
Cellular Targets of HIV-1 Protease: Just the Tip of the Iceberg?




Abstract
:1. Introduction
1.1. HIV-1 PR Structure and Function
1.2. Sequence Specificity
1.3. PR Activation and Virion Maturation
2. Cellular Targets of HIV-1 Protease
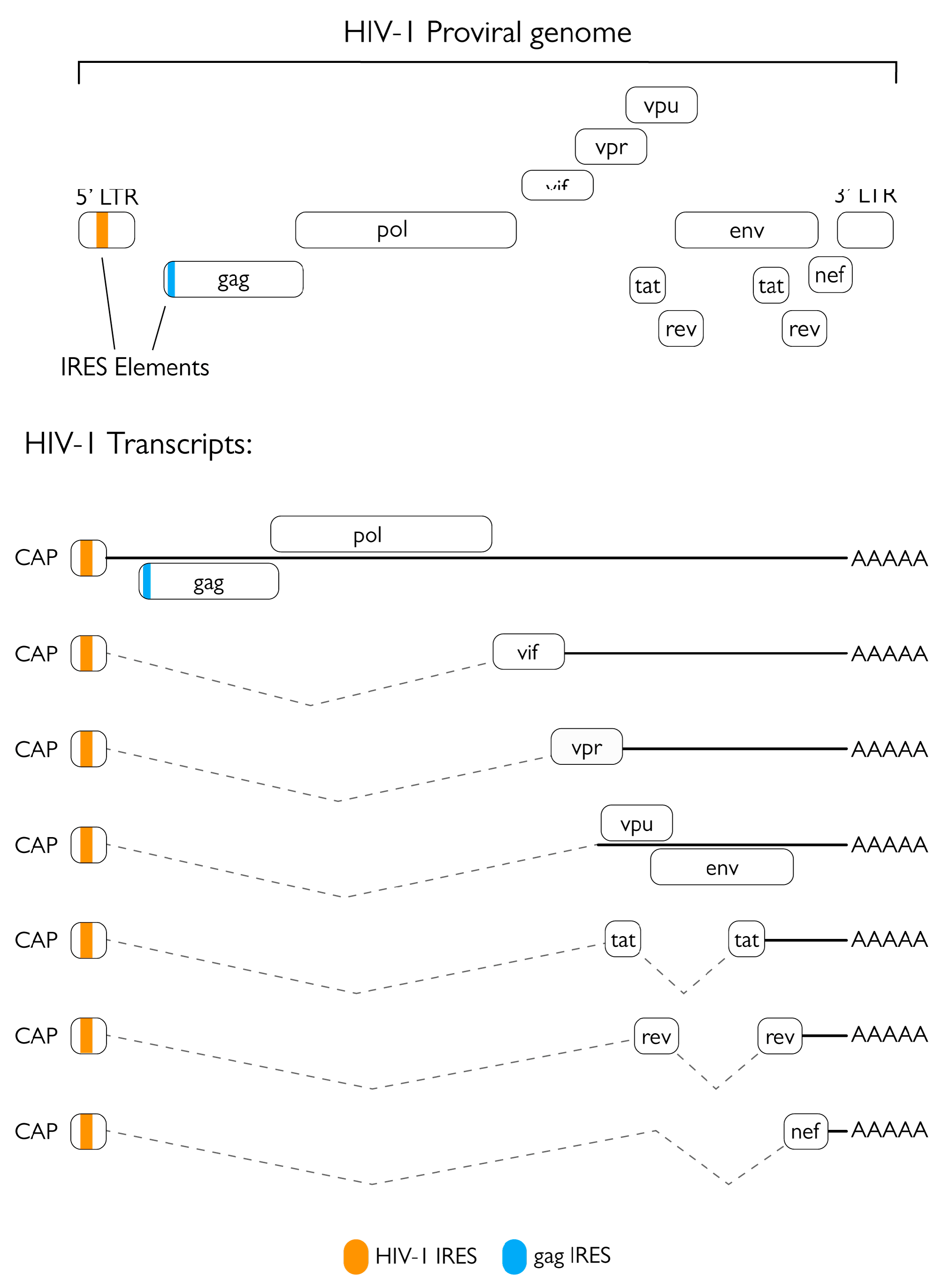
2.1. Role of the Protease in Protein Synthesis Modulation
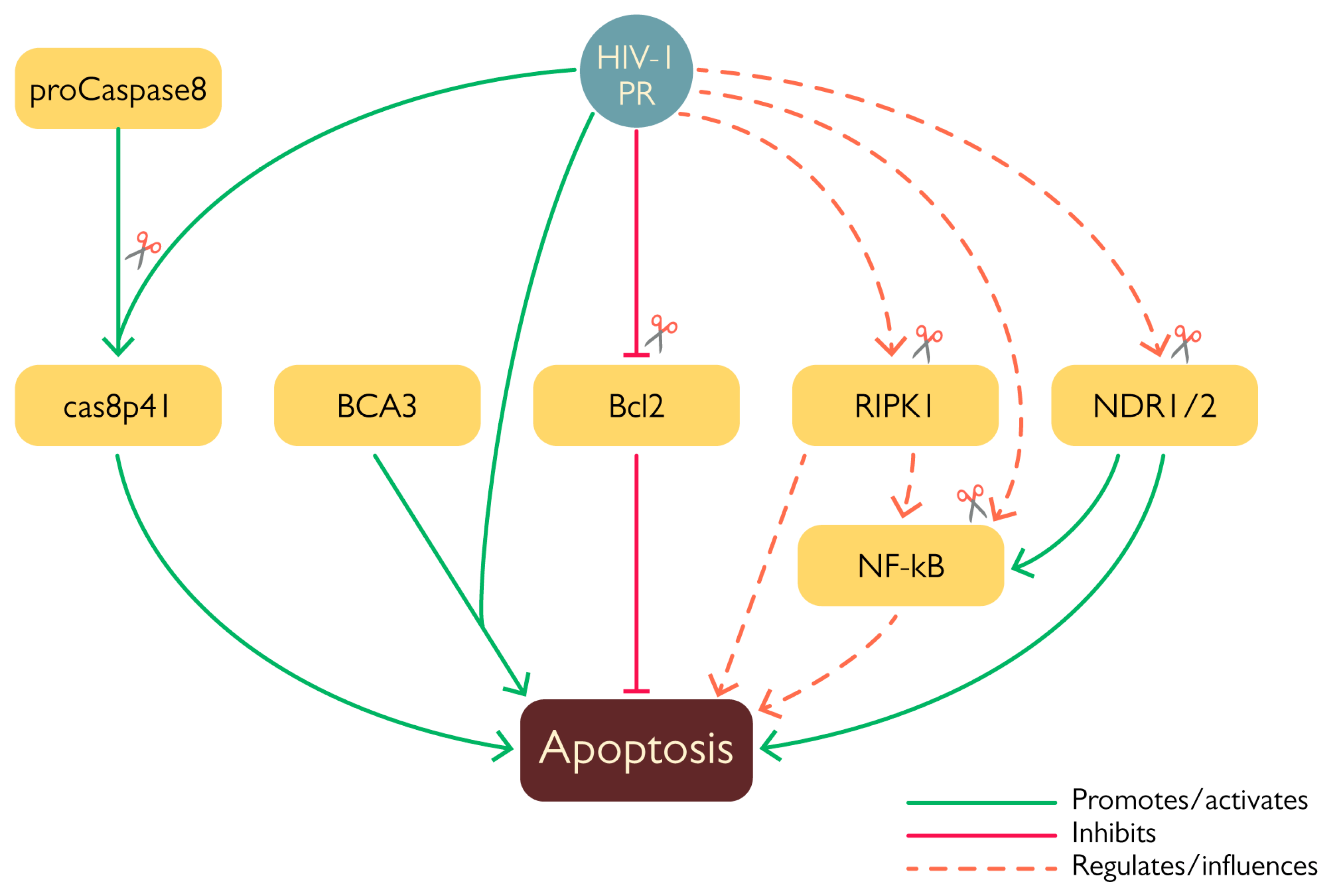
2.2. PR and Apoptosis
2.3. Effect of PR on Innate Defenses
2.4. Cleavage of Virion-Incorporated Restriction Factors
2.5. PR-Mediated Cytoskeleton Modulation
2.6. Cleavage of Host Factors by HIV-2 PR
2.7. HIV-1 PR Inhibitors
2.8. HIV-1 PI Resistance Mutations
2.9. Implication of HIV-1 PR Mediated Host Factor Cleavage for Antiviral Therapy
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS Pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IN DANGER: UNAIDS Global AIDS Update 2022; Joint United Nations Programme on HIV/AIDS: Geneva, Switzerland, 2022.
- Groopman, J.E. Zidovudine Intolerance. Rev. Infect. Dis. 1990, 12 (Suppl. S5), S500–S506. [Google Scholar] [CrossRef] [PubMed]
- Larder, B.A.; Darby, G.; Richman, D.D. HIV with Reduced Sensitivity to Zidovudine (AZT) Isolated During Prolonged Therapy. Science 1989, 243, 1731–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochhauser, D.; Harris, A.L. Drug Resistance. Br. Med. Bull. 1991, 47, 178–196. [Google Scholar] [CrossRef]
- Tomasselli, A.G.; Thaisrivongs, S.; Heinrikson, R.L. Discovery and Design of HIV Protease Inhibitors as Drugs for Treatment of Aids; Elsevier: Amsterdam, The Netherlands, 1996; Volume 2, ISBN 1559386932. [Google Scholar]
- Collier, A.C.; Coombs, R.W.; Schoenfeld, D.A.; Bassett, R.L.; Timpone, J.; Baruch, A.; Jones, M.; Facey, K.; Whitacre, C.; McAuliffe, V.J.; et al. Treatment of Human Immunodeficiency Virus Infection with Saquinavir, Zidovudine, and Zalcitabine. AIDS Clinical Trials Group. N. Engl. J. Med. 1996, 334, 1011–1017. [Google Scholar] [CrossRef]
- Simon, V.; Ho, D.D.; Abdool Karim, Q. HIV/AIDS Epidemiology, Pathogenesis, Prevention, and Treatment. Lancet 2006, 368, 489–504. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, M.R.; Rojo, D.R.; Von Lindern, J.J.; O’Brien, W.A. HIV-1 Replication Cycle. Clin. Lab. Med. 2002, 22, 611–635. [Google Scholar] [CrossRef]
- Balachandran, A.; Wong, R.; Stoilov, P.; Pan, S.; Blencowe, B.; Cheung, P.; Harrigan, P.R.; Cochrane, A. Identification of Small Molecule Modulators of HIV-1 Tat and Rev Protein Accumulation. Retrovirology 2017, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Checkley, M.A.; Luttge, B.G.; Freed, E.O. HIV-1 Envelope Glycoprotein Biosynthesis, Trafficking, and Incorporation. J. Mol. Biol. 2011, 410, 582–608. [Google Scholar] [CrossRef] [Green Version]
- Freed, E.O. HIV-1 Gag Proteins: Diverse Functions in the Virus Life Cycle. Virology 1998, 251, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.; Tachedjian, G.; Mak, J. The Packaging and Maturation of the HIV-1 Pol Proteins. Curr. HIV Res. 2005, 3, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Kirchhoff, F. Immune Evasion and Counteraction of Restriction Factors by HIV-1 and Other Primate Lentiviruses. Cell Host Microbe 2010, 8, 55–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef Promotes Infection by Excluding SERINC5 from Virion Incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 Restrict HIV-1 Infectivity and Are Counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Yamada, E.; Yoshikawa, R.; Nakano, Y.; Misawa, N.; Koyanagi, Y.; Sato, K. Impacts of Humanized Mouse Models on the Investigation of HIV-1 Infection: Illuminating the Roles of Viral Accessory Proteins in Vivo. Viruses 2015, 7, 1373–1390. [Google Scholar] [CrossRef] [Green Version]
