEDLINE | Cohort study | Quantify messenger RNA (mRNA) levels of the ecto-nucleotide pyrophosphatase/phosphodiesterase family member 2 (ENPP2) in swabs of nasopharyngeal and autotaxin (ATX) protein levels in the serum of two cohorts of patients with COVID-19. | (9/11) | Increased serum levels of autotaxin (ATX) were detected in critically ill patients with COVID-19 and were correlated with corresponding increases in serum levels of IL-6 and biomarkers of endothelial damage, suggesting an interaction between the ATX/lysophosphatidic acid (LPA) axis with hyperinflammation and associated vascular dysfunction in COVID-19. Ecto-nucleotide mRNA expression family member 2 pyrophosphatase/phosphodiesterase or autotaxin (ENPP2/ATX) in mild and severe COVID-19 patients was compared to uninfected individuals. Increased serum ATX concentrations were identified in ICU patients compared to ward patients, indicating an association of ATX with disease severity. |
| Ren et al., 2021 [84]. | PUBMED | In vitro assay | Know how specific SARS-CoV-2-encoded proteins, particularly the Nucleocapsid (N) protein, contribute to viral pathogenicity. | * | Although SARS-CoV-2 N does not directly cause apoptosis, it can increase the amount of apoptosis brought on by SARS-CoV-2 M by enhancing their association with Phosphoinositide-Dependent Kinase-1 (PDK1), which, in turn, inhibits their connection with PKB/Akt and downstream signaling. |
| McAleavy et al., 2022 [85]. | MEDLINE | Clinical trial | To determine the molecular mechanisms involved with COVID-19-induced mortality. | (12/13) | Activin A and its pathway marker, FLRG (follistatin-related gene, also called FLSTL3) and PAI1, were elevated in critically ill patients relative to critically ill patients or healthy controls. Patients with elevated levels of activin A, activin B, and FLRG at hospital admission were associated with more severe outcomes of COVID-19, including the need for mechanical ventilation and mortality. Activin A was considered a biomarker for critically ill patients in the ICU. |
| Author and Year | Randomization | Allocation Concealment | Blinding | Sample Size Calculation | Standardization of Specimen | Standardized Preparation (Single Operator) | Statistical Analysis | Reporting of Data |
|---|---|---|---|---|---|---|---|---|
| Li et al., 2020 [70]. | Moderate Not mentioned | Low Allocation concealment was mentioned | High Not mentioned | High Not mentioned | Low All specimens standardized | Low Spacemen preparation and mixing methods reported clearly | Moderate The statistical analysis software was not mentioned | Low All outcomes reported |
| Wang et al., 2020 [76]. | High Not mentioned | Low Allocation concealment was mentioned | Low Blinding was mentioned | High Not mentioned | Low All specimens standardized | Low Spacemen preparation and mixing methods reported clearly | Moderate The statistical analysis software was not mentioned | Low All outcomes reported |
| Nishitsuji et al., 2022 [80]. | High Not mentioned | Low Allocation concealment was mentioned | Low Blinding was mentioned | High Not mentioned | Low All specimens standardized | Low Spacemen preparation and mixing methods reported clearly | Moderate The statistical analysis software was not mentioned | Low All outcomes reported |
| Ren et al., 2021 [84]. | Moderate Not mentioned | Low Allocation concealment was mentioned | Moderate Not mentioned | High Not mentioned | Low All specimens standardized | Low Spacemen preparation and mixing methods reported clearly | Moderate The statistical analysis software was not mentioned | Low All outcomes reported |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, M.J.A.; Ribeiro, L.R.; Gouveia, M.I.M.; Marcelino, B.d.R.; Santos, C.S.d.; Lima, K.V.B.; Lima, L.N.G.C. Hyperinflammatory Response in COVID-19: A Systematic Review. Viruses 2023, 15, 553. https://doi.org/10.3390/v15020553
Silva MJA, Ribeiro LR, Gouveia MIM, Marcelino BdR, Santos CSd, Lima KVB, Lima LNGC. Hyperinflammatory Response in COVID-19: A Systematic Review. Viruses. 2023; 15(2):553. https://doi.org/10.3390/v15020553
Chicago/Turabian StyleSilva, Marcos Jessé Abrahão, Layana Rufino Ribeiro, Maria Isabel Montoril Gouveia, Beatriz dos Reis Marcelino, Carolynne Silva dos Santos, Karla Valéria Batista Lima, and Luana Nepomuceno Gondim Costa Lima. 2023. "Hyperinflammatory Response in COVID-19: A Systematic Review" Viruses 15, no. 2: 553. https://doi.org/10.3390/v15020553
APA StyleSilva, M. J. A., Ribeiro, L. R., Gouveia, M. I. M., Marcelino, B. d. R., Santos, C. S. d., Lima, K. V. B., & Lima, L. N. G. C. (2023). Hyperinflammatory Response in COVID-19: A Systematic Review. Viruses, 15(2), 553. https://doi.org/10.3390/v15020553