- Freed, E.O. HIV-1 Replication. Somat. Cell Mol. Genet. 2001, 26, 13–33. [Google Scholar] [CrossRef]
- Taube, R.; Peterlin, M. Lost in Transcription: Molecular Mechanisms That Control HIV Latency. Viruses 2013, 5, 902–927. [Google Scholar] [CrossRef] [Green Version]
- Fanales-Belasio, E.; Raimondo, M.; Suligoi, B.; Buttò, S. HIV Virology and Pathogenetic Mechanisms of Infection: A Brief Overview. Ann. Ist. Super Sanità 2010, 46, 5–14. [Google Scholar] [CrossRef]
- Engelman, A.; Cherepanov, P. The Structural Biology of HIV-1: Mechanistic and Therapeutic Insights. Nat. Rev. Microbiol. 2012, 10, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Bryant, M.; Ratner, L. Myristoylation-Dependent Replication and Assembly of Human Immunodeficiency Virus 1. Proc. Natl. Acad. Sci. USA 1990, 87, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin Inhibits Retrovirus Release and Is Antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The Interferon-Induced Protein BST-2 Restricts HIV-1 Release and Is Downregulated from the Cell Surface by the Viral Vpu Protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Balachandran, R.; Ho, M.; Enrico, A.; Rinaldo, C. Cell-to-Cell Transmission of Human Immunodeficiency Virus Type 1 in the Presence of Azidothymidine and Neutralizing Antibody. J. Virol. 1989, 63, 2361–2365. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Orensteint, J.; Dimitrov, D.; Martin, M. Cell-to-Cell Spread of HIV-1 Occurs within Minutes and May Not Involve the Participation of Virus Particles. Virology 1992, 186, 712–724. [Google Scholar] [CrossRef] [PubMed]
- Carr, J.M.; Hocking, H.; Li, P.; Burrell, C.J. Rapid and Efficient Cell-to-Cell Transmission of Human Immunodeficiency Virus Infection from Monocyte-Derived Macrophages to Peripheral Blood Lymphocytes. Virology 1999, 265, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Piguet, V.; Sattentau, Q. Dangerous Liaisons at the Virological Synapse. J. Clin. Investig. 2004, 114, 605–610. [Google Scholar] [CrossRef] [Green Version]
- Agosto, L.M.; Zhong, P.; Munro, J.; Mothes, W. Highly Active Antiretroviral Therapies Are Effective against HIV-1 Cell-to-Cell Transmission. PLoS Pathog. 2014, 10, e1003982. [Google Scholar] [CrossRef]
- Titanji, B.K.; Aasa-Chapman, M.; Pillay, D.; Jolly, C. Protease Inhibitors Effectively Block Cell-to-Cell Spread of HIV-1 between T Cells. Retrovirology 2013, 10, 161. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Nkeze, J.; Zhao, R.Y. Effects of HIV-1 Protease on Cellular Functions and Their Potential Applications in Antiretroviral Therapy. Cell Biosci. 2012, 2, 32. [Google Scholar] [CrossRef] [Green Version]
- Mager, P.P. The Active Site of HIV-1 Protease. Med. Res. Rev. 2001, 21, 348–353. [Google Scholar] [CrossRef]
- Konvalinka, J.; Kräusslich, H.G.; Müller, B. Retroviral Proteases and Their Roles in Virion Maturation. Virology 2015, 479–480, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Wang, J.; Chen, Z.; Wang, G.; Shao, Q.; Shi, J.; Zhu, W. Structural Insights into HIV-1 Protease Flap Opening Processes and Key Intermediates. RSC Adv. 2017, 7, 45121–45128. [Google Scholar] [CrossRef] [Green Version]
- Scott, W.R.P.; Schiffer, C.A. Curling of Flap Tips in HIV-1 Protease as a Mechanism for Substrate Entry and Tolerance of Drug Resistance. Structure 2000, 8, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Trylska, J.; Tozzini, V.; Chang, C.E.A.; McCammon, J.A. HIV-1 Protease Substrate Binding and Product Release Pathways Explored with Coarse-Grained Molecular Dynamics. Biophys. J. 2007, 92, 4179–4187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laco, G.S. HIV-1 Protease Substrate-Groove: Role in Substrate Recognition and Inhibitor Resistance. Biochimie 2015, 118, 90–103. [Google Scholar] [CrossRef]
- Impens, F.; Timmerman, E.; Staes, A.; Moens, K.; Ariën, K.K.; Verhasselt, B.; Vandekerckhove, J.; Gevaert, K. A Catalogue of Putative HIV-1 Protease Host Cell Substrates. Biol. Chem. 2012, 393, 915–931. [Google Scholar] [CrossRef]
- Lawal, M.M.; Sanusi, Z.K.; Govender, T.; Maguire, G.E.M.; Honarparvar, B.; Kruger, H.G. From Recognition to Reaction Mechanism: An Overview on the Interactions between HIV-1 Protease and Its Natural Targets. Curr. Med. Chem. 2018, 25, 2514–2549. [Google Scholar] [CrossRef]
- Lee, S.K.; Potempa, M.; Swanstrom, R. The Choreography of HIV-1 Proteolytic Processing and Virion Assembly. J. Biol. Chem. 2012, 287, 40867–40874. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Louis, J.M.; Aniana, A.; Suh, J.Y.; Clore, G.M. Visualizing Transient Events in Amino-Terminal Autoprocessing of HIV-1 Protease. Nature 2008, 455, 693–696. [Google Scholar] [CrossRef] [Green Version]
- Agniswamy, J.; Sayer, J.M.; Weber, I.T.; Louis, J.M. Terminal Interface Conformations Modulate Dimer Stability Prior to Amino Terminal Autoprocessing of HIV-1 Protease. Biochemistry 2012, 51, 1041–1050. [Google Scholar] [CrossRef]
- Kaplan, A.H.; Swanstrom, R. Human Immunodeficiency Virus Type 1 Gag Proteins Are Processed in Two Cellular Compartments. Proc. Natl. Acad. Sci. USA 1991, 88, 4528–4532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jäger, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K.; et al. Global Landscape of HIV-Human Protein Complexes. Nature 2012, 481, 365–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, R.N.; Reed, J.C.; Chanda, S.K. HIV-1 Protease Cleaves the Serine-Threonine Kinases RIPK1 and RIPK2. Retrovirology 2015, 12, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devroe, E.; Silver, P.A.; Engelman, A. HIV-1 Incorporates and Proteolytically Processes Human NDR1 and NDR2 Serine-Threonine Kinases. Virology 2005, 331, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Nie, Z.; Bren, G.D.; Vlahakis, S.R.; Schimnich, A.A.; Brenchley, J.M.; Trushin, S.A.; Warren, S.; Schnepple, D.J.; Kovacs, C.M.; Loutfy, M.R.; et al. Human Immunodeficiency Virus Type 1 Protease Cleaves Procaspase 8 In Vivo. J. Virol. 2007, 81, 6947–6956. [Google Scholar] [CrossRef] [Green Version]
- Ventoso, I.; Blanco, R.; Perales, C.; Carrasco, L. HIV-1 Protease Cleaves Eukaryotic Initiation Factor 4G and Inhibits Cap-Dependent Translation. Proc. Natl. Acad. Sci. USA 2001, 98, 12966–12971. [Google Scholar] [CrossRef] [Green Version]
- Strack, P.R.; Frey, M.W.; Rizzo, C.J.; Cordova, B.; George, H.J.; Meade, R.; Ho, S.P.; Corman, J.; Tritch, R.; Korant, B.D. Apoptosis Mediated by HIV Protease Is Preceded by Cleavage of Bcl-2. Proc. Natl. Acad. Sci. USA 1996, 93, 9571–9576. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, E.; Castelló, A.; Menéndez-Arias, L.; Carrasco, L. HIV Protease Cleaves Poly(A)-Binding Protein. Biochem. J. 2006, 396, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Del Pino, J.; Jiménez, J.L.; Ventoso, I.; Castelló, A.; Muñoz-Fernández, M.Á.; de Haro, C.; Berlanga, J.J. GCN2 Has Inhibitory Effect on Human Immunodeficiency Virus-1 Protein Synthesis and Is Cleaved upon Viral Infection. PLoS ONE 2012, 7, e47272. [Google Scholar] [CrossRef]
- Rivière, Y.; Blank, V.; Kourilsky, P.; Israël, A. Processing of the Precursor of NF-ΚB by the HIV-1 Protease during Acute Infection. Nature 1991, 350, 625–626. [Google Scholar] [CrossRef]
- Jeremiah, S.S.; Miyakawa, K.; Matsunaga, S.; Nishi, M.; Kudoh, A.; Takaoka, A.; Sawasaki, T.; Ryo, A. Cleavage of TANK-Binding Kinase 1 by HIV-1 Protease Triggers Viral Innate Immune Evasion. Front. Microbiol. 2021, 12, 643407. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amorim, R.; Costa, S.M.; Cavaleiro, N.P.; da Silva, E.E.; da Costa, L.J. HIV-1 Transcripts Use IRES-Initiation under Conditions Where Cap-Dependent Translation Is Restricted by Poliovirus 2A Protease. PLoS ONE 2014, 9, e88619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelló, A.; Franco, D.; Moral-López, P.; Berlanga, J.J.; Álvarez, E.; Wimmer, E.; Carrasco, L. HIV-1 Protease Inhibits Cap-and Poly(A)-Dependent Translation upon EIF4GI and PABP Cleavage. PLoS ONE 2009, 4, e7997. [Google Scholar] [CrossRef] [Green Version]
- Brasey, A.; Lopez-Lastra, M.; Ohlmann, T.; Beerens, N.; Berkhout, B.; Darlix, J.-L.; Sonenberg, N. The Leader of Human Immunodeficiency Virus Type 1 Genomic RNA Harbors an Internal Ribosome Entry Segment That Is Active during the G2/M Phase of the Cell Cycle. J. Virol. 2003, 77, 3939–3949. [Google Scholar] [CrossRef] [Green Version]
- Vallejos, M.; Carvajal, F.; Pino, K.; Navarrete, C.; Ferres, M.; Huidobro-Toro, J.P.; Sargueil, B.; López-Lastra, M. Functional and Structural Analysis of the Internal Ribosome Entry Site Present in the MRNA of Natural Variants of the HIV-1. PLoS ONE 2012, 7, e35031. [Google Scholar] [CrossRef]
- Buck, C.B.; Shen, X.; Egan, M.A.; Pierson, T.C.; Walker, C.M.; Siliciano, R.F. The Human Immunodeficiency Virus Type 1 Gag Gene Encodes an Internal Ribosome Entry Site. J. Virol. 2001, 75, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Plank, T.D.M.; Whitehurst, J.T.; Kieft, J.S. Cell Type Specificity and Structural Determinants of IRES Activity from the 5’ Leaders of Different HIV-1 Transcripts. Nucleic Acids Res. 2013, 41, 6698–6714. [Google Scholar] [CrossRef] [Green Version]
- Baboonian, C.; Dalgleish, A.; Bountiff, L.; Gross, J.; Oroszlan, S.; Rickett, G.; Smith-Burchnell, C.; Troke, P.; Merson, J. HIV-1 Proteinase Is Required for Synthesis of pro-Viral DNA. Biochem. Biophys. Res. Commun. 1991, 179, 17–24. [Google Scholar] [CrossRef]
- Jacobsen, H.; Ahlborn-laake, L.; Gugel, R.; Mous, J.A.N. Progression of Early Steps of Human Immunodeficiency Virus Type 1 Replication in the Presence of an Inhibitor of Viral Protease A-I. J. Virol. 1992, 66, 5087–5091. [Google Scholar] [CrossRef] [Green Version]
- Nagy, K.; Young, M.; Baboonian, C.; Merson, J.; Whittle, P.; Oroszlan, S. Antiviral Activity of Human Immunodeficiency Virus Type 1 Protease Inhibitors in a Single Cycle of Infection: Evidence for a Role of Protease in the Early Phase. J. Virol. 1994, 68, 757–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, A.H.; Manchester, M.; Smith, T.; Yang, Y.L.; Swanstrom, R. Conditional Human Immunodeficiency Virus Type 1 Protease Mutants Show No Role for the Viral Protease Early in Virus Replication. J. Virol. 1996, 70, 5840–5844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, H.; Maeda, Y.; Mitsuya, H. HIV-1 Protease Does Not Play a Critical Role in the Early Stages of HIV-1 Infection. Antivir. Res. 1997, 36, 107–113. [Google Scholar] [CrossRef]
- Stefanidou, M.; Herrera, C.; Armanasco, N.; Shattock, R.J. Saquinavir Inhibits Early Events Associated with Establishment of HIV-1 Infection: Potential Role for Protease Inhibitors in Prevention. Antimicrob. Agents Chemother. 2012, 56, 4381–4390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monette, A.; Valiente-Echeverría, F.; Rivero, M.; Cohen, É.A.; Lopez-Lastra, M. Dual Mechanisms of Translation Initiation of the Full-Length HIV-1 MRNA Contribute to Gag Synthesis. PLoS ONE 2013, 8, e68108. [Google Scholar] [CrossRef] [Green Version]
- Vichalkovski, A.; Gresko, E.; Cornils, H.; Hergovich, A.; Schmitz, D.; Hemmings, B.A. NDR Kinase Is Activated by RASSF1A/MST1 in Response to Fas Receptor Stimulation and Promotes Apoptosis. Curr. Biol. 2008, 18, 1889–1895. [Google Scholar] [CrossRef] [Green Version]
- Gougeon, M.L. Apoptosis as an HIV Strategy to Escape Immune Attack. Nat. Rev. Immunol. 2003, 3, 392–404. [Google Scholar] [CrossRef]
- Baum, E.Z.; Bebernitz, G.A.; Gluzman, Y. Isolation of Mutants of Human Immunodeficiency Virus Protease Based on the Toxicity of the Enzyme in Escherichia Coli. Proc. Natl. Acad. Sci. USA 1990, 87, 5573–5577. [Google Scholar] [CrossRef] [Green Version]
- M’Barek, N.B.; Audoly, G.; Raoult, D.; Gluschankof, P. HIV-2 Protease Resistance Defined in Yeast Cells. Retrovirology 2006, 3, 58. [Google Scholar] [CrossRef] [Green Version]
- Konvalinka, J.; Litterst, M.A.; Welker, R.; Kottler, H.; Rippmann, F.; Heuser, A.M.; Kräusslich, H.G. An Active-Site Mutation in the Human Immunodeficiency Virus Type 1 Proteinase (PR) Causes Reduced PR Activity and Loss of PR-Mediated Cytotoxicity without Apparent Effect on Virus Maturation and Infectivity. J. Virol. 1995, 69, 7180–7186. [Google Scholar] [CrossRef] [Green Version]
- Rumlová, M.; Křížová, I.; Keprová, A.; Hadravová, R.; Doležal, M.; Strohalmová, K.; Pichová, I.; Hájek, M.; Ruml, T. HIV-1 Protease-Induced Apoptosis. Retrovirology 2014, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Rumlová, M.; Křížová, I.; Zelenka, J.; Weber, J.; Ruml, T. Does BCA3 Play a Role in the HIV-1 Replication Cycle? Viruses 2018, 10, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartier, C.; Hemonnot, B.; Gay, B.; Bardy, M.; Sanchiz, C.; Devaux, C.; Briant, L. Active CAMP-Dependent Protein Kinase Incorporated within Highly Purified HIV-1 Particles Is Required for Viral Infectivity and Interacts with Viral Capsid Protein. J. Biol. Chem. 2003, 278, 35211–35219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baichwal, V.R.; Baeuerle, P.A. Apoptosis: Activate NF-ΚB or Die? Curr. Biol. 1997, 7, R94–R96. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Ong, N.; An, H.; Zheng, Y. The Emerging Roles of NDR1/2 in Infection and Inflammation. Front. Immunol. 2020, 11, 534. [Google Scholar] [CrossRef] [Green Version]
- Wen, M.; Ma, X.; Cheng, H.; Jiang, W.; Xu, X.; Zhang, Y.; Zhang, Y.; Guo, Z.; Yu, Y.; Xu, H.; et al. Stk38 Protein Kinase Preferentially Inhibits TLR9-Activated Inflammatory Responses by Promoting MEKK2 Ubiquitination in Macrophages. Nat. Commun. 2015, 6, 7617. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Wang, D.; Li, N.; Gao, P.; Zhang, M.; Zhang, Y. Hippo Kinase NDR2 Inhibits IL-17 Signaling by Promoting Smurf1-Mediated MEKK2 Ubiquitination and Degradation. Mol. Immunol. 2019, 105, 131–136. [Google Scholar] [CrossRef]
- Liu, Z.; Qin, Q.; Wu, C.; Li, H.; Shou, J.; Yang, Y.; Gu, M.; Ma, C.; Lin, W.; Zou, Y.; et al. Downregulated NDR1 Protein Kinase Inhibits Innate Immune Response by Initiating an MiR146a-STAT1 Feedback Loop. Nat. Commun. 2018, 9, 2789. [Google Scholar] [CrossRef] [Green Version]
- Beignon, A.S.; McKenna, K.; Skoberne, M.; Manches, O.; DaSilva, I.; Kavanagh, D.G.; Larsson, M.; Gorelick, R.J.; Lifson, J.D.; Bhardwaj, N. Endocytosis of HIV-1 Activates Plasmacytoid Dendritic Cells via Toll-like Receptor-Viral RNA Interactions. J. Clin. Investig. 2005, 115, 3265–3275. [Google Scholar] [CrossRef] [Green Version]
- Jakobsen, M.R.; Bak, R.O.; Andersen, A.; Berg, R.K.; Jensen, S.B.; Jin, T.; Laustsen, A.; Hansen, K.; Østergaard, L.; Fitzgerald, K.A.; et al. IFI16 Senses DNA Forms of the Lentiviral Replication Cycle and Controls HIV-1 Replication. Proc. Natl. Acad. Sci. USA 2013, 110, E4571–E4580. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Wu, J.; Wu, Y.T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP Synthase Is an Innate Immune Sensor of HIV and Other Retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahaye, X.; Gentili, M.; Silvin, A.; Conrad, C.; Picard, L.; Jouve, M.; Zueva, E.; Maurin, M.; Nadalin, F.; Knott, G.J.; et al. NONO Detects the Nuclear HIV Capsid to Promote CGAS-Mediated Innate Immune Activation. Cell 2018, 175, 488–501. [Google Scholar] [CrossRef] [Green Version]
- Yoh, S.M.; Schneider, M.; Seifried, J.; Soonthornvacharin, S.; Akleh, R.E.; Olivieri, K.C.; De Jesus, P.D.; Ruan, C.; De Castro, E.; Ruiz, P.A.; et al. PQBP1 Is a Proximal Sensor of the CGAS-Dependent Innate Response to HIV-1. Cell 2015, 161, 1293–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Shi, H.; Wu, J.; Zhang, X.; Sun, L.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Containing Mixed Phosphodiester Linkages Is an Endogenous High-Affinity Ligand for STING. Mol. Cell 2013, 51, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Solis, M.; Nakhaei, P.; Jalalirad, M.; Lacoste, J.; Douville, R.; Arguello, M.; Zhao, T.; Laughrea, M.; Wainberg, M.A.; Hiscott, J. RIG-I-Mediated Antiviral Signaling Is Inhibited in HIV-1 Infection by a Protease-Mediated Sequestration of RIG-I. J. Virol. 2011, 85, 1224–1236. [Google Scholar] [CrossRef] [Green Version]
- Jurczyszak, D.; Zhang, W.; Terry, S.N.; Kehrer, T.; Bermúdez González, M.C.; McGregor, E.; Mulder, L.C.F.; Eckwahl, M.J.; Pan, T.; Simon, V. HIV Protease Cleaves the Antiviral M6A Reader Protein YTHDF3 in the Viral Particle. PLoS Pathog. 2020, 16, e1008305. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, D.; Richards, C.M.; Carpenter, M.A.; Wang, J.; Ikeda, T.; Becker, J.T.; Cheng, A.Z.; McCann, J.L.; Shaban, N.M.; Salamango, D.J.; et al. Genetic and Mechanistic Basis for APOBEC3H Alternative Splicing, Retrovirus Restriction, and Counteraction by HIV-1 Protease. Nat. Commun. 2018, 9, 4137. [Google Scholar] [CrossRef] [Green Version]
- Engeland, C.E.; Oberwinkler, H.; Schumann, M.; Krause, E.; Muller, G.A.; Krausslich, H.-G. The Cellular Protein Lyric Interacts with HIV-1 Gag. J. Virol. 2011, 85, 13322–13332. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Zhang, Y.; Xu, Q.; Zheng, H.; Wu, X.; Qiu, J.; Zhang, Z.; Wang, W.; Shao, Y.; Xing, H.Q. HIV-1 Tat Inhibits EAAT-2 through AEG-1 Upregulation in Models of HIV-Associated Neurocognitive Disorder. Oncotarget 2017, 8, 39922–39934. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.P.; Koyuncu, O.O.; Enquist, L.W. Subversion of the Actin Cytoskeleton during Viral Infection. Nat. Rev. Microbiol. 2011, 9, 427–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honer, B.; Shoeman, R.L.; Traub, P. Human Immunodeficiency Virus Type 1 Protease Microinjected into Cultured Human Skin Fibroblasts Cleaves Vimentin and Affects Cytoskeletal and Nuclear Architecture. J. Cell Sci. 1991, 100, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Shoeman, R.L.; Sachse, C.; Höner, B.; Mothes, E.; Kaufmann, M.; Traub, P. Cleavage of Human and Mouse Cytoskeletal and Sarcomeric Proteins by Human Immunodeficiency Virus Type 1 Protease: Actin, Desmin, Myosin, and Tropomyosin. Am. J. Pathol. 1993, 142, 221–230. [Google Scholar] [PubMed]
- Adams, L.D.; Tomasselli, A.G.; Robbins, P.; Moss, B.; Heinrikson, R.L. HIV-1 Protease Cleaves Actin During Acute Infection of Human T-Lymphocytes. AIDS Res. Hum. Retroviruses 1992, 8, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Stephens, C.; Naghavi, M.H. The Host Cytoskeleton: A Key Regulator of Early HIV-1 Infection. FEBS J. 2022. [CrossRef]
- Matarrese, P.; Malorni, W. Human Immunodeficiency Virus (HIV)-1 Proteins and Cytoskeleton: Partners in Viral Life and Host Cell Death. Cell Death Differ. 2005, 12, 932–941. [Google Scholar] [CrossRef] [Green Version]
- Nyamweya, S.; Hegedus, A.; Jaye, A.; Rowland-Jones, S.; Flanagan, K.L.; Macallan, D.C. Comparing HIV-1 and HIV-2 Infection: Lessons for Viral Immunopathogenesis. Rev. Med. Virol. 2013, 23, 221–240. [Google Scholar] [CrossRef]
- Gustchina, A.; Weber, I.T. Comparative Analysis of the Sequences and Structures of HIV-1 and HIV-2 Proteases. Proteins Struct. Funct. Bioinform. 1991, 10, 325–339. [Google Scholar] [CrossRef]
- Tözsér, J.; Weber, I.T.; Gustchina, A.; Bláha, I.; Copeland, T.D.; Oroszlan, S.; Louis, J.M. Kinetic and Modeling Studies of S3-S3′ Subsites of HIV Proteinases. Biochemistry 1992, 31, 4793–4800. [Google Scholar] [CrossRef]
- Álvarez, E.; Menéndez-Arias, L.; Carrasco, L. The Eukaryotic Translation Initiation Factor 4GI Is Cleaved by Different Retroviral Proteases. J. Virol. 2003, 77, 12392–12400. [Google Scholar] [CrossRef] [Green Version]
- Roberts, N.A.; Martin, J.A.; Kinchington, D.; Broadhurst, A.V.; Craig, J.C.; Duncan, I.B.; Galpin, S.A.; Handa, B.K.; Kay, J.; Kröhn, A.; et al. Rational Design of Peptide-Based HIV Proteinase Inhibitors. Science 1990, 248, 358–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, S.R.; Strohbach, J.W.; Tommasi, R.A.; Aristoff, P.A.; Johnson, P.D.; Skulnick, H.I.; Dolak, L.A.; Seest, E.P.; Tomich, P.K.; Bohanon, M.J.; et al. Tipranavir (PNU-140690): A Potent, Orally Bioavailable Nonpeptidic HIV Protease Inhibitor of the 5,6-Dihydro-4-Hydroxy-2-Pyrone Sulfonamide Class. J. Med. Chem. 1998, 41, 3467–3476. [Google Scholar] [CrossRef] [PubMed]
- Mallolas, J. Darunavir Stands Up as Preferred HIV Protease Inhibitor. Aids Rev. 2017, 19, 105–112. [Google Scholar]
- Consolidated Guidelines on HIV Prevention, Testing, Treatment, Service Delivery and Monitoring: Recommendations for a Public Health Approach; World Health Organization: Geneva, Switzerland, 2021.
- Wensing, A.M.J.; van Maarseveen, N.M.; Nijhuis, M. Fifteen Years of HIV Protease Inhibitors: Raising the Barrier to Resistance. Antivir. Res. 2010, 85, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Doyon, L.; Croteau, G.; Thibeault, D.; Poulin, F.; Pilote, L.; Lamarre, D. Second Locus Involved in Human Immunodeficiency Virus Type 1 Resistance to Protease Inhibitors. J. Virol. 1996, 70, 3763–3769. [Google Scholar] [CrossRef] [Green Version]
- Fun, A.; Wensing, A.M.J.; Verheyen, J.; Nijhuis, M. Human Immunodeficiency Virus Gag and Protease: Partners in Resistance. Retrovirology 2012, 9, 63. [Google Scholar] [CrossRef] [Green Version]
- Nijhuis, M.; Van Maarseveen, N.M.; Lastere, S.; Schipper, P.; Coakley, E.; Glass, B.; Rovenska, M.; De Jong, D.; Chappey, C.; Goedegebuure, I.W.; et al. A Novel Substrate-Based HIV-1 Protease Inhibitor Drug Resistance Mechanism. PLoS Med. 2007, 4, e36. [Google Scholar] [CrossRef]
- Maguire, M.F.; Guinea, R.; Griffin, P.; Macmanus, S.; Elston, R.C.; Wolfram, J.; Richards, N.; Hanlon, M.H.; Porter, D.J.T.; Wrin, T.; et al. Changes in Human Immunodeficiency Virus Type 1 Gag at Positions L449 and P453 Are Linked to I50V Protease Mutants in Vivo and Cause Reduction of Sensitivity to Amprenavir and Improved Viral Fitness in Vitro. J. Virol. 2002, 76, 7398–7406. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, B.; Louis, J.M.; Reed, C.C.; Adomat, J.M.; Krouse, J.; Wang, Y.F.; Harrison, R.W.; Weber, I.T. Structural and Kinetic Analysis of Drug Resistant Mutants of HIV-1 Protease. Eur. J. Biochem. 1999, 263, 238–244. [Google Scholar] [CrossRef]
- Kožíšek, M.; Henke, S.; Šašková, K.G.; Jacobs, G.B.; Schuch, A.; Buchholz, B.; Müller, V.; Kräusslich, H.G.; Řezáčová, P.; Konvalinka, J.; et al. Mutations in HIV-1 Gag and Pol Compensate for the Loss of Viral Fitness Caused by a Highly Mutated Protease. Antimicrob. Agents Chemother. 2012, 56, 4320–4330. [Google Scholar] [CrossRef] [Green Version]
- Su, C.T.T.; Koh, D.W.S.; Gan, S.K.E. Reviewing HIV-1 Gag Mutations in Protease Inhibitors Resistance: Insights for Possible Novel Gag Inhibitor Designs. Molecules 2019, 24, 3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolli, M.; Lastere, S.; Schiffer, C.A. Co-Evolution of Nelfinavir-Resistant HIV-1 Protease and the P1-P6 Substrate. Virology 2006, 347, 405–409. [Google Scholar] [CrossRef]
- Côté, H.C.F.; Brumme, Z.L.; Harrigan, P.R. Human Immunodeficiency Virus Type 1 Protease Cleavage Site Mutations Associated with Protease Inhibitor Cross-Resistance Selected by Indinavir, Ritonavir, and/or Saquinavir. J. Virol. 2001, 75, 589–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbour, J.D.; Wrin, T.; Grant, R.M.; Martin, J.N.; Segal, M.R.; Petropoulos, C.J.; Deeks, S.G. Evolution of Phenotypic Drug Susceptibility and Viral Replication Capacity during Long-Term Virologic Failure of Protease Inhibitor Therapy in Human Immunodeficiency Virus-Infected Adults. J. Virol. 2002, 76, 11104–11112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Lu, W.; Li, F. Pharmacological Intervention of HIV-1 Maturation. Acta Pharm. Sin. B 2015, 5, 493–499. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Cleavage Sequence |
|---|---|
| HIV-1 cleavage sites | |
| Gag (MA-CA) | SQNY^PIVQ [39] |
| Gag (CA-SP1) | ARVL^AEAM [39] |
| Gag (SP1-NC) | ATIM^MQRG [39] |
| Gag (NC-SP2) | RQAN^FLGK [39] |
| Gag (SP2-p6) | PGNF^LQSR [39] |
| Gag-Pol (TF-PR) | SFNF^PQIT [39] |
| Gag-Pol (PR-RT) | TLNF^PISP [39] |
| Gag-Pol (RT-RH) | AETF^YVDG [39] |
| Gag-Pol (RH-IN) | RKIL^FLDG [39] |
| Host protein cleavage sites | |
| RIPK1 | PQVL^YQNN [45] |
| NDR1 | KDWV^FINY [46] |
| NDR2 | KDWV^FlNY [46] |
| proCaspase8 | PKVF^FIQA [47] |
| eIF4GI | KIIA^TVLM [48] |
| ATVL^MTED [48] | |
| RFSA^LQQA [48] | |
| eIF3d | RRNM^LQFN [44] |
| Bcl2 | RRDF^AEMS [49] |
| PABP | GFVS^FERH [50] |
| PRVM^STQR [50] | |
| GCN2 | GQDY^VETV [51] |
| NF-κB1 | HYGF^PTYG [52] |
| TBK1 | SNTL^VEMT [53] |
| Protease Inhibitor | FDA Approval (Year) | Notable Resistance Mutations |
|---|---|---|
| Saquinavir | 1995 | G48V, L90M |
| Ritonavir | 1996 | Used as pharmacokinetic enhancer |
| Indinavir | 1996 | M46I/L, V82A/F/T, I84V |
| Nelfinavir | 1997 | D30N, L90M |
| Fosamprenavir | 1999 | I50V, I84V |
| Lopinavir | 2000 | V32I, I47V/A, L76V, V82A/F/T/S |
| Atazanavir | 2003 | I50L, I84V, N88S |
| Tipranavir | 2005 | I47V, Q58E, T74P, V82L/T, N83, I84V |
| Darunavir | 2006 | I47V, I50V, I54M/L, V76V, I84V |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Centazzo, M.; Manganaro, L.; Alvisi, G. Cellular Targets of HIV-1 Protease: Just the Tip of the Iceberg? Viruses 2023, 15, 712. https://doi.org/10.3390/v15030712
Centazzo M, Manganaro L, Alvisi G. Cellular Targets of HIV-1 Protease: Just the Tip of the Iceberg? Viruses. 2023; 15(3):712. https://doi.org/10.3390/v15030712
Chicago/Turabian StyleCentazzo, Matteo, Lara Manganaro, and Gualtiero Alvisi. 2023. "Cellular Targets of HIV-1 Protease: Just the Tip of the Iceberg?" Viruses 15, no. 3: 712. https://doi.org/10.3390/v15030712
APA StyleCentazzo, M., Manganaro, L., & Alvisi, G. (2023). Cellular Targets of HIV-1 Protease: Just the Tip of the Iceberg? Viruses, 15(3), 712. https://doi.org/10.3390/v15030